
FNIP1 Deficiency: Pathophysiology and Clinical Manifestations of a Rare Syndromic Primary Immunodeficiency


Abstract
1. Introduction
2. Structure and Function of FNIP1
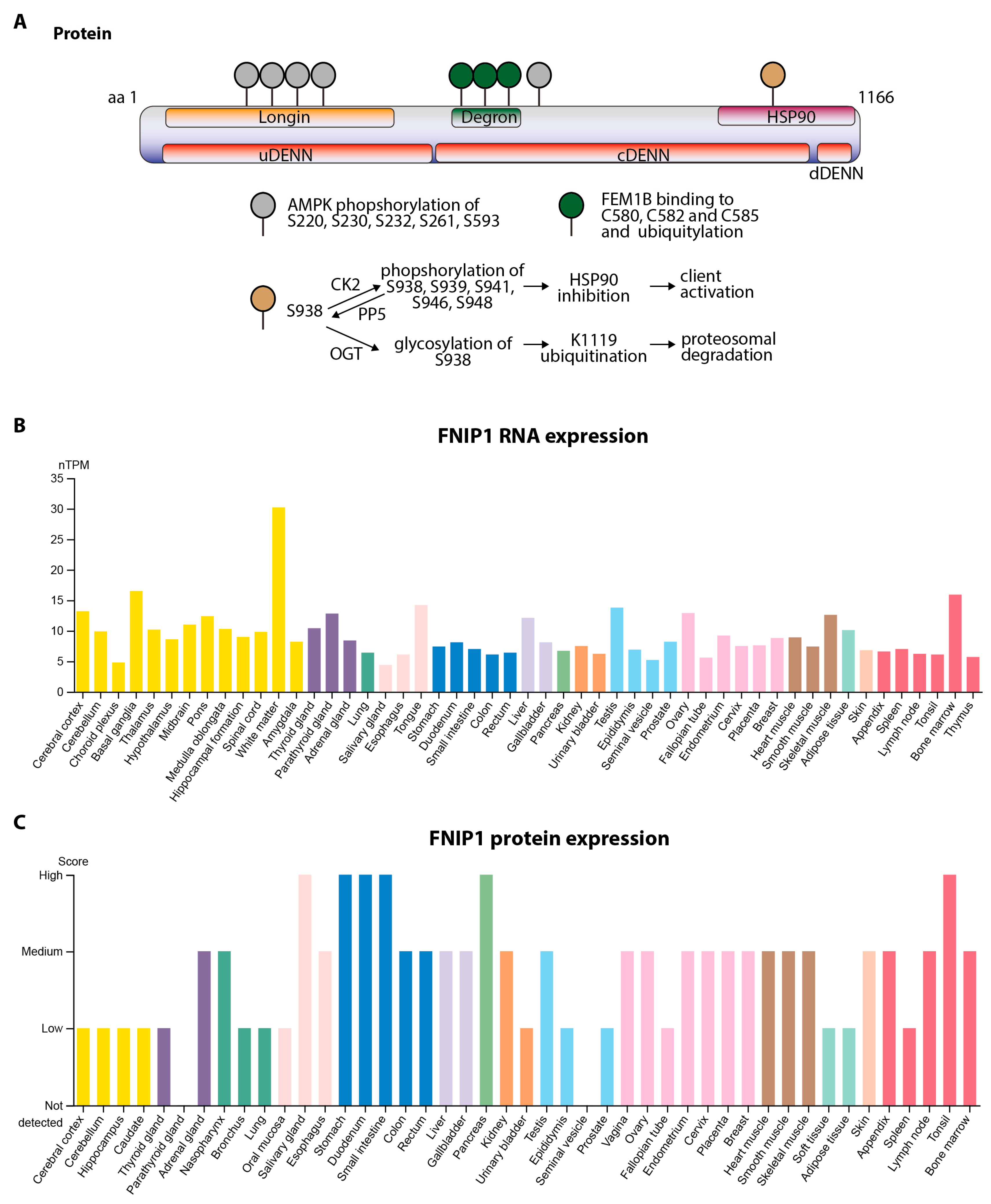
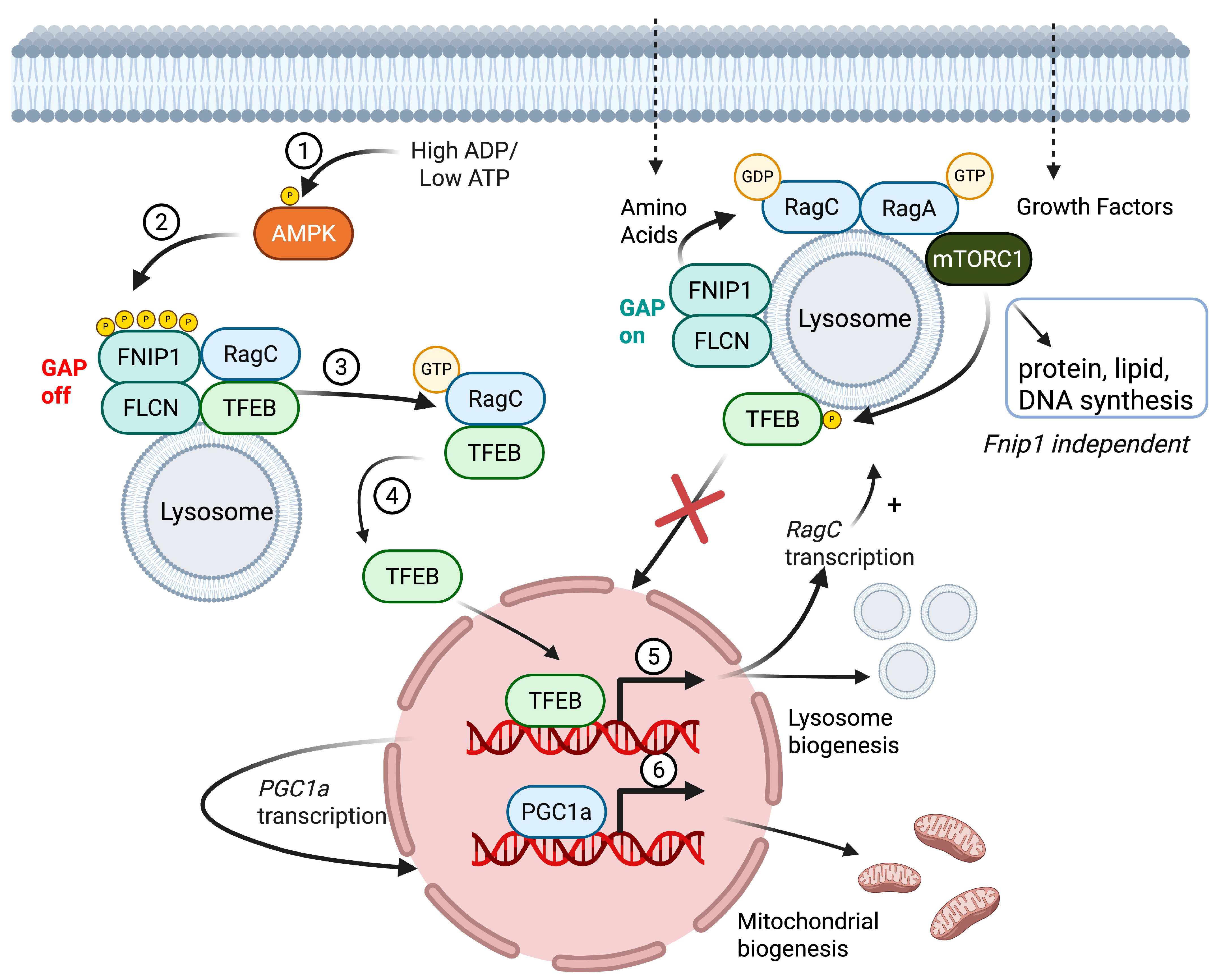
2.1. Protein Domains, Post-Translational Modifications, Subcellular Localization, and Functional Roles of FNIP1
2.2. Transcript Variants and Tissue-Specific Expression
2.3. Fnip1−/− Models
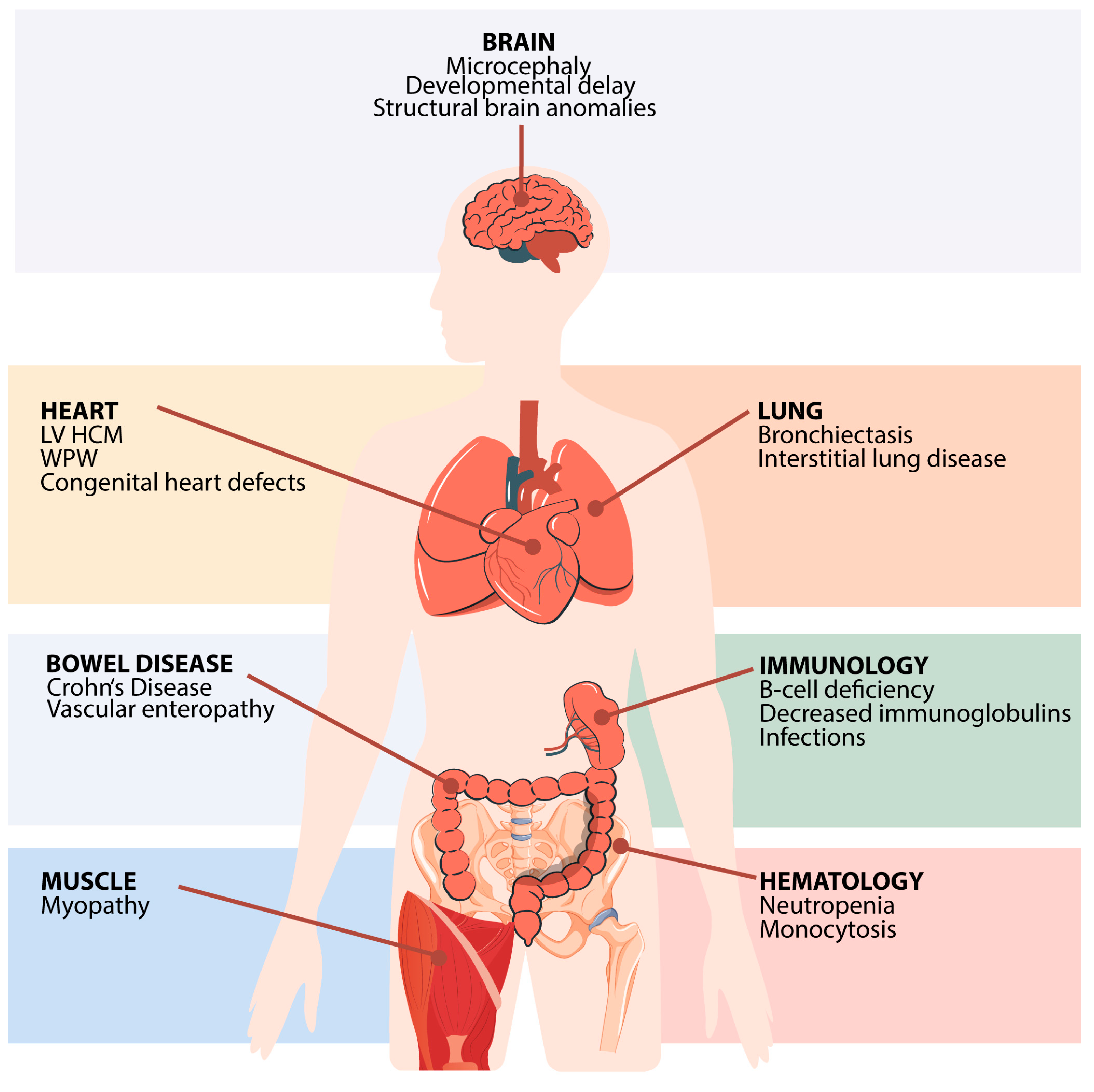
3. Clinical Features of FNIP1 Deficiency
3.1. Germline FNIP1 Mutations and Disease
3.2. Hematological and Immunological Findings
3.3. Heart Disease
3.4. Infectious Susceptibility
3.5. Pulmonary Involvement
3.6. Muscular Abnormalities
3.7. Central Nervous System (CNS) and Dysmorphic Features
3.8. Gastrointestinal and Renal Manifestations
3.9. Treatment and Clinical Outcomes
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- IJspeert, H.; Edwards, E.S.J.; O’Hehir, R.E.; Dalm, V.A.S.H.; van Zelm, M.C. Update on inborn errors of immunity. J. Allergy Clin. Immunol. 2024, 155, 470–751. [Google Scholar] [CrossRef] [PubMed]
- Saettini, F.; Herriot, R.; Prada, E.; Nizon, M.; Zama, D.; Marzollo, A.; Romaniouk, I.; Lougaris, V.; Cortesi, M.; Morreale, A.; et al. Prevalence of Immunological Defects in a Cohort of 97 Rubinstein-Taybi Syndrome Patients. J. Clin. Immunol. 2020, 40, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Saettini, F.; Guerra, F.; Mauri, M.; Salter, C.G.; Adam, M.P.; Adams, D.; Baple, E.L.; Barredo, E.; Bhatia, S.; Borkhardt, A.; et al. Biallelic PI4KA Mutations Disrupt B-Cell Metabolism and Cause B-Cell Lymphopenia and Hypogammaglobulinemia. J. Clin. Immunol. 2024, 45, 15. [Google Scholar] [CrossRef]
- Rossini, L.; Ricci, S.; Montin, D.; Azzari, C.; Gambineri, E.; Tellini, M.; Conti, F.; Pession, A.; Saettini, F.; Naviglio, S.; et al. Immunological Aspects of Kabuki Syndrome: A Retrospective Multicenter Study of the Italian Primary Immunodeficiency Network (IPINet). J. Clin. Immunol. 2024, 44, 105. [Google Scholar] [CrossRef] [PubMed]
