
Teriflunomide Preserves Neuronal Activity and Protects Mitochondria in Brain Slices Exposed to Oxidative Stress

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results
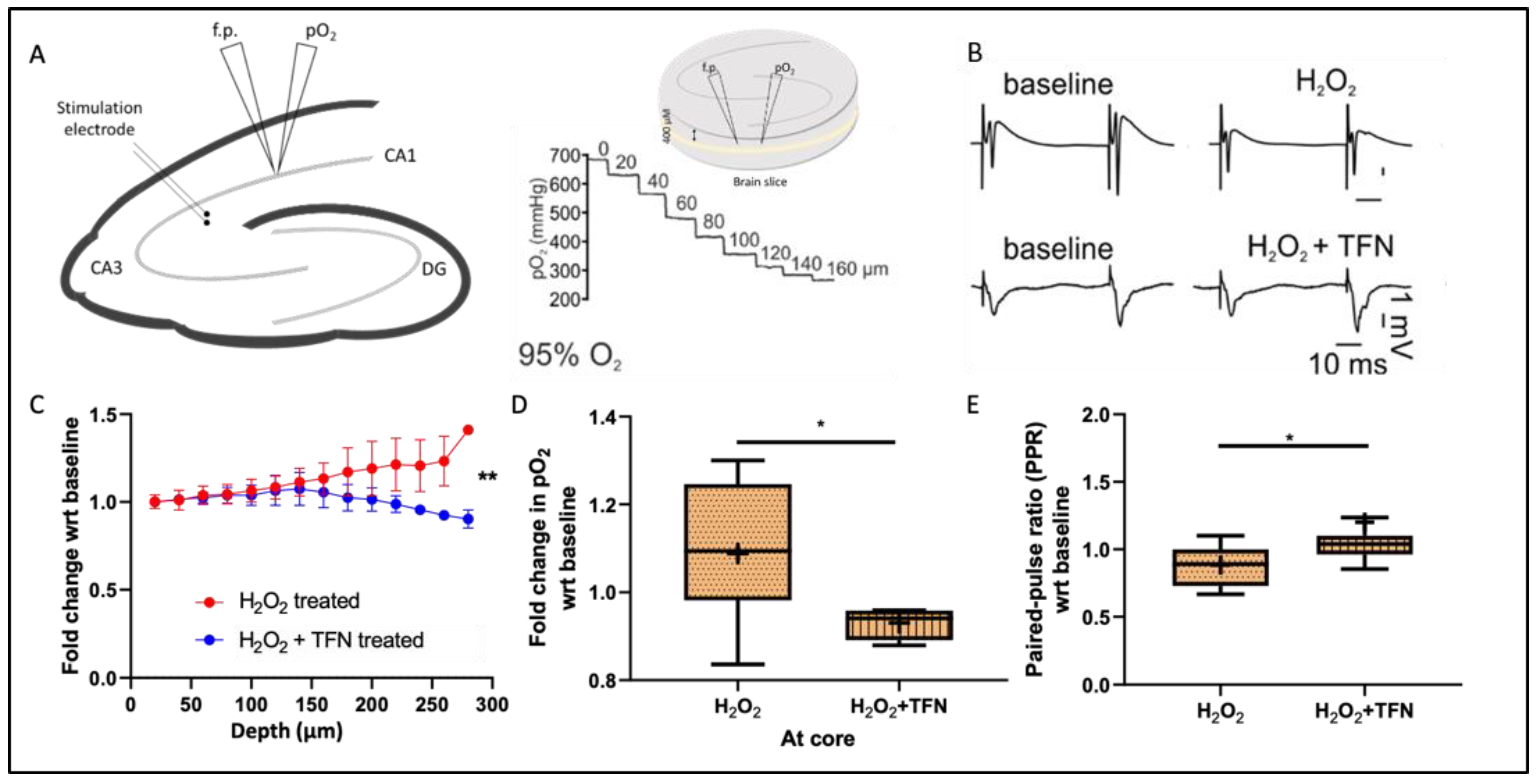
2.1. TFN Restores Tissue Respiration during Oxidative Stress
2.2. TFN Prevents Oxidative Stress-Mediated