
In Silico Design of Quantitative Polymerase Chain Reaction (qPCR) Assay Probes for Prostate Cancer Diagnosis, Prognosis, and Personalised Treatment


Abstract
1. Introduction
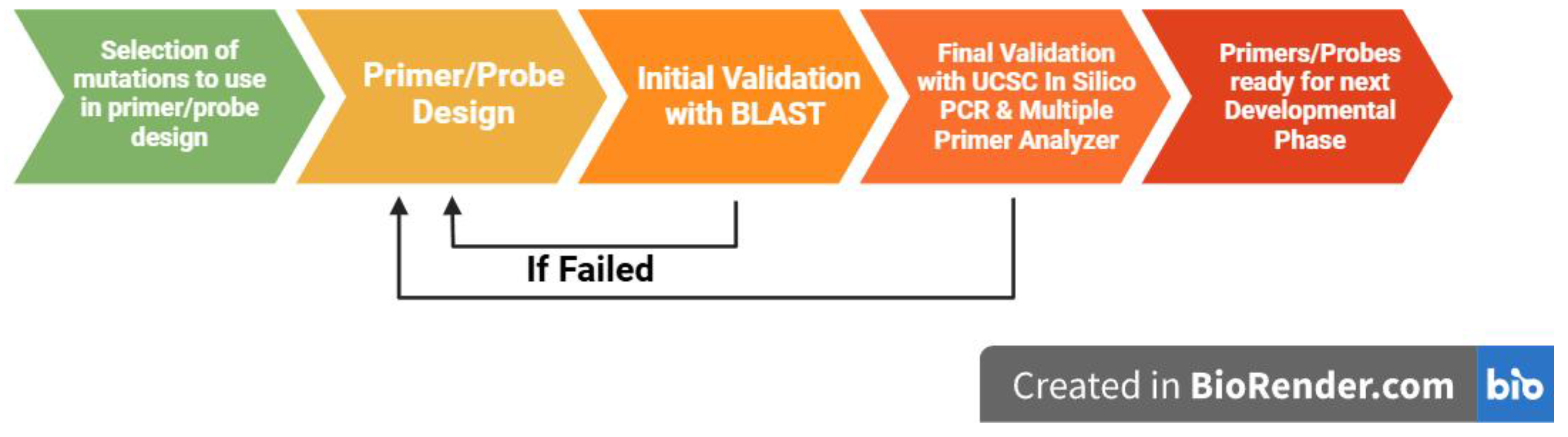
2. Methods
2.1. Mutational Data
2.2. Primer Design
2.3. Probe Design
2.4. In Silico Probe Validation
3. Results
3.1. Mutational Data
3.2. qPCR Primers

{kind=link}
| ID | OLIGO | Length | Tm | GC% | Sequence | Amplicon Length | BLAST e-Value | Hairpin Tm | Hairpin ΔG |
|---|---|---|---|---|---|---|---|---|---|
| AR1 | Forward Primer | 24 | 62 | 45.8 | TCTCCCTCTTATTGTTCCCTACAG | 84 | 2.00 × 10−4 | 29.6 | −0.33 |
| Reverse Primer | 22 | 62 | 45.5 | GCTCACCATGTGTGACTTGATT | 0.002 | 52.4 | −2.14 | ||
| Wild-Type Probe | 26 | 68 | 50 | TGCGAGAGAGCTGCATCAGTTCACTT | 1.00 × 10−5 | 42.3 | −2.06 | ||
| Mutant Probe | 24 | 69 | 58 | TGCGAGAGAGCTGCATCAGTTCGC | 2.00 × 10−4 | 43.8 | −2.95 | ||
| AR2 Wild-Type | Forward Primer | 19 | 62 | 52.6 | ACCTCCTTGTCAACCCTGT | 106 | 0.055 | 17.2 | 0.48 |
| Reverse Primer | 23 | 62 | 43.5 | GCTCACCATGTGTGACTTGATTA | 6.00 × 10−4 | 52.4 | −2.14 | ||
| Wild-Type Probe | 30 | 70 | 50 | TTGTTCCCTACAGATTGCGAGAGAGCTGCA | 8.00 × 10−8 | 33.4 | −1.27 | ||
| AR2 Mutant | Forward Primer | 24 | 62 | 45.8 | TCTCCCTCTTATTGTTCCCTACAG | 84 | 2.00 × 10−4 | 29.6 | −0.33 |
| Reverse Primer | 22 | 62 | 45.5 | GCTCACCATGTGTGACTTGATT | 2.00 × 10−3 | 52.4 | −2.14 | ||
| Mutant Probe | 30 | 68 | 43 | AGAGCTGTATCAGTTCACTTTTGACCTGCT | 2.00 × 10−5 | 42.3 | −2.06 | ||
| AR3 | Forward Primer | 21 | 62 | 52.4 | CATTGAGCCAGGTGTAGTGTG | 100 | 5.00 × 10−3 | 7.2 | 1.18 |
| Reverse Primer | 20 | 62 | 50 | AAGCTGTCTCTCTCCCAGTT | 2.10 × 10−2 | 31.7 | −0.62 | ||
| Wild-Type Probe | 30 | 70 | 53 | CCTTTGCAGCCTTGCTCTCTAGCCTCAATG | 8.00 × 10−8 | 49.7 | −2.39 | ||
| Mutant Probe | 30 | 70 | 53 | CCTTTGCAGCCTTGCACTCTAGCCTCAATG | 2.00 × 10−5 | 52.6 | −2.71 | ||
| AR4 | Forward Primer | 21 | 62 | 52.4 | GACCAGATGGCTGTCATTCAG | 98 | 5.00 × 10−3 | 43.4 | −1.6 |
| Reverse Primer | 22 | 62 | 45.5 | AAGTAGAGCATCCTGGAGTTGA | 2.00 × 10−3 | 20.4 | 0.21 | ||
| Wild-Type Probe | 24 | 69 | 54 | ATGGGCTGGCGATCCTTCACCAAT | 2.00 × 10−4 | 33.8 | −0.61 | ||
| Mutant Probe | 24 | 69 | 54 | TGGTGTTTGCCATGGGCTAGCGAT | 4.20 × 10−2 | 50 | −2.16 |
| ID | OLIGO | Length | Tm | GC% | Sequence | Amplicon Length | BLAST e-Value | Hairpin ΔG | Hairpin Tm |
|---|---|---|---|---|---|---|---|---|---|
| ATM1 | Forward Primer | 22 | 60 | 40.9 | GTTGCTTGGTTCTTTGTTTGTC | 265 | 2.00 × 10−3 | 1.86 | −8.2 |
| Reverse Primer | 19 | 60 | 52.6 | CGCACCTGGCCTTAATTTC | 5.50 × 10−2 | −0.84 | 38.5 | ||
| Wild-Type Probe | 29 | 68 | 45 | AGATTGCATCTGGCTTTTTCCTGCGATTG | 3.00 × 10−7 | −1.44 | 45 | ||
| Mutant Probe | 29 | 69 | 48 | AGATTGCATCTGGCTTTTTCCTGCGATGG | 5.00 × 10−6 | −1.6 | 39.6 | ||
| ATM2 | Forward Primer | 21 | 57 | 42.9 | GAGTTGGGAGTTACATATTGG | 163 | 0.005 | 0.41 | 17.7 |
| Reverse Primer | 20 | 59 | 45 | CACCACAGCCATACAAACTA | 0.021 | 1.17 | −11.2 | ||
| Wild-Type Probe | 30 | 64 | 43 | CTTAGAAATCTACAGAAGTATAGGGGAGCC | 8.00 × 10−8 | −0.64 | 30.8 | ||
| Mutant Probe | 30 | 64 | 43 | CCTTAGAAATCTACAGAAGTATAGGGGAGC | 3.00 × 10−7 | −0.34 | 31.2 | ||
| ATM3 | Forward Primer | 25 | 60 | 40 | GAACTTCTGAAACCACTATCGTAAG | 198 | 5.00 × 10−5 | 0.54 | 18.7 |
| Reverse Primer | 25 | 61 | 40 | GGTTTTTCACTACATGAAGGACATG | 5.00 × 10−5 | −3.1 | 52.6 | ||
| Wild-Type Probe | 30 | 68 | 47 | CCACAGCAATGTGTGTTCTTTGTATCGTCG | 8.00 × 10−8 | −4.49 | 64.4 | ||
| Mutant Probe | 30 | 66 | 43 | CCACAACAATGTGTGTTCTTTGTATCGTCG | 2.00 × 10−5 | −3.01 | 51.1 | ||
| ATM4 | Forward Primer | 22 | 59 | 40.9 | GGACTCAACATAGGCTTAATGA | 217 | 2.00 × 10−3 | −0.96 | 36.1 |