- Saettini, F.; Guerra, F.; Fazio, G.; Bugarin, C.; McMillan, H.J.; Ohtake, A.; Ardissone, A.; Itoh, M.; Giglio, S.; Cappuccio, G.; et al. Antibody Deficiency in Patients with Biallelic KARS1 Mutations. J. Clin. Immunol. 2023, 43, 2115–2125. [Google Scholar] [CrossRef]
- Niehues, T.; Özgür, T.T.; Bickes, M.; Waldmann, R.; Schöning, J.; Bräsen, J.; Hagel, C.; Ballmaier, M.; Klusmann, J.-H.; Niedermayer, A.; et al. Mutations of the gene FNIP1 associated with a syndromic autosomal recessive immunodeficiency with cardiomyopathy and pre-excitation syndrome. Eur. J. Immunol. 2020, 50, 1078–1080. [Google Scholar] [CrossRef]
- Saettini, F.; Poli, C.; Vengoechea, J.; Bonanomi, S.; Orellana, J.C.; Fazio, G.; Rodriguez, F.H.; Noguera, L.P.; Booth, C.; Jarur-Chamy, V.; et al. Absent B cells, agammaglobulinemia, and hypertrophic cardiomyopathy in folliculin-interacting protein 1 deficiency. Blood 2021, 137, 493–499. [Google Scholar] [CrossRef]
- Tangye, S.G.; Al-Herz, W.; Bousfiha, A.; Cunningham-Rundles, C.; Franco, J.L.; Holland, S.M.; Klein, C.; Morio, T.; Oksenhendler, E.; Picard, C.; et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2022, 42, 1473–1507. [Google Scholar] [CrossRef]
- Moreno-Corona, N.; Valagussa, A.; Thouenon, R.; Fischer, A.; Kracker, S. A Case Report of Folliculin-Interacting Protein 1 Deficiency. J. Clin. Immunol. 2023, 43, 1751–1753. [Google Scholar] [CrossRef]
- Ulaş, S.; Naiboğlu, S.; Özyilmaz, İ.; Öztürk Demir, A.G.; Turan, I.; Yuzkan, S.; Ayaz, A.; Çeliksoy, M.H. Clinical and Immunologic Features of a Patient with Homozygous FNIP1 Variant. J. PediatrHematol. Oncol. 2024, 46, e472–e475. [Google Scholar] [CrossRef]
- Spivak, I.; Lev, A.; Simon, A.J.; Barel, O.; Somekh, I.; Somech, R. A novel mutation in FNIP1 associated with a syndromic immunodeficiency and cardiomyopathy. Immunogenetics 2024, 77, 2. [Google Scholar] [CrossRef]