Depression of Synaptic Transmission
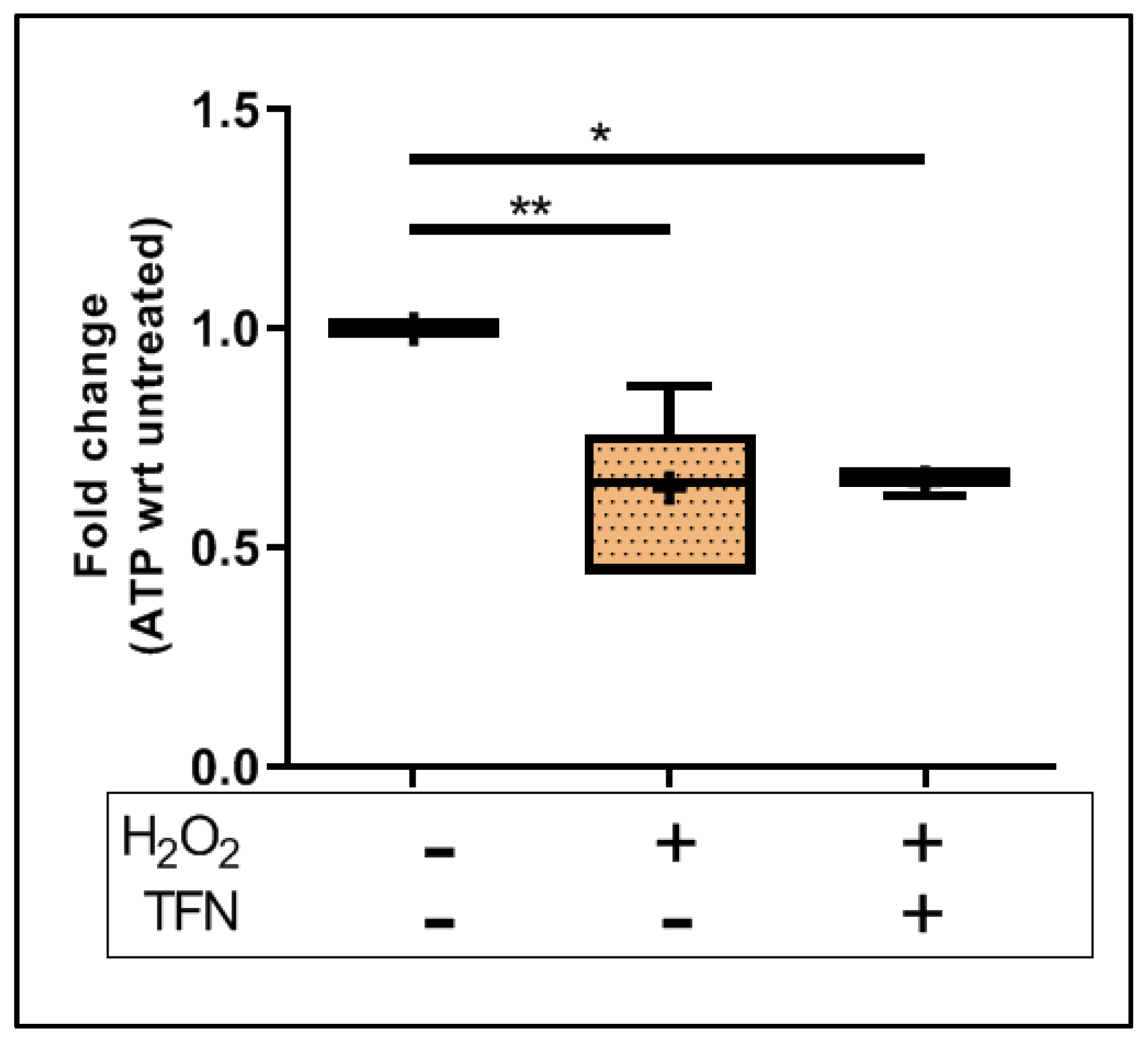
2.3. TFN Does Not Prevent Oxidative Stress-Mediated ATP Decrease in Acute Hippocampal Slices