| Reverse Primer | 21 | 59 | 47.6 | CCAGAGAAATCCAGAGGAAAG | 5.00 × 10−3 | −0.7 | 36.9 | ||
| Wild-Type Probe | 30 | 68 | 47 | ATATCCATCATCCGAAAGGAGCCAAAACCC | 8.00 × 10−8 | −2.6 | 56.5 | ||
| Mutant Probe | 30 | 67 | 43 | ATATCCATCATCTGAAAGGAGCCAAAACCC | 2.00 × 10−5 | −1.33 | 34.9 | ||
| ATM5 Wild Type | Forward Primer | 24 | 62 | 41.7 | CTCAGACTGACGGATTAAATTCCA | 198 | 2.00 × 10−4 | −1.21 | 38.2 |
| Reverse Primer | 25 | 60 | 40 | GGAATCCACTAGTTCTGTTATGATG | 5.00 × 10−5 | −0.14 | 26.4 | ||
| Wild-Type Probe | 30 | 69 | 47 | TCCAAGGCTATTCAGTGTGCGAGGTAATCT | 8.00 × 10−8 | −0.11 | 25.9 | ||
| ATM5 Mutant | Forward Primer | 22 | 60 | 45.5 | GTTACCAAAGGATGCTGTTCTC | 243 | 2.00 × 10−3 | −0.47 | 33.7 |
| Reverse Primer | 21 | 60 | 42.9 | GAAAGTGTTGGACTTGGTTGT | 5.00 × 10−3 | −1.91 | 49.9 | ||
| Mutant Probe | 28 | 66 | 43 | TAAGGCTATTCAGTGTGCGAGGTAATCT | 5.00 × 10−6 | 0.09 | 23.8 |
| ID | OLIGO | Length | Tm | GC% | Sequence | Amplicon Length | BLAST e-Value | Hairpin ΔG | Hairpin Tm |
|---|---|---|---|---|---|---|---|---|---|
| PTEN1 | Forward Primer | 23 | 62 | 43.5 | TGACCAATGGCTAAGTGAAGATG | 133 | 6.00 × 10−4 | 0.16 | 22.4 |
| Reverse Primer | 20 | 62 | 50 | TAGGGCCTCTTGTGCCTTTA | 2.10 × 10−2 | −2.07 | 47.6 | ||
| Wild-Type Probe | 30 | 68 | 43 | AGGGACGAACTGGTGTAATGATATGTGCAT | 8.00 × 10−8 | 0.13 | 21.9 | ||
| Mutant Probe | 30 | 69 | 47 | AGGGACGAACTGGTGTAAGGATATGTGCAT | 2.00 × 10−5 | 0.13 | 21.9 | ||
| PTEN2 | Forward Primer | 23 | 60 | 43.5 | CTCTGTATTAGTGGCATCACAAGT | 336 | 2.00 × 10−4 | −2.13 | 47.6 |
| Reverse Primer | 23 | 60 | 47 | AGTACATTCATACCTACCTCTGC | 6.00 × 10−4 | 0.11 | 23.7 | ||
| Wild-Type Probe | 30 | 69 | 47 | AGTTGTGCTGAAAGACATTATGACACCGCC | 8.00 × 10−8 | −0.51 | 36.5 | ||
| Mutant Probe | 30 | 68 | 43 | AATTGTGCTGAAAGACATTATGACACCGCC | 1.00 × 10−6 | −0.51 | 36.5 | ||
| PTEN3 | Forward Primer | 20 | 60 | 50 | CATAACCCACCACAGCTAGA | 261 | 2.10 × 10−2 | 2.89 | −68.7 |
| Reverse Primer | 21 | 60 | 47.6 | TCAGATCCAGGAAGAGGAAAG | 0.005 | −0.61 | 34.3 | ||
| Wild-Type Probe | 26 | 68 | 50 | AAAGCTGGAAAGGGACGAACTGGTGT | 1.00 × 10−5 | −1 | 40.9 | ||
| Mutant Probe | 25 | 69 | 52 | AAGCTGGAAACGGACGAACTGGTGT | 0.012 | −1.17 | 44.3 | ||
| PTEN4 | Forward Primer | 22 | 63 | 50 | GTTTGTGGTCTGCCAGCTAAAG | 97 | 0.002 | −1.17 | 40.7 |
| Reverse Primer | 21 | 63 | 52.4 | CGGCTGAGGGAACTCAAAGTA | 0.005 | −3.18 | 58 | ||
| Wild-Type Probe | 24 | 69 | 58 | TTCAGGACCCACACGACGGGAAGA | 2.00 × 10−4 | −2.43 | 55.3 | ||
| Mutant Probe | 25 | 69 | 56 | TCAGGACCCACATGACGGGAAGACA | 0.012 | −2.43 | 55.3 | ||
| PTEN5 | Forward Primer | 22 | 60 | 45.5 | GACCAATGGCTAAGTGAAGATG | 164 | 2.00 × 10−3 | 0.16 | 22.4 |
| Reverse Primer | 21 | 60 | 47.6 | TTGTCTCTGGTCCTTACTTCC | 0.005 | 0.06 | 24.1 | ||
| Wild-Type Probe | 26 | 68 | 50 | AAAGCTGGAAAGGGACGAACTGGTGT | 1.00 × 10−5 | −1 | 40.9 | ||
| Mutant Probe | 30 | 68 | 43 | AGCTGGAAAGGGATGAACTGGTGTAATGAT | 8.00 × 10−8 | −1 | 40.9 | ||
| PTEN6 | Forward Primer | 22 | 60 | 45.5 | GACCAATGGCTAAGTGAAGATG | 164 | 2.00 × 10−3 | 0.16 | 22.4 |
| Reverse Primer | 21 | 60 | 47.6 | TTGTCTCTGGTCCTTACTTCC | 5.00 × 10−3 | 0.06 | 24.1 | ||
| Wild-Type Probe | 26 | 68 | 50 | AAAGCTGGAAAGGGACGAACTGGTGT | 1.00 × 10−5 | −1 | 40.9 | ||
| Mutant Probe | 30 | 68 | 43 | AGCTGGAAAGGGACAAACTGGTGTAATGAT | 8.00 × 10−8 | −1.66 | 34.4 | ||
| PTEN7 Wild Type | Forward Primer | 21 | 62 | 47.6 | TTTCTGTCCACCAGGGAGTAA | 148 | 5.00 × 10−3 | −1.47 | 42.4 |
| Reverse Primer | 21 | 62 | 47.6 | CCGCCACTGAACATTGGAATA | 5.00 × 10−3 | −1.29 | 41.3 | ||
| Wild-Type Probe | 24 | 68 | 54 | TTCCCAGTCAGAGGCGCTATGTGT | 2.00 × 10−4 | 0.22 | 22.2 | ||