- Park, H.; Staehling, K.; Tsang, M.; Appleby, M.W.; Brunkow, M.E.; Margineantu, D.; Hockenbery, D.M.; Habib, T.; Liggitt, H.D.; Carlson, G.; et al. Disruption of Fnip1 reveals a metabolic checkpoint controlling B lymphocyte development. Immunity 2012, 36, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Keller, J.R.; Sun, H.W.; Resch, W.; Kuchen, S.; Suh, H.C.; Hasumi, H.; Hasumi, Y.; Kieffer-Kwon, K.R.; Gallego Gonzalez, C.; et al. The folliculin-FNIP1 pathway deleted in human Birt-Hogg-Dube syndrome is required for murine B-cell development. Blood 2012, 120, 1254–1261. [Google Scholar] [CrossRef]
- Malik, N.; Ferreira, B.I.; Hollstein, P.E.; Curtis, S.D.; Trefts, E.; Weiser Novak, S.; Yu, J.; Gilson, R.; Hellberg, K.; Fang, L.; et al. Induction of lysosomal and mitochondrial biogenesis by AMPK phosphorylation of FNIP1. Science 2023, 380, eabj5559. [Google Scholar] [CrossRef]
- Jansen, R.M.; Hurley, J.H. Longin domain GAP complexes in nutrient signalling, membrane traffic and neurodegeneration. FEBS Lett. 2023, 597, 750–761. [Google Scholar] [CrossRef] [PubMed]
- Zeng, F.; Cao, J.; Li, W.; Zhou, Y.; Yuan, X. FNIP1: A key regulator of mitochondrial function. Biomed. Pharmacother. 2024, 177, 117146. [Google Scholar] [CrossRef]
- Petit, C.S.; Roczniak-Ferguson, A.; Ferguson, S.M. Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of Rag GTPases. J. Cell Biol. 2013, 202, 1107–1122. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.E.; Fromm, S.A.; Fu, Y.; Yokom, A.L.; Kim, D.J.; Thelen, A.M.; Young, L.N.; Lim, C.Y.; Samelson, A.J.; Hurley, J.H.; et al. Structural mechanism of a Rag GTPase activation checkpoint by the lysosomal folliculin complex. Science 2019, 366, 971–977. [Google Scholar] [CrossRef]
- Mao, Y.; Jin, Z.; Yang, J.; Xu, D.; Zhao, L.; Kiram, A.; Yin, Y.; Zhou, D.; Sun, Z.; Xiao, L.; et al. Muscle-bone cross-talk through the FNIP1-TFEB-IGF2 axis is associated with bone metabolism in human and mouse. Sci. Transl. Med. 2024, 16, eadk9811. [Google Scholar] [CrossRef]
- Xiao, L.; Yin, Y.; Sun, Z.; Liu, J.; Jia, Y.; Yang, L.; Mao, Y.; Peng, S.; Xie, Z.; Fang, L.; et al. AMPK phosphorylation of FNIP1 (S220) controls mitochondrial function and muscle fuel utilization during exercise. Sci. Adv. 2024, 10, eadj2752. [Google Scholar] [CrossRef]