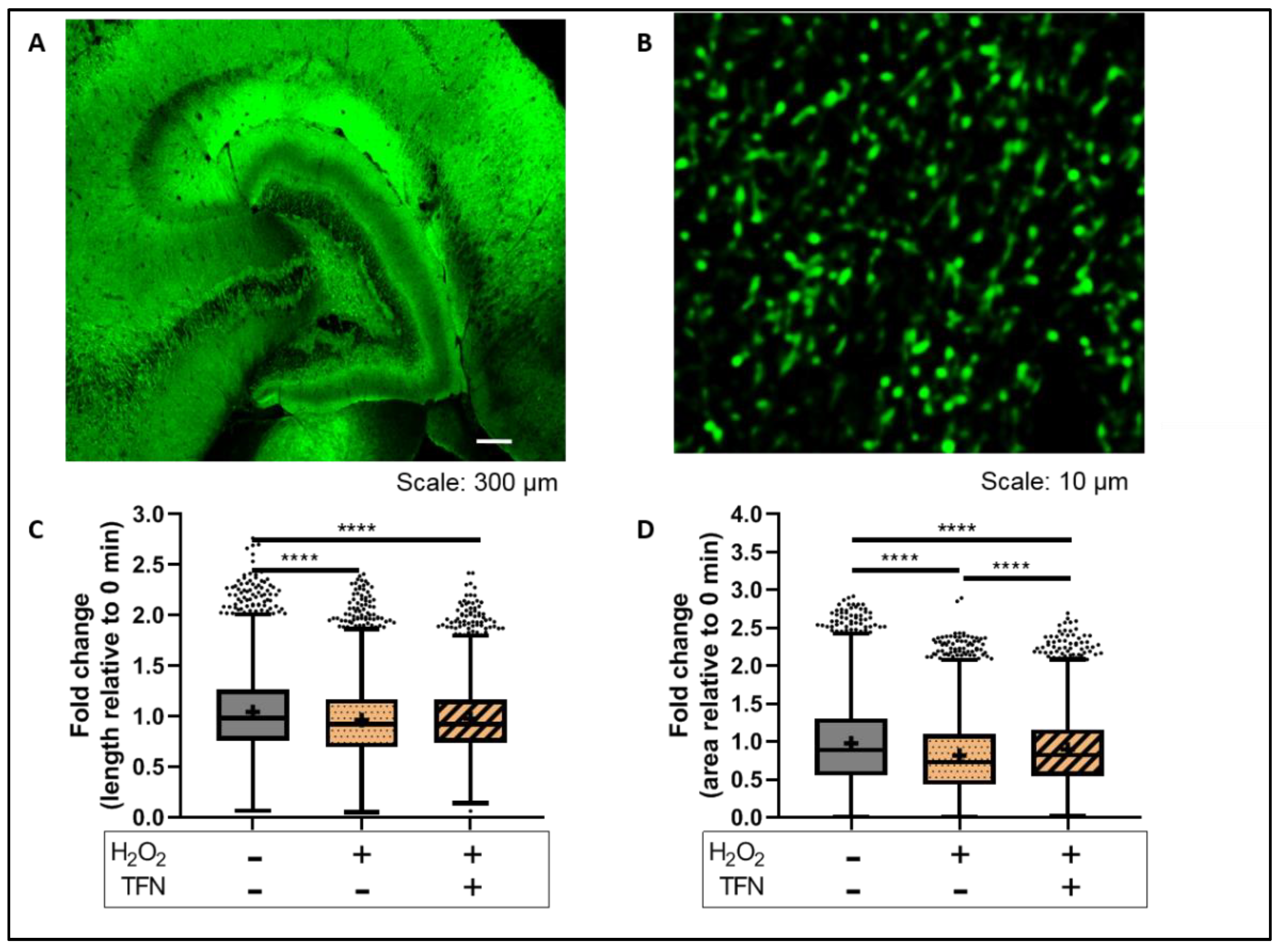
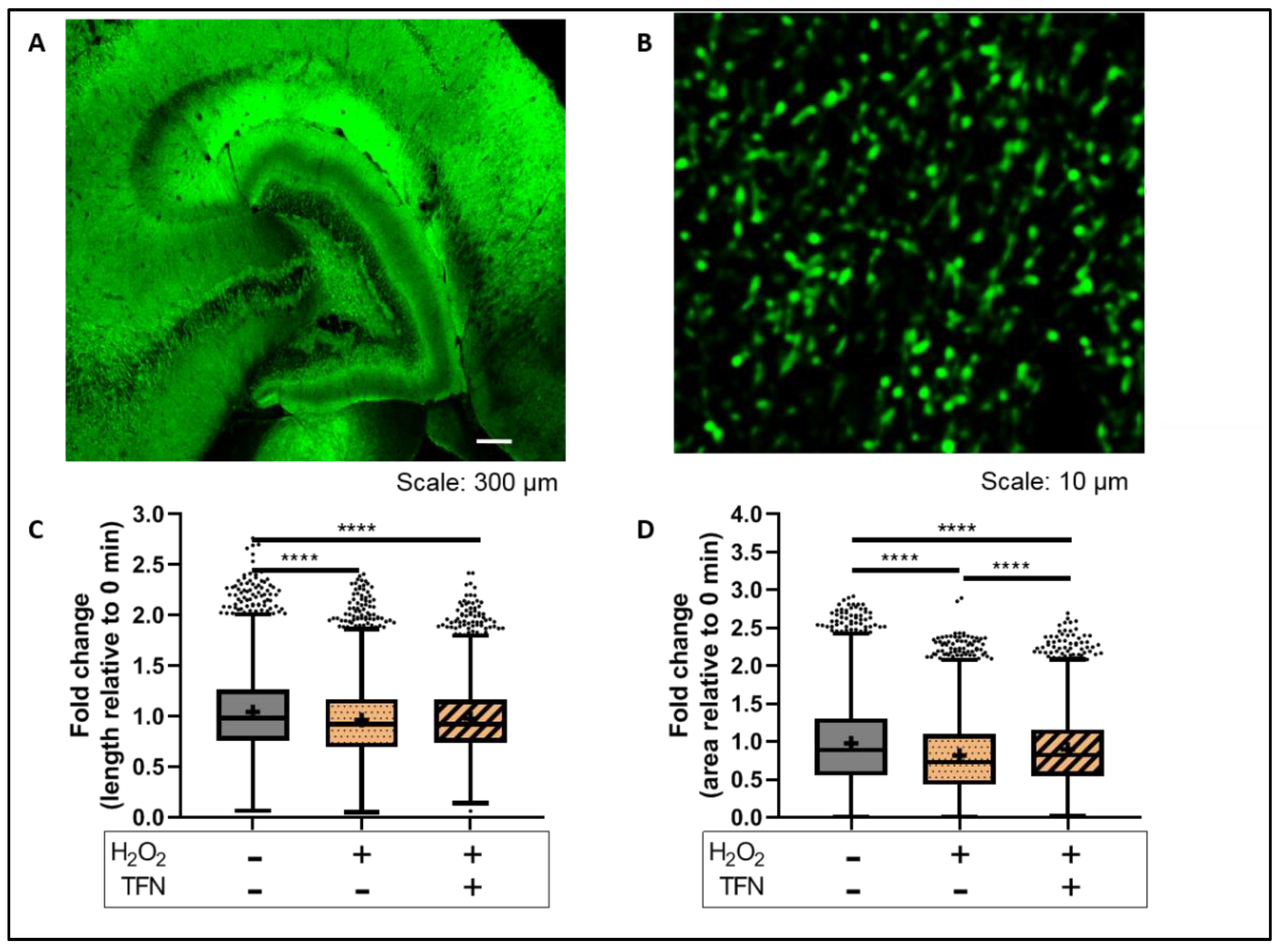
2.4. TFN Prevented Oxidative Stress-Induced Decrease in Mitochondrial Area