| PTEN7 Mutant | Forward Primer | 24 | 62 | 41.7 | CGACCCAGTTACCATAGCAATTTA | 155 | 2.00 × 10−4 | 1.24 | 5.8 |
| Reverse Primer | 25 | 62 | 40 | TCCAGATGATTCTTTAACAGGTAGC | 5.00 × 10−5 | −0.06 | 26.4 | ||
| Mutant Probe | 30 | 69 | 53 | CAGGGAGTAACTATTCCCAGTCAGAGGTGC | 5.00 × 10−6 | −4.14 | 52.9 | ||
| PTEN8 Wild Type | Forward Primer | 21 | 62 | 47.6 | TTTCTGTCCACCAGGGAGTAA | 148 | 5.00 × 10−3 | −1.47 | 42.4 |
| Reverse Primer | 21 | 62 | 47.6 | CCGCCACTGAACATTGGAATA | 5.00 × 10−3 | −1.29 | 41.3 | ||
| Wild-Type Probe | 24 | 68 | 54 | TTCCCAGTCAGAGGCGCTATGTGT | 2.00 × 10−4 | 0.22 | 22.2 | ||
| PTEN8 Mutant | Forward Primer | 24 | 62 | 41.7 | CGACCCAGTTACCATAGCAATTTA | 155 | 2.00 × 10−4 | 1.24 | 5.8 |
| Reverse Primer | 25 | 62 | 40 | TCCAGATGATTCTTTAACAGGTAGC | 5.00 × 10−5 | −0.06 | 26.4 | ||
| Mutant Probe | 30 | 69 | 50 | AGGGAGTAACTATTCCCAGTCAGAGGCACT | 5.00 × 10−6 | −3.53 | 50.2 | ||
| PTEN9 | Forward Primer | 23 | 60 | 43.5 | CTCTGTATTAGTGGCATCACAAG | 417 | 6.00 × 10−4 | −2.13 | 47.6 |
| Reverse Primer | 23 | 60 | 43.5 | CTCACTCGATAATCTGGATGACT | 6.00 × 10−4 | 0.36 | 17.6 | ||
| Wild-Type Probe | 30 | 68 | 43.5 | TGACACCGCCAAATTTAATTGCAGAGGTAG | 8.00 × 10−8 | −0.31 | 29.8 | ||
| Mutant Probe | 30 | 66 | 40 | TGACACCGCCAAATTTAATTGCAGAGTTAG | 1.00 × 10−6 | −0.27 | 28.4 | ||
| PTEN10 Wild-Type | Forward Primer | 20 | 60 | 50 | GCTACGACCCAGTTACCATA | 172 | 0.021 | 0.37 | 17.9 |
| Reverse Primer | 25 | 60 | 40 | CACTGGTCTATAATCCAGATGATTC | 5.00 × 10−5 | −3.68 | 56.2 | ||
| Wild-Type Probe | 30 | 68 | 53 | CCACCAGGGAGTAACTATTCCCAGTCAGAG | 8.00 × 10−8 | −3.14 | 52.9 | ||
| PTEN10 Mutant | Forward Primer | 21 | 61 | 52.4 | GCTACGACCCAGTTACCATAG | 130 | 0.005 | 0.37 | 17.9 |
| Reverse Primer | 22 | 60 | 40.9 | ATAATACACATAGCGCCTCTGA | 0.002 | −0.14 | 27.4 | ||
| Mutant Probe | 30 | 67 | 47 | TTCTGTCCACCAAGGAGTAACTATTCCCAG | 2.00 × 10−5 | −1.28 | 49.3 | ||
| PTEN11 Wild Type | Forward Primer | 21 | 62 | 47.6 | TTTCTGTCCACCAGGGAGTAA | 148 | 0.005 | −1.47 | 42.4 |
| Reverse Primer | 21 | 62 | 47.6 | CCGCCACTGAACATTGGAATA | 0.005 | −1.29 | 41.3 | ||
| Wild-Type Probe | 24 | 68 | 54 | TTCCCAGTCAGAGGCGCTATGTGT | 2.00 × 10−4 | 0.22 | 22.2 | ||
| PTEN11 Mutant | Forward Primer | 20 | 60 | 50 | GCTACGACCCAGTTACCATA | 172 | 0.021 | 0.37 | 17.9 |
| Reverse Primer | 25 | 60 | 40 | CACTGGTCTATAATCCAGATGATTC | 5.00 × 10−5 | −3.68 | 56.2 | ||
| Mutant Probe | 27 | 66 | 48 | CTATTCCCAGTTAGAGGCGCTATGTGT | 0.001 | −0.19 | 28.4 | ||
| PTEN12 | Forward Primer | 23 | 60 | 43.5 | GGACGAACTGGTGTAATGATATG | 153 | 6.00 × 10−4 | 0.13 | 21.9 |
| Reverse Primer | 21 | 60 | 47.6 | TCAGATCCAGGAAGAGGAAAG | 0.005 | −0.61 | 34.3 | ||
| Wild-Type Probe | 30 | 66 | 40 | TTAAAGGCACAAGAGGCCCTAGATTTCTAT | 8.00 × 10−8 | −2.28 | 40.4 | ||
| Mutant Probe | 30 | 66 | 40 | TAAAAGGCACAAGAGGCCCTAGATTTCTAT | 1.00 × 10−6 | −2.28 | 40.4 | ||
| PTEN13 | Forward Primer | 24 | 61 | 41.7 | GCTTGAGATCAAGATTGCAGATAC | 202 | 2.00 × 10−4 | −3.45 | 55.3 |
| Reverse Primer | 19 | 61 | 52.6 | TCGTGTGGGTCCTGAATTG | 5.50 × 10−2 | 1.1 | −2.5 | ||
| Wild-Type Probe | 27 | 67 | 44 | TAACCATGCAGATCCTCAGTTTGTGGT | 4.00 × 10−6 | −1.1 | 38.2 | ||
| Mutant Probe | 27 | 68 | 48 | ATGCAAATCCTCAGTTTGTGGTCTGCC | 1.00 × 10−3 | −1.76 | 40.2 |
| ID | OLIGO | Length | Tm | GC% | Sequence | Amplicon Length | BLAST e-Value | Hairpin ΔG | Hairpin Tm |
|---|---|---|---|---|---|---|---|---|---|
| TP53_1 | Forward Primer | 22 | 63 | 50 | CAGTGTGATGATGGTGAGGAT | 109 | 5.00 × 10−3 | 0.76 | 8.9 |
| Reverse Primer | 22 | 62 | 50 | TATCTCCTAGGTTGGCTCTGAC | 2.00 × 10−3 | −0.06 | 26 | ||
| Wild-Type Probe | 24 | 69 | 54 | CGCCCATGCAGGAACTGTTACACA | 2.00 × 10−4 | −2.72 | 57.3 | ||