- Manford, A.G.; Rodríguez-Pérez, F.; Shih, K.Y.; Shi, Z.; Berdan, C.A.; Choe, M.; Titov, D.V.; Nomura, D.K.; Rape, M. A Cellular Mechanism to Detect and Alleviate Reductive Stress. Cell 2020, 183, 46–61.e21. [Google Scholar] [CrossRef] [PubMed]
- Manford, A.G.; Mena, E.L.; Shih, K.Y.; Gee, C.L.; McMinimy, R.; Martínez-González, B.; Sherriff, R.; Lew, B.; Zoltek, M.; Rodríguez-Pérez, F.; et al. Structural basis and regulation of the reductive stress response. Cell 2021, 184, 5375–5390.e16. [Google Scholar] [CrossRef] [PubMed]
- Woodford, M.R.; Dunn, D.M.; Blanden, A.R.; Capriotti, D.; Loiselle, D.; Prodromou, C.; Panaretou, B.; Hughes, P.F.; Smith, A.; Ackerman, W.; et al. The FNIP co-chaperones decelerate the Hsp90 chaperone cycle and enhance drug binding. Nat. Commun. 2016, 7, 12037. [Google Scholar] [CrossRef] [PubMed]
- Sager, R.A.; Woodford, M.R.; Backe, S.J.; Makedon, A.M.; Baker-Williams, A.J.; DiGregorio, B.T.; Loiselle, D.R.; Haystead, T.A.; Zachara, N.E.; Prodromou, C.; et al. Post-translational Regulation of FNIP1 Creates a Rheostat for the Molecular Chaperone Hsp90. Cell Rep. 2019, 26, 1344–1356.e5. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Karlsson, M.J.; Zhong, W.; Tebani, A.; Pou, C.; Mikes, J.; Lakshmikanth, T.; Forsström, B.; Edfors, F.; Odeberg, J.; et al. A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science 2019, 366, eaax9198. [Google Scholar] [CrossRef]
- Ramirez, J.A.; Iwata, T.; Park, H.; Tsang, M.; Kang, J.; Cui, K.; Lakshmikanth, T.; Forsström, B.; Edfors, F.; Odeberg, J.; et al. Folliculin Interacting Protein 1 Maintains Metabolic Homeostasis during B Cell Development by Modulating AMPK, mTORC1, and TFE3. J. Immunol. 2019, 203, 2899–2908. [Google Scholar] [CrossRef]
- Park, H.; Tsang, M.; Iritani, B.M.; Bevan, M.J. Metabolic regulator Fnip1 is crucial for iNKT lymphocyte development. Proc. Natl. Acad. Sci. USA 2014, 111, 7066–7071. [Google Scholar] [CrossRef]
- Siggs, O.M.; Stockenhuber, A.; Deobagkar-Lele, M.; Bull, K.R.; Crockford, T.L.; Kingston, B.L.; Crawford, G.; Anzilotti, C.; Steeples, V.; Ghaffari, S.; et al. Mutation of Fnip1 is associated with B-cell deficiency, cardiomyopathy, and elevated AMPK activity. Proc. Natl. Acad. Sci. USA 2016, 113, E3706–E3715. [Google Scholar] [CrossRef]