in Acute Hippocampal Slices
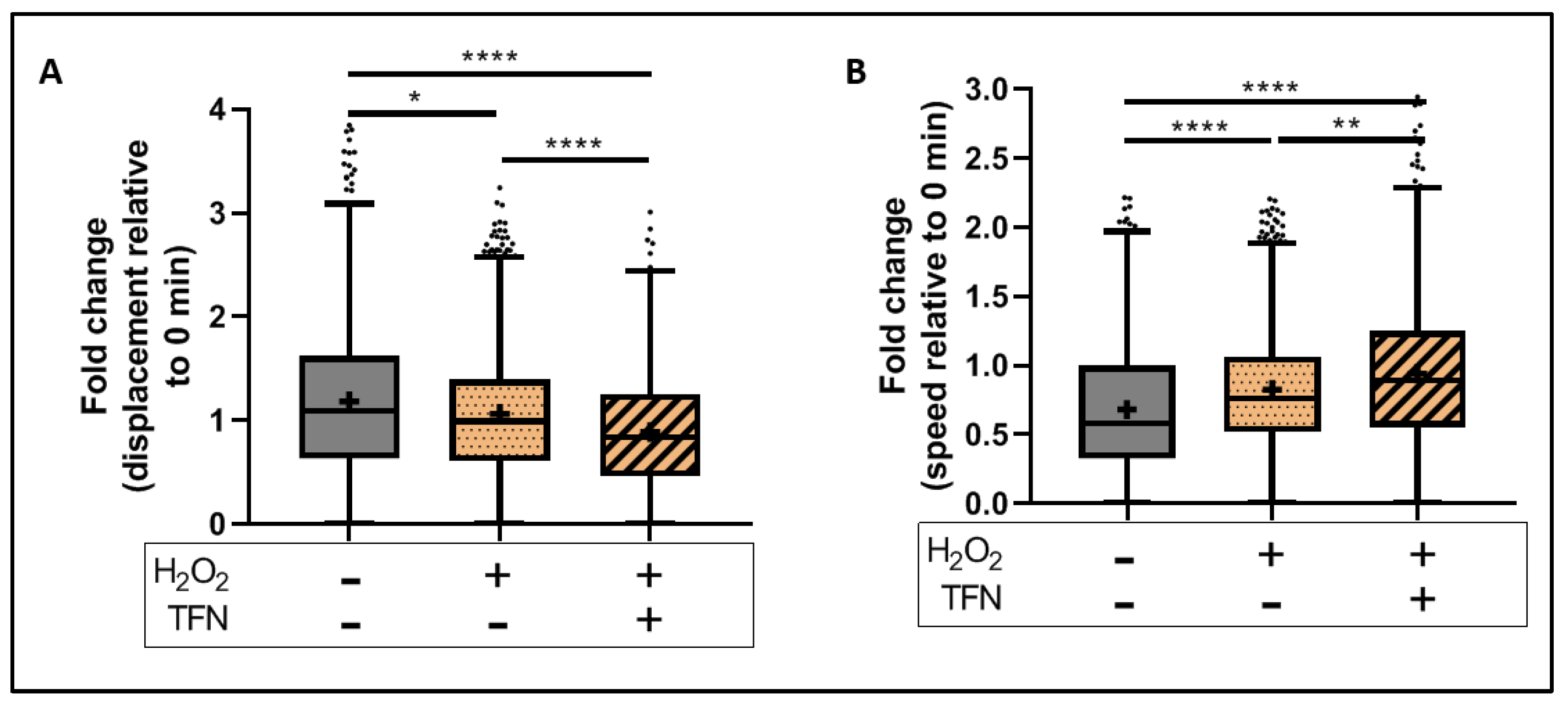
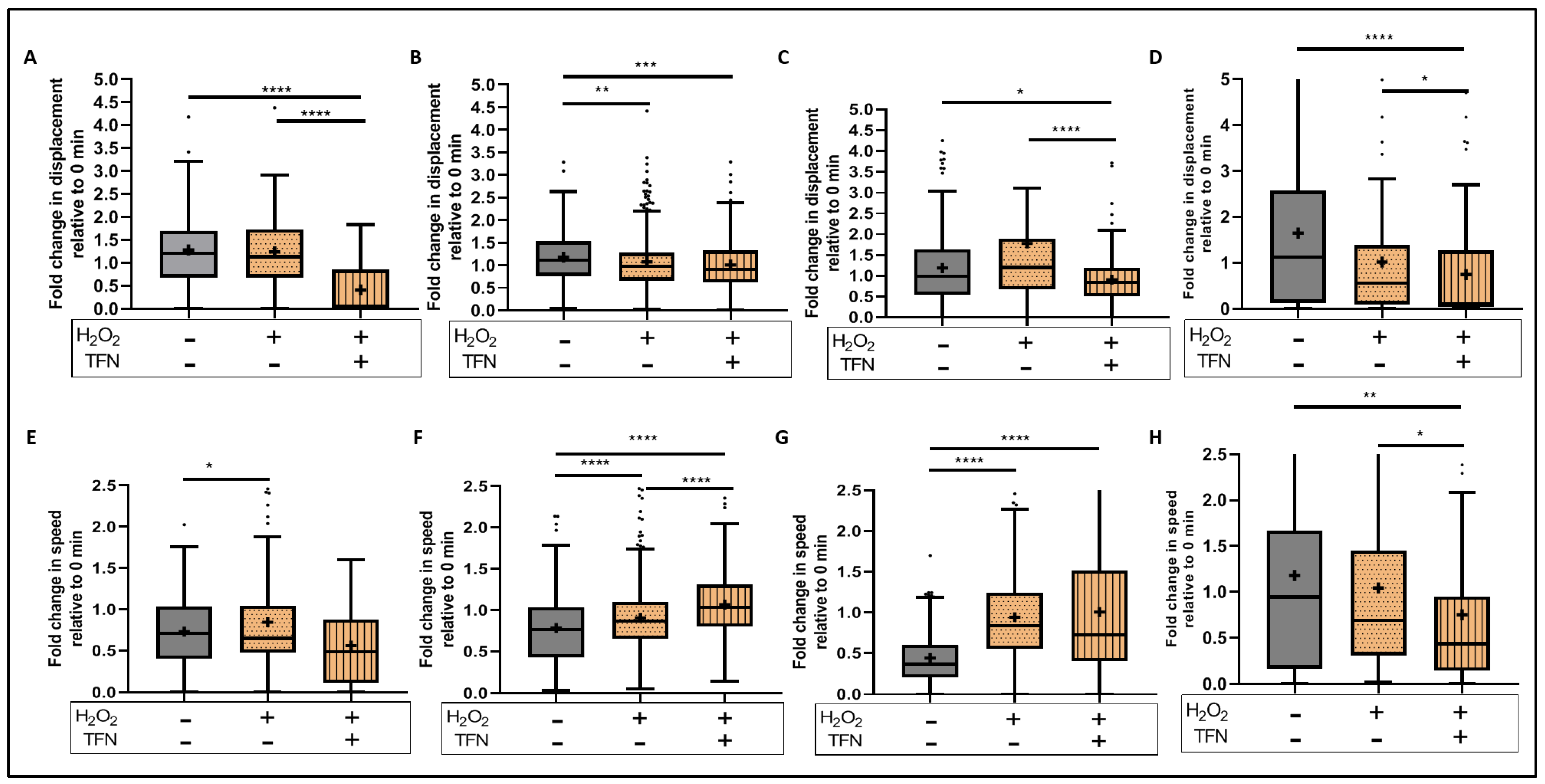
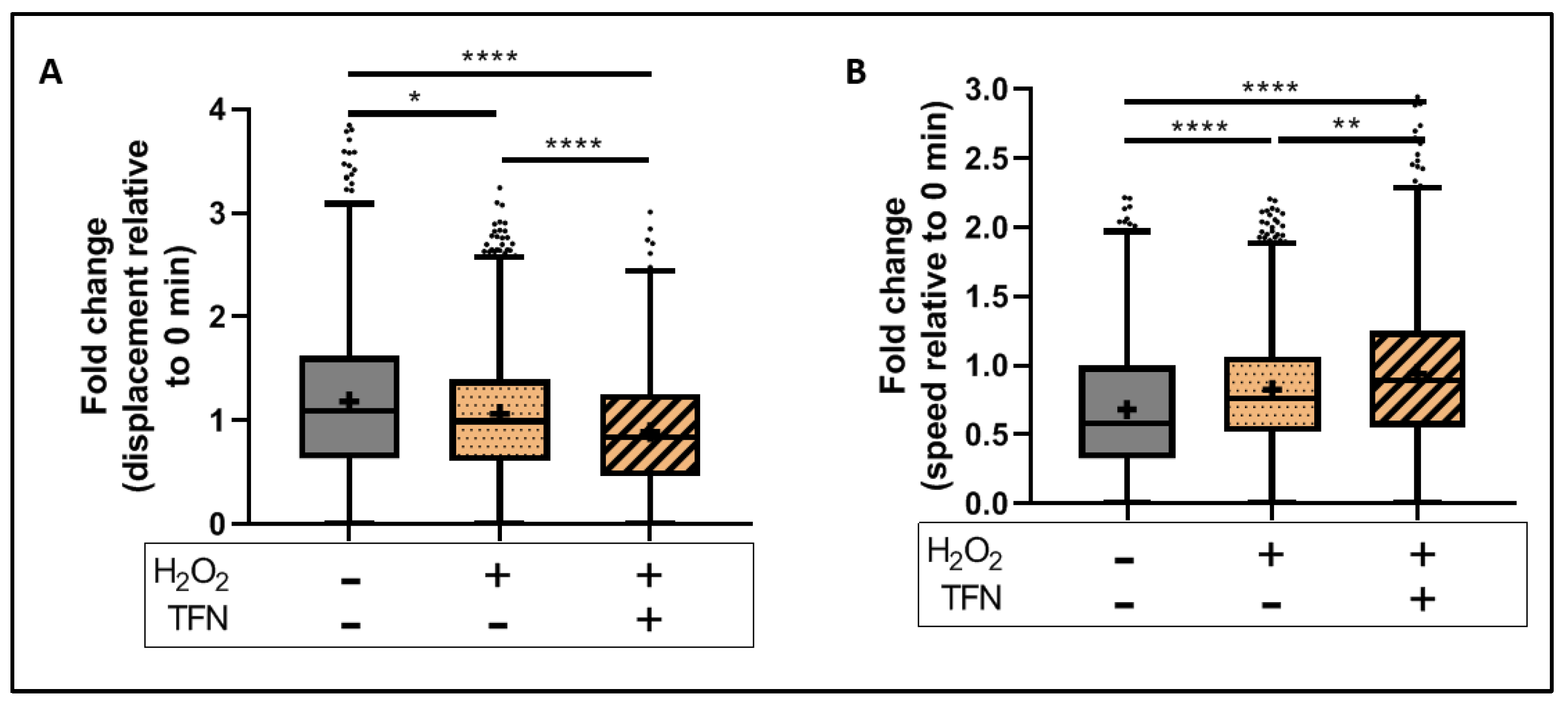