| Mutant Probe | 22 | 69 | 54 | ATGCCGGCCATGCAGGAACTGTTA | 0.042 | −2.72 | 57.3 | ||
| TP53_2 | Forward Primer | 22 | 62 | 50 | CAGTGTGATGATGGTGAGGATG | 127 | 0.002 | 0.76 | 8.9 |
| Reverse Primer | 22 | 62 | 50 | CTCATCTTGGGCCTGTGTTATC | 0.002 | 0.87 | 7.8 | ||
| Wild-Type Probe | 30 | 69 | 50 | AGTTGTAGTGGATGGTGGTACAGTCAGAGC | 8.00 × 10−8 | −0.3 | 28.2 | ||
| Mutant Probe | 30 | 69 | 50 | TGGTTGTAGTGGATGGTGGTACAGTCAGAG | 1.00 × 10−6 | −0.3 | 28.2 | ||
| TP53_3 | Forward Primer | 20 | 62 | 50 | AGACTTGGCTGTCCCAGAAT | 83 | 0.021 | −1.18 | 40.7 |
| Reverse Primer | 22 | 62 | 45.5 | ATCTTCTGTCCCTTCCCAGAAA | 0.002 | −1.86 | 40.7 | ||
| Wild-Type Probe | 24 | 69 | 58 | ACGGTTTCCGTCTGGGCTTCTTGC (Anti-Sense) | 2.00 × 10−4 | −3.15 | 45.8 | ||
| Mutant Probe | 24 | 69 | 58 | ACGGTTTCCCTCTGGGCTTCTTGC (Anti-Sense) | 0.042 | −1.17 | 43.3 | ||
| TP53_4 Wild-Type | Forward Primer | 21 | 60 | 47.6 | CATGAAGGCAGGATGAGAATG | 184 | 0.005 | −0.7 | 40.2 |
| Reverse Primer | 18 | 60 | 50 | TGAGCGCTTCGAGATGTT | 0.22 | −0.61 | 35.5 | ||
| Wild-Type Probe | 26 | 68 | 54 | AGTTCCAAGGCCTCATTCAGCTCTCG | 1.00 × 10−5 | −0.27 | 28.9 | ||
| TP53_4 Mutant | Forward Primer | 18 | 60 | 50 | AAGGCCTCATTCAGCTCT | 272 | 0.22 | −0.27 | 28.9 |
| Reverse Primer | 18 | 60 | 50 | ATTGCACCATTGCACTCC | 0.22 | −2.45 | 51.2 | ||
| Mutant Probe | 24 | 68 | 58 | CAGAACATCTCGAAGCGCTCACGC | 3.00 × 10−3 | −2.96 | 61.9 | ||
| TP53_5 | Forward Primer | 17 | 62 | 58.8 | AGCCTGGGCATCCTTGA | 273 | 0.86 | −1.74 | 46 |
| Reverse Primer | 19 | 61 | 58.9 | CTGGGCAACAGAGTGAGAC | 0.055 | −1.69 | 49.4 | ||
| Wild-Type Probe | 28 | 68 | 50 | CCTCATTCAGCTCTCGGAACATCTCGAA | 1.00 × 10−6 | −0.57 | 33.6 | ||
| Mutant Probe | 27 | 69 | 59 | CAAACATCTCGAAGCGCTCACGCCCAC | 7.00 × 10−5 | −3.27 | 62.1 | ||
| TP53_6 | Forward Primer | 23 | 61 | 43.5 | GACTTAGTACCTGAAGGGTGAAA | 106 | 6.00 × 10−4 | −2.5 | 53.6 |
| Reverse Primer | 21 | 61 | 47.6 | CCTTTCCTTGCCTCTTTCCTA | 5.00 × 10−3 | 3.78 | −251.2 | ||
| Wild-Type Probe | 30 | 68 | 50 | TCTCCATCCAGTGGTTTCTTCTTTGGCTGG | 8.00 × 10−8 | −1.99 | 44.3 | ||
| Mutant Probe | 30 | 68 | 50 | ATCCATCCAGTGGTTTCTTCTTTGGCTGGG | 3.00 × 10−7 | −2.47 | 46.6 |
3.3. qPCR Probes
3.4. Primer/Probe Validation
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gandaglia, G.; Leni, R.; Bray, F.; Fleshner, N.; Freedland, S.J.; Kibel, A.; Stattin, P.; Van Poppel, H.; La Vecchia, C. Epidemiology and Prevention of Prostate Cancer. Eur. Urol. Oncol. 2021, 4, 877–892. [Google Scholar] [CrossRef]
- Sekhoacha, M.; Riet, K.; Motloung, P.; Gumenku, L.; Adegoke, A.; Mashele, S. Prostate Cancer Review: Genetics, Diagnosis, Treatment Options, and Alternative Approaches. Molecules 2022, 27, 5730. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J. Biomarkers for prostate cancer: Prostate-specific antigen and beyond. Clin. Chem. Lab. Med. (CCLM) 2020, 58, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Wasim, S.; Lee, S.-Y.; Kim, J. Complexities of Prostate Cancer. Int. J. Mol. Sci. 2022, 23, 14257. [Google Scholar] [CrossRef] [PubMed]
- Giona, S. The Epidemiology of Prostate Cancer. In Prostate Cancer; Exon Publications: Brisbane, QLD, Australia, 2021; Volume 1, pp. 1–16. [Google Scholar] [CrossRef]
- Jamroze, A.; Chatta, G.; Tang, D.G. Androgen receptor (AR) heterogeneity in prostate cancer and therapy resistance. Cancer Lett. 2021, 518, 1–9. [Google Scholar] [CrossRef]
- Formaggio, N.; Rubin, M.A.; Theurillat, J.-P. Loss and revival of androgen receptor signaling in advanced prostate cancer. Oncogene 2021, 40, 1205–1216. [Google Scholar] [CrossRef]