- Reyes, N.L.; Banks, G.B.; Tsang, M.; Margineantu, D.; Gu, H.; Djukovic, D.; Chan, J.; Torres, M.; Liggitt, H.D.; Hirenallur-S, D.K.; et al. Fnip1 regulates skeletal muscle fiber type specification, fatigue resistance, and susceptibility to muscular dystrophy. Proc. Natl. Acad. Sci. USA 2015, 112, 424–429. [Google Scholar] [CrossRef]
- Yin, Y.; Xu, D.; Mao, Y.; Xiao, L.; Sun, Z.; Liu, J.; Zhou, D.; Xu, Z.; Liu, L.; Fu, T.; et al. FNIP1 regulates adipocyte browning and systemic glucose homeostasis in mice by shaping intracellular calcium dynamics. J. Exp. Med. 2022, 219, e20212491. [Google Scholar] [CrossRef]
- Centini, R.; Tsang, M.; Iwata, T.; Park, H.; Delrow, J.; Margineantu, D.; Iritani, B.M.; Gu, H.; Liggitt, H.D.; Kang, J.; et al. Loss of Fnip1 alters kidney developmental transcriptional program and synergizes with TSC1 loss to promote mTORC1 activation and renal cyst formation. PLoS ONE 2018, 13, e0197973. [Google Scholar] [CrossRef]
- Sun, Z.; Yang, L.; Kiram, A.; Yang, J.; Yang, Z.; Xiao, L.; Yin, Y.; Liu, J.; Mao, Y.; Zhou, D.; et al. FNIP1 abrogation promotes functional revascularization of ischemic skeletal muscle by driving macrophage recruitment. Nat. Commun. 2023, 14, 7136. [Google Scholar] [CrossRef]
- Yan, M.; Audet-Walsh, E.; Manteghi, S.; Dufour, C.R.; Walker, B.; Baba, M.; St-Pierre, J.; Giguère, V.; Pause, A. Chronic AMPK activation via loss of FLCN induces functional beige adipose tissue through PGC-1alpha/ERRalpha. Genes Dev. 2016, 30, 1034–1046. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.S.; Linehan, W.M. FLCN: The causative gene for Birt-Hogg-Dube syndrome. Gene 2018, 640, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Hasumi, Y.; Baba, M.; Ajima, R.; Hasumi, H.; Valera, V.A.; Klein, M.E.; Haines, D.C.; Merino, M.J.; Hong, S.-B.; Yamaguchi, T.P.; et al. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc. Natl. Acad. Sci. USA 2009, 106, 18722–18727. [Google Scholar] [CrossRef] [PubMed]