2.5. TFN Did Not Prevent Oxidative Stress-Mediated Alterations in Mitochondrial Motility in Acute Hippocampal Slices
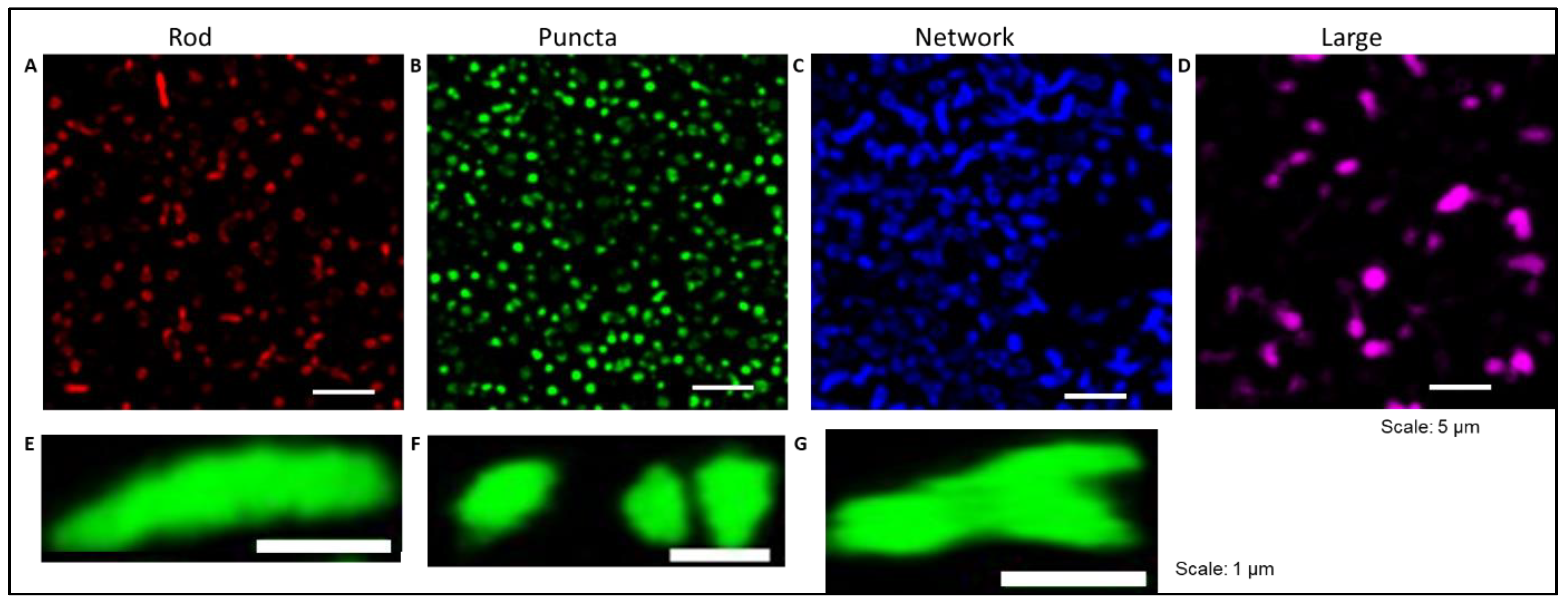