- Aurilio, G.; Cimadamore, A.; Mazzucchelli, R.; Lopez-Beltran, A.; Verri, E.; Scarpelli, M.; Massari, F.; Cheng, L.; Santoni, M.; Montironi, R. Androgen Receptor Signaling Pathway in Prostate Cancer: From Genetics to Clinical Applications. Cells 2020, 9, 2653. [Google Scholar] [CrossRef]
- Kaur, H.; Salles, D.C.; Murali, S.; Hicks, J.L.; Nguyen, M.; Pritchard, C.C.; De Marzo, A.M.; Lanchbury, J.S.; Trock, B.J.; Isaacs, W.B.; et al. Genomic and Clinicopathologic Characterization of ATM deficient Prostate Cancer. Clin. Cancer Res. 2020, 26, 4869–4881. [Google Scholar] [CrossRef]
- Neeb, A.; Herranz, N.; Arce-Gallego, S.; Miranda, S.; Buroni, L.; Yuan, W.; Athie, A.; Casals, T.; Carmichael, J.; Rodrigues, D.N.; et al. Advanced Prostate Cancer with ATM Loss: PARP and ATR Inhibitors. Eur. Urol. 2021, 79, 200–211. [Google Scholar] [CrossRef]
- Lee, J.-H.; Paull, T.T. Cellular functions of the protein kinase ATM and their relevance to human disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 796–814. [Google Scholar] [CrossRef]
- Rafiei, S.; Fitzpatrick, K.; Liu, D.; Cai, M.-Y.; Elmarakeby, H.A.; Park, J.; Ricker, C.; Kochupurakkal, B.S.; Choudhury, A.D.; Hahn, W.C.; et al. ATM Loss Confers Greater Sensitivity to ATR Inhibition Than PARP Inhibition in Prostate Cancer. Cancer Res. 2020, 80, 2094–2100. [Google Scholar] [CrossRef] [PubMed]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kim, S.; Li, L.; Kemp, C.J.; Jiang, C.; Lü, J. Proteomic and transcriptomic profiling of PTEN gene-knockout mouse model of prostate cancer. Prostate 2020, 80, 588–605. [Google Scholar] [CrossRef] [PubMed]
- Turnham, D.J.; Bullock, N.; Dass, M.S.; Staffurth, J.N.; Pearson, H.B. The PTEN Conundrum: How to Target PTEN-Deficient Prostate Cancer. Cells 2020, 9, 2342. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Hodakoski, C.; Barrows, D.; Mense, S.M.; Parsons, R.E. PTEN function: The long and the short of it. Trends Biochem. Sci. 2014, 39, 183–190. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef]
- Lin, J.; Sampath, D.; Nannini, M.A.; Lee, B.B.; Degtyarev, M.; Oeh, J.; Savage, H.; Guan, Z.; Hong, R.; Kassees, R.; et al. Targeting Activated Akt with GDC-0068, a Novel Selective Akt Inhibitor That Is Efficacious in Multiple Tumor Models. Clin. Cancer Res. 2013, 19, 1760–1772. [Google Scholar] [CrossRef]
- Chipidza, F.E.; Alshalalfa, M.; Mahal, B.A.; Karnes, R.J.; Liu, Y.; Davicioni, E.; Martin, N.E.; Mouw, K.W.; Feng, F.Y.; Nguyen, P.L.; et al. Development and Validation of a Novel TP53 Mutation Signature That Predicts Risk of Metastasis in Primary Prostate Cancer. Clin. Genitourin. Cancer 2021, 19, 246–254. [Google Scholar] [CrossRef]
- Mateo, J.; Seed, G.; Bertan, C.; Rescigno, P.; Dolling, D.; Figueiredo, I.; Miranda, S.; Nava Rodrigues, D.; Gurel, B.; Clarke, M.; et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. J. Clin. Investig. 2020, 130, 1743–1751. [Google Scholar] [CrossRef]
- Nientiedt, C.; Endris, V.; Jenzer, M.; Mansour, J.; Sedehi, N.T.P.; Pecqueux, C.; Volckmar, A.-L.; Leichsenring, J.; Neumann, O.; Kirchner, M.; et al. High prevalence of DNA damage repair gene defects and TP53 alterations in men with treatment-naïve metastatic prostate cancer –Results from a prospective pilot study using a 37 gene panel. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 17–27. [Google Scholar] [CrossRef]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018, 50, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Lu, L.-y.; Wang, X.-q.; Li, B.-x.; Kelly, K.; Lin, H.-s. Gambogic Acid Induces Cell Apoptosis and Inhibits MAPK Pathway in PTEN-p53 Prostate Cancer Cells In Vitro and Ex Vivo. Chin. J. Integr. Med. 2018, 24, 109–116. [Google Scholar] [CrossRef]