- Hasumi, H.; Baba, M.; Hasumi, Y.; Lang, M.; Huang, Y.; Oh, H.F.; Matsuo, M.; Merino, M.J.; Yao, M.; Ito, Y.; et al. Folliculin-interacting proteins Fnip1 and Fnip2 play critical roles in kidney tumor suppression in cooperation with Flcn. Proc. Natl. Acad. Sci. USA 2015, 112, E1624–E1631. [Google Scholar] [CrossRef]
- Tsun, Z.Y.; Bar-Peled, L.; Chantranupong, L.; Zoncu, R.; Wang, T.; Kim, C.; Spooner, E.; Sabatini, D.M. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol. Cell. 2013, 52, 495–505. [Google Scholar] [CrossRef]
- Napolitano, G.; Di Malta, C.; Esposito, A.; de Araujo, M.E.G.; Pece, S.; Bertalot, G.; Matarese, M.; Benedetti, V.; Zampelli, A.; Stasyk, T.; et al. A substrate-specific mTORC1 pathway underlies Birt-Hogg-Dube syndrome. Nature 2020, 585, 597–602. [Google Scholar] [CrossRef]
- Hernandez-Trujillo, V.P.; Scalchunes, C.; Cunningham-Rundles, C.; Ochs, H.D.; Bonilla, F.A.; Paris, K.; Yel, L.; Sullivan, K.E. Autoimmunity and inflammation in X-linked agammaglobulinemia. J. Clin. Immunol. 2014, 34, 627–632. [Google Scholar] [CrossRef]
- Lougaris, V.; Soresina, A.; Baronio, M.; Montin, D.; Martino, S.; Signa, S.; Volpi, S.; Zecca, M.; Marinoni, M.; Baselli, L.A.; et al. Long-term follow-up of 168 patients with X-linked agammaglobulinemia reveals increased morbidity and mortality. J. Allergy Clin. Immunol. 2020, 146, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Bosch, J.V.W.T.; Hlaváčková, E.; Derpoorter, C.; Fischer, U.; Saettini, F.; Ghosh, S.; Farah, R.; Bogaert, D.; Wagener, R.; Loeffen, J.; et al. How to recognize inborn errors of immunity in a child presenting with a malignancy: Guidelines for the pediatric hemato-oncologist. Pediatr. Hematol. Oncol. 2023, 40, 131–146. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Summary | Niehues, 2020 [6] | Saettini, 2021 [7] | Moreno-Corona, 2023 [9] | Ulaş, 2024 [10] | Spivak, 2025 [11] | |
|---|---|---|---|---|---|---|
| Number of patients | 10 | 3 | 3 | 1 | 1 | 2 |
| Study design | WES, n = 1 Sanger sequencing, n = 2 | WES, n = 3 | WES = 1 | WES = 1 | WES = 1 | |
| Consanguinity | 5/10 | 3/3 | 1/3 | 0/1 | 1/1 | 0/2 ** |