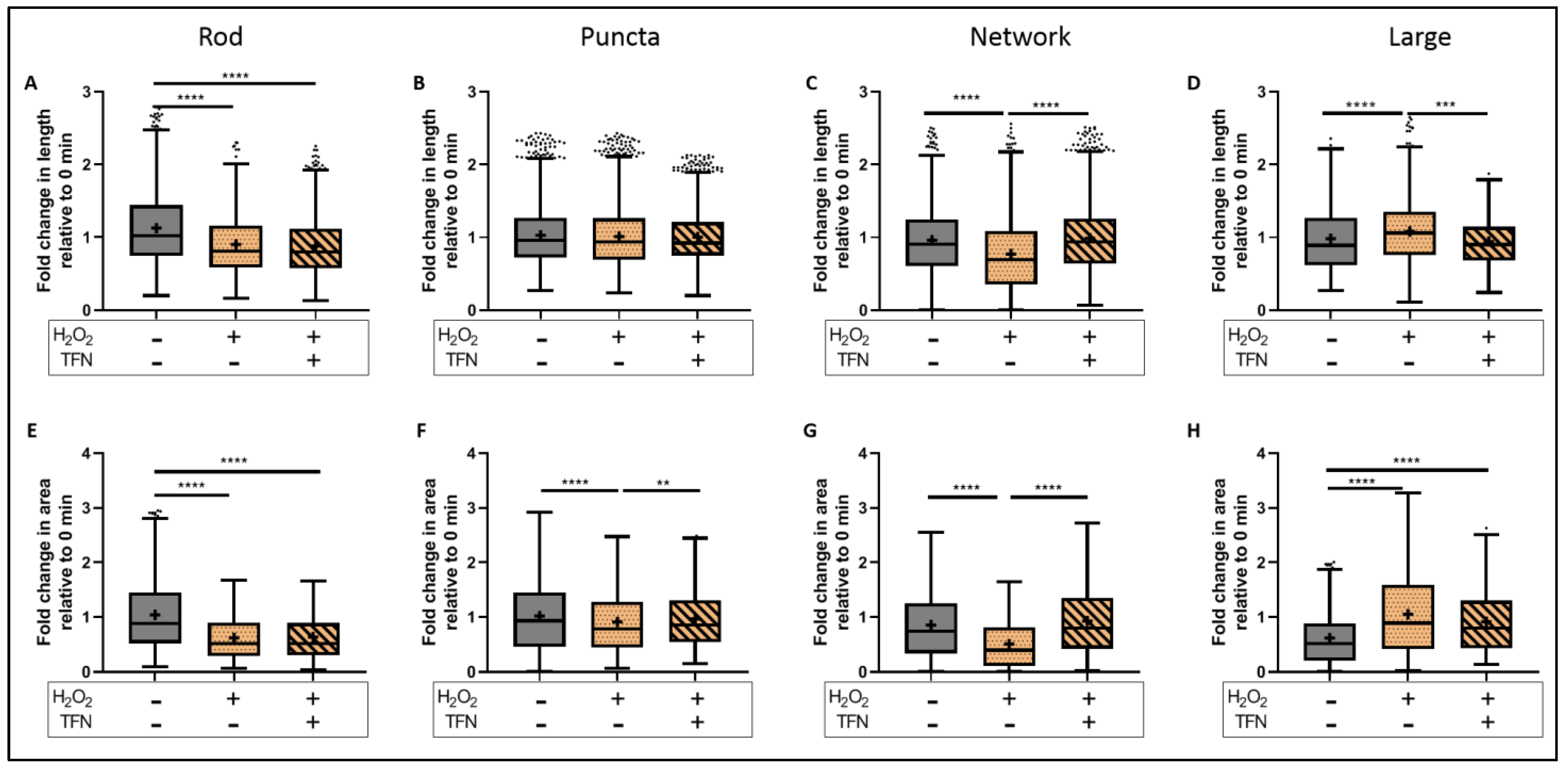
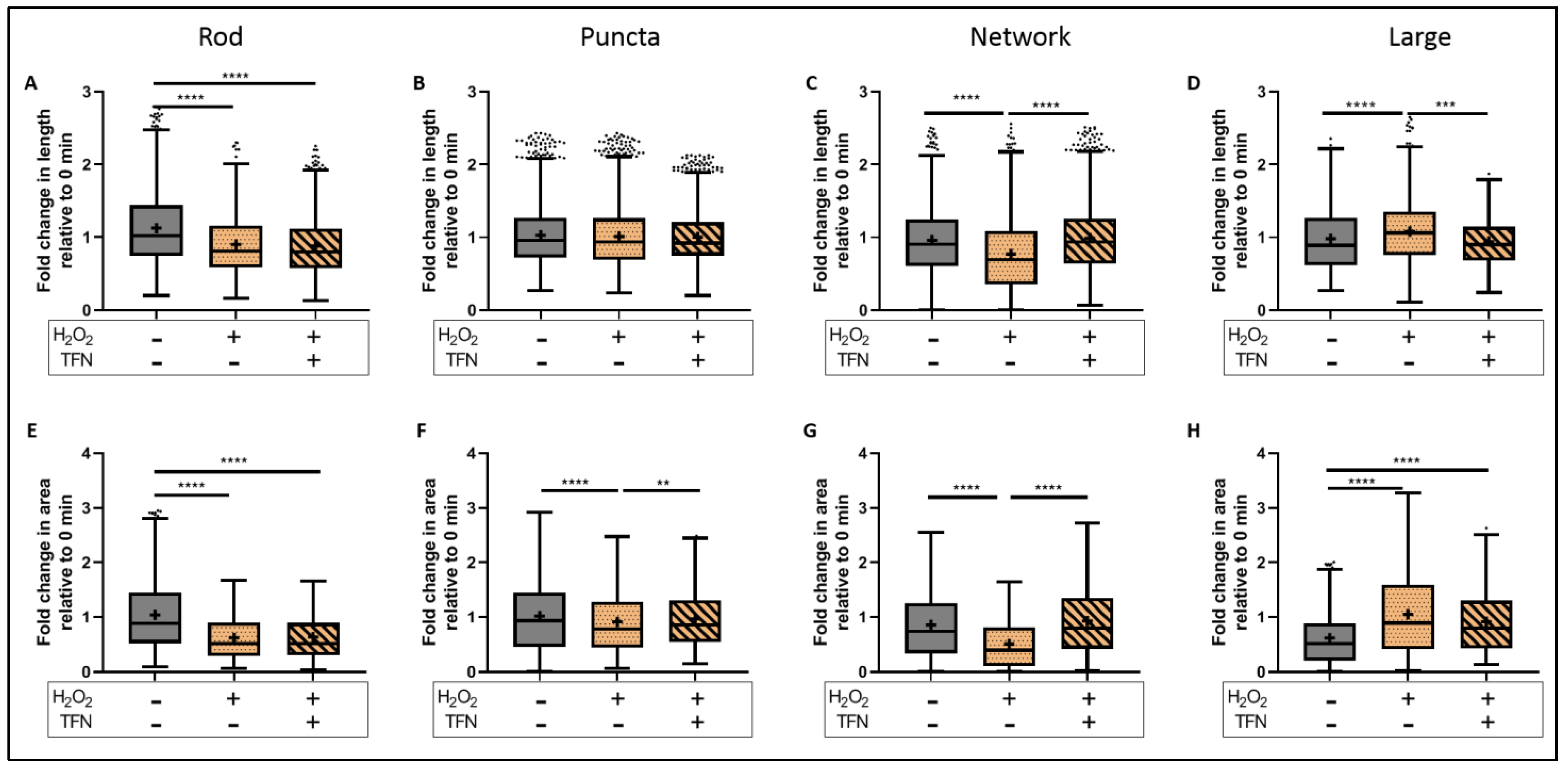
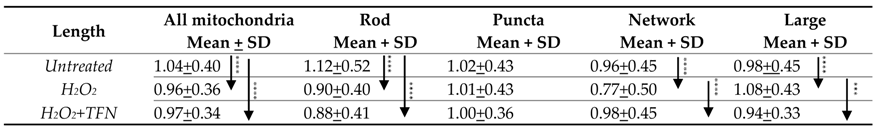
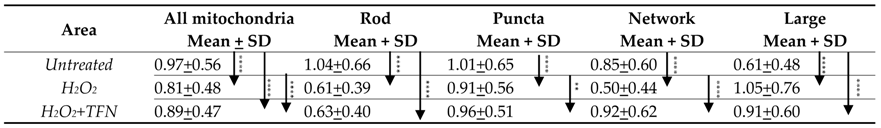
2.6. TFN Prevented the Oxidative Stress-Promoted Decrease in Length of Network Mitochondria and Size of Puncta-Shaped and Network Mitochondria in Acute Hippocampal Slices