- Perez, G.; Barber, G.P.; Benet-Pages, A.; Casper, J.; Clawson, H.; Diekhans, M.; Fischer, C.; Gonzalez, J.N.; Hinrichs, A.S.; Lee, C.M.; et al. The UCSC Genome Browser database: 2025 update. Nucleic Acids Res. 2025, 53, 1243–1249. [Google Scholar] [CrossRef]
- Prekovic, S.; Van Royen, M.E.; Voet, A.R.D.; Geverts, B.; Houtman, R.; Melchers, D.; Zhang, K.Y.J.; Van Den Broeck, T.; Smeets, E.; Spans, L.; et al. The Effect of F877L and T878A Mutations on Androgen Receptor Response to Enzalutamide. Mol. Cancer Ther. 2016, 15, 1702–1712. [Google Scholar] [CrossRef]
- Rahbaran, M.; Doabsari, M.H.; Sharifi, F.; Afshari, M.; Hasanzad, M. The Risk of Prostate Cancer Associated with Common Mutations of AR Gene in Iranian Population: A Perspective Study of Personalized Treatment. Res. Sq. 2021, 1–14. [Google Scholar]
- Khan, A.; Mao, Y.; Tahreem, S.; Wei, D.-Q.; Wang, Y. Structural and molecular insights into the mechanism of resistance to enzalutamide by the clinical mutants in androgen receptor (AR) in castration-resistant prostate cancer (CRPC) patients. Int. J. Biol. Macromol. 2022, 218, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, V.; Wetterskog, D.; Sharabiani, M.T.A.; Grande, E.; Fernandez-Perez, M.P.; Jayaram, A.; Salvi, S.; Castellano, D.; Romanel, A.; Lolli, C.; et al. Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: A multi-institution correlative biomarker study. Ann. Oncol. 2017, 28, 1508–1516. [Google Scholar] [CrossRef]
- Wise, H.M.; Hermida, M.A.; Leslie, N.R. Prostate cancer, PI3K, PTEN and prognosis. Clin. Sci. 2017, 131, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef]
- Richards, J.E.; Hawley, R.S. Chapter 5—We Are All Mutants: How Mutation Alters Function. Hum. Genome 2011, 3, 143–195. [Google Scholar] [CrossRef]
- Imada, E.L.; Sanchez, D.F.; Dinalankara, W.; Vidotto, T.; Ebot, E.M.; Tyekucheva, S.; Franco, G.R.; Mucci, L.A.; Loda, M.; Schaeffer, E.M.; et al. Transcriptional landscape of PTEN loss in primary prostate cancer. BMC Cancer 2021, 21, 856. [Google Scholar] [CrossRef]
- Molinari, F.; Frattini, M. Functions and Regulation of the PTEN Gene in Colorectal Cancer. Front. Oncol. 2014, 3, 326. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.N.; Dawson, J.E.; Krieger, J.; Thacker, S.; Bahar, I.; Eng, C. Structural and Dynamic Effects of PTEN C-Terminal Tail Phosphorylation. J. Chem. Inf. Model. 2022, 62, 4175–4190. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Garcia, V.; Tawil, Y.; Wise, H.M.; Leslie, N.R. Mechanisms of PTEN loss in cancer: It’s all about diversity. Semin. Cancer Biol. 2019, 59, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.D. PTEN-PI3K pathway alterations in advanced prostate cancer and clinical implications. Prostate 2022, 82, S60–S72. [Google Scholar] [CrossRef]
- Kruczek, K.; Ratterman, M.; Tolzien, K.; Sulo, S.; Lestingi, T.M.; Nabhan, C. A phase II study evaluating the toxicity and efficacy of single-agent temsirolimus in chemotherapy-naïve castration-resistant prostate cancer. Br. J. Cancer 2013, 109, 1711–1716. [Google Scholar] [CrossRef]
- Hotte, S.J.; Chi, K.N.; Joshua, A.M.; Tu, D.; Macfarlane, R.J.; Gregg, R.W.; Ruether, J.D.; Basappa, N.S.; Finch, D.; Salim, M.; et al. A Phase II Study of PX-866 in Patients With Recurrent or Metastatic Castration-resistant Prostate Cancer: Canadian Cancer Trials Group Study IND205. Clin. Genitourin. Cancer 2019, 17, 201–208. [Google Scholar] [CrossRef]
- Choudhury, A.D.; Higano, C.S.; De Bono, J.S.; Cook, N.; Rathkopf, D.E.; Wisinski, K.B.; Martin-Liberal, J.; Linch, M.; Heath, E.I.; Baird, R.D.; et al. A Phase I Study Investigating AZD8186, a Potent and Selective Inhibitor of PI3Kβ/δ, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2022, 28, 2257–2269. [Google Scholar] [CrossRef]