| Age at the time of the description | P1: 7 months P2: 14 years P3: 8 months | P1: 3 years P2: 9 years P3: 25 years | 30 years | 11 months | II. 4: 1 year II. 1: 2 months | |
| Complete Blood Count | ||||||
| Anemia | 3/4 | NA | 2/3 | NA | 1/1 | NA |
| White blood cells | ||||||
| Leukocytosis | 1/9 | 0/3 | 1/3 | NA | 0/1 | 0/2 |
| Leukopenia | 2/9 | 0/3 | 1/3 | NA | 1/1 | 0/2 |
| Neutropenia | 6/10 | 2/3 | 2/3 | 1/1 | 1/1 | 0/2 |
| ALC, normal | 5/9 | 0/3 | 3/3 | NA | 0/1 | 2/2 |
| ALC, increased | 2/9 | 2/3 | 0/3 | NA | 0/1 | 0/2 |
| Monocytosis | 3/6 | 1/3 | 2/3 | NA | NA | NA |
| Monocytopenia | 1/6 | 1/3 | 0/3 | NA | NA | NA |
| Platelets, normal | 3/3 | NA | 3/3 | NA | NA | NA |
| Lymphocyte Subsets | ||||||
| T-cells | ||||||
| Increased CD3+ | 6/9 | 3/3 | 2/3 | 1/1 | 0/1 * | 0/1 |
| Increased CD4+ | 5/9 | 3/3 | 2/3 | 0/1 | 0/1 * | 1/1 |
| Increased CD8+ | 7/9 | 3/3 | 2/3 | 1/1 | 0/1 * | 1/1 |
| B-cells | ||||||
| Decreased B-cells | 8/9 | 2/3 | 3/3 | 1/1 | 1/1 | 1/1 |
| B-cells < 2% | 6/9 | 0/3 | 3/3 | 1/1 | 1/1 | 1/1 |
| NK cells, normal | 7/8 | 3/3 | 3/3 | NA | 0/1 * | 1/1 * |
| Decreased NKT cells | 1/3 | NA | 1/3 | NA | NA | NA |
| Immunoglobulin | ||||||
| IgG | 9/10 | 2/3 | 3/3 | 1/1 | 1/1 | 2/2 |
| IgA | 8/10 | 1/3 | 3/3 | 1/1 | 1/1 | 2/2 |
| IgM | 9/10 | 2/3 | 3/3 | 1/1 | 1/1 | 2/2 |
| Vaccine responses, low/absent | 3/3 | 2/2 | 1/1 | NA | NA | NA |
| T-cell proliferation, defective | 1/6 | 0/3 | 1/3 | NA | NA | NA |
| Bone Marrow | ||||||
| BM B-cells | 2/2 | 1/1 B-cell maturation defect at the pre-B-cell stage with an increase in CD34−CD10+ CD21low-B-cell precursors | One out of one, no-maturation block; increase in earlier maturation stages (pro-B and pre-B1) with a significant reduction in immature B-cells. | NA | NA | NA |
| BM myeloid | 1/1 | NA | One out of one, delayed granulocyte maturation; no overt arrest. | NA | NA | NA |
| Infections | 9/10 | 3/3 | 3/3 | 1/1 | 1/1 | 1/2 |
| Onset < 1 year | 9/10 | 3/3 | 3/3 | 1/1 | 1/1 | 1/2 |
| Heart disease | 10/10 | 3/3 | 3/3 | 1/1 | 1/1 | 2/2 |
| LV HCM | 10/10 | 3/3 | 3/3 | 1/1 | 1/1 | 2/2 |
| WPW | 5/10 | 2/3 | 1/3 | 1/1 | 1/1 | 0/2 |