- Aghajanian, C.; Bell-McGuinn, K.M.; Burris, H.A.; Siu, L.L.; Stayner, L.-A.; Wheler, J.J.; Hong, D.S.; Kurkjian, C.; Pant, S.; Santiago-Walker, A.; et al. A phase I, open-label, two-stage study to investigate the safety, tolerability, pharmacokinetics, and pharmacodynamics of the oral AKT inhibitor GSK2141795 in patients with solid tumors. Investig. New Drugs 2018, 36, 1016–1025. [Google Scholar] [CrossRef]
- Shahbandi, A.; Nguyen, H.D.; Jackson, J.G. TP53 Mutations and Outcomes in Breast Cancer: Reading beyond the Headlines. Trends Cancer 2020, 6, 98–110. [Google Scholar] [CrossRef]
- Nathan, C.-A.; Khandelwal, A.R.; Wolf, G.T.; Rodrigo, J.P.; Mäkitie, A.A.; Saba, N.F.; Forastiere, A.A.; Bradford, C.R.; Ferlito, A. TP53 mutations in head and neck cancer. Mol. Carcinog. 2022, 61, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Canale, M.; Andrikou, K.; Priano, I.; Cravero, P.; Pasini, L.; Urbini, M.; Delmonte, A.; Crinò, L.; Bronte, G.; Ulivi, P. The Role of TP53 Mutations in EGFR-Mutated Non-Small-Cell Lung Cancer: Clinical Significance and Implications for Therapy. Cancers 2022, 14, 1143. [Google Scholar] [CrossRef]
- Maxwell, K.N.; Cheng, H.H.; Powers, J.; Gulati, R.; Ledet, E.M.; Morrison, C.; Le, A.; Hausler, R.; Stopfer, J.; Hyman, S.; et al. Inherited TP53 Variants and Risk of Prostate Cancer. Eur. Urol. 2022, 81, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Kaur, H.B.; Lu, J.; Guedes, L.B.; Maldonado, L.; Reitz, L.; Barber, J.R.; De Marzo, A.M.; Tomlins, S.A.; Sfanos, K.S.; Eisenberger, M.; et al. TP53 missense mutation is associated with increased tumor-infiltrating T cells in primary prostate cancer. Hum. Pathol. 2019, 87, 95–102. [Google Scholar] [CrossRef]
- Song, H.; Hollstein, M.; Xu, Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat. Cell Biol. 2007, 9, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.H.; Antonarakis, E.S. Emerging treatments for metastatic castration-resistant prostate cancer: Immunotherapy, PARP inhibitors, and PSMA-targeted approaches. Cancer Treat. Res. Commun. 2020, 23, 100164. [Google Scholar] [CrossRef]
- Papanicolau-Sengos, A.; Yang, Y.; Pabla, S.; Lenzo, F.L.; Kato, S.; Kurzrock, R.; DePietro, P.; Nesline, M.; Conroy, J.; Glenn, S.; et al. Identification of targets for prostate cancer immunotherapy. Prostate 2019, 79, 498–505. [Google Scholar] [CrossRef]
- Patel, V.G.; Oh, W.K. The Evolving Landscape of Immunotherapy in Advanced Prostate Cancer. Immunotherapy 2019, 11, 903–912. [Google Scholar] [CrossRef]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.-K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 1370–1384. [Google Scholar] [CrossRef]
- Teroerde, M.; Nientiedt, C.; Duensing, A.; Hohenfellner, M.; Stenzinger, A.; Duensing, S. Revisiting the Role of p53 in Prostate Cancer. In Prostate Cancer; Exon Publications: Brisbane, QLD, Australia, 2021; Volume 1. [Google Scholar]
- Gonen-Korkmaz, C.; Sevin, G.; Gokce, G.; Arun, M.Z.; Yildirim, G.; Reel, B.; Kaymak, A.; Ogut, D. Analysis of tumor necrosis factor α-induced and nuclear factor κB-silenced LNCaP prostate cancer cells by RT-qPCR. Exp. Ther. Med. 2014, 8, 1695–1700. [Google Scholar] [CrossRef] [PubMed]
- Latil, A.; Bièche, I.; Vidaud, D.; Lidereau, R.; Berthon, P.; Cussenot, O.; Vidaud, M. Evaluation of Androgen, Estrogen (ERα and ERβ), and Progesterone Receptor Expression in Human Prostate Cancer by Real-Time Quantitative Reverse Transcription-Polymerase Chain Reaction Assays. Cancer Res. 2001, 61, 1919–1926. [Google Scholar] [PubMed]
- Uygur, B.; Abramo, K.; Leikina, E.; Vary, C.; Liaw, L.; Wu, W.-S. SLUG is a direct transcriptional repressor of PTEN tumor suppressor. Prostate 2015, 75, 907–916. [Google Scholar] [CrossRef] [PubMed]
| Gene | Mutation ID | Consequence | Chromosome | Position | Reference Allele | Alternative Allele | Code |
|---|---|---|---|---|---|---|---|
| AR | rs137852578 | Missense | X | 67723710 | A | G | AR1 |
| rs137852581 | Missense | X | 67723701 | C | T | AR2 | |
| rs864622007 | Missense | X | 67711621 | T | A | AR3 | |
| rs2147530924 | Stop-Gained | X | 67717559 | G | A | AR4 | |
| ATM | rs1565399936 | Stop-Gained | 11 | 108259026 | T | G | ATM1 |
| rs1438576066 | Frameshift | 11 | 108315831 | C | CC | ATM2 | |
| rs1555084931 | Splice Acceptor Variant | 11 | 108271250 | G | A | ATM3 | |
| rs747727055 | Missense | 11 | 108245000 | C | T | ATM4 | |
| rs876660315 | Frameshift | 11 | 108244097 | C | - | ATM5 | |
| PTEN | rs1085308046 | Missense | 10 | 87933160 | T | G | PTEN1 |
| rs1114167621 | Splice Acceptor Variant | 10 | 87931045 | G | A | PTEN2 | |
| rs1114167645 | Missense | 10 | 87933143 | G | C | PTEN3 | |
| rs121909219 | Stop-Gained | 10 | 87957915 | C | T | PTEN4 | |
| rs121909224 | Stop-Gained | 10 | 87933147 | C | T | PTEN5 | |
| rs121909229 | Missense | 10 | 87933148 | G | A | PTEN6 | |
| rs121913293 | Missense | 10 | 87952142 | C | T | PTEN7 | |
| rs121913294 | Missense | 10 | 87952143 | G | A | PTEN8 | |
| rs587776667 | Splice Donor Variant | 10 | 87931090 | G | T | PTEN9 | |
| rs786204862 | Splice Acceptor Variant | 10 | 87952117 | G | A | PTEN10 | |
| rs786204864 | Stop-Gained | 10 | 87952136 | C | T | PTEN11 | |
| rs786204933 | Stop-Gained | 10 | 87933196 | T | A | PTEN12 | |
| rs876661024 | Splice Acceptor Variant | 10 | 87957852 | G | A | PTEN13 | |
| TP53 | rs985033810 | Missense | 17 | 7674232 | C | G | TP53-1 |
| rs587782289 | Missense | 17 | 7674257 | A | G | TP53-2 | |
| rs11540654 | Missense | 17 | 7676040 | C | G | TP53-3 | |
| rs730882029 | Stop-Gained | 17 | 7670685 | G | A | TP53-4 | |
| rs1131691022 | Frameshift | 17 | 7670686 | GA | A | TP53-5 | |
| rs876659384 | Stop-Gained | 17 | 7673552 | C | A | TP53-6 |
| Gene | Assay Uses |
|---|---|
| AR | Personalised treatments involving enzalutamide/arbiraterone acetate |
| ATM | Personalised treatments involving ATR inhibitors |
| PTEN | Primary diagnosis; treatment monitoring; personalised treatments involving PI3K inhibitors |
| TP53 | Personalised treatments involving immunotherapy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilson, T.K.; Zishiri, O.T. In Silico Design of Quantitative Polymerase Chain Reaction (qPCR) Assay Probes for Prostate Cancer Diagnosis, Prognosis, and Personalised Treatment. Curr. Issues Mol. Biol. 2025, 47, 292. https://doi.org/10.3390/cimb47040292
Wilson TK, Zishiri OT. In Silico Design of Quantitative Polymerase Chain Reaction (qPCR) Assay Probes for Prostate Cancer Diagnosis, Prognosis, and Personalised Treatment. Current Issues in Molecular Biology. 2025; 47(4):292. https://doi.org/10.3390/cimb47040292
Chicago/Turabian StyleWilson, Trevor Kenneth, and Oliver Tendayi Zishiri. 2025. "In Silico Design of Quantitative Polymerase Chain Reaction (qPCR) Assay Probes for Prostate Cancer Diagnosis, Prognosis, and Personalised Treatment" Current Issues in Molecular Biology 47, no. 4: 292. https://doi.org/10.3390/cimb47040292
APA StyleWilson, T. K., & Zishiri, O. T. (2025). In Silico Design of Quantitative Polymerase Chain Reaction (qPCR) Assay Probes for Prostate Cancer Diagnosis, Prognosis, and Personalised Treatment. Current Issues in Molecular Biology, 47(4), 292. https://doi.org/10.3390/cimb47040292