| Other + | 4/10 | 0/3 | 1/3 | 0/1 | 1/1 | 2/2 |
| Chronic Lung disease | 5/8 | 2/3 | 3/3 | NA | NA | 0/2 |
| CNS | ||||||
| Microcephaly | 4/10 | 2/3 | 1/3 | 0/1 | 1/1 | 0/2 |
| Developmental or cognitive delay | 6/10 | 2/3 | 1/3 | 1/1 | 1/1 | 1/2 |
| Brain abnormalities | 2/3 | NA | 1/1 | NA | 1/1 | 0/1 |
| Muscle | 4/8 | 3/3 | 0/3 | NA | NA | 1/2 |
| Bowel disease | 4/10 | 1/3 | 1/3 | 1/1 | 0/1 | 1/2 |
| IBD | 1/10 | 0/3 | 1/3 | 0/1 | 0/1 | 0/2 |
| Other ++ | 3/10 | 1/3 | 0/3 | 1/1 | 0/1 | 1/2 |
| Dysmorphic features | 2/10 | 0/3 | 0/3 | NA | 1/1 | 1/2 |
| Kidney | 0/9 | 0/3 | 0/3 | NA | 0/1 | 0/2 |
| Treatment | ||||||
| IgRT | 9/10 | 3/3 | 3/3 | 1/1 | 1/1 | 1/1 |
| G-CSF | 2/10 | 0/3 | 1/3 | 1/1 | 0/1 | 0/2 |
| Antibiotic prophylaxis | 1/10 | 0/3 | 1/3 | 0/1 | 0/1 | 0/2 |
| Oxygen | 1/10 | 0/3 | 1/3 | 0/1 | 0/1 | 0/2 |
| Status at the time of the description, alive | 6/10 | 2/3 | 2/3 | 0/1 | 1/1 | 1/2 |
| Cause of death | P2: sudden death | P2: pneumonia | Heart failure associated with SARS-CoV-2 infection | II. 1: cardiogenic shock | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roncareggi, S.; Iritani, B.M.; Saettini, F. FNIP1 Deficiency: Pathophysiology and Clinical Manifestations of a Rare Syndromic Primary Immunodeficiency. Curr. Issues Mol. Biol. 2025, 47, 290. https://doi.org/10.3390/cimb47040290
Roncareggi S, Iritani BM, Saettini F. FNIP1 Deficiency: Pathophysiology and Clinical Manifestations of a Rare Syndromic Primary Immunodeficiency. Current Issues in Molecular Biology. 2025; 47(4):290. https://doi.org/10.3390/cimb47040290
Chicago/Turabian StyleRoncareggi, Samuele, Brian M. Iritani, and Francesco Saettini. 2025. "FNIP1 Deficiency: Pathophysiology and Clinical Manifestations of a Rare Syndromic Primary Immunodeficiency" Current Issues in Molecular Biology 47, no. 4: 290. https://doi.org/10.3390/cimb47040290
APA StyleRoncareggi, S., Iritani, B. M., & Saettini, F. (2025). FNIP1 Deficiency: Pathophysiology and Clinical Manifestations of a Rare Syndromic Primary Immunodeficiency. Current Issues in Molecular Biology, 47(4), 290. https://doi.org/10.3390/cimb47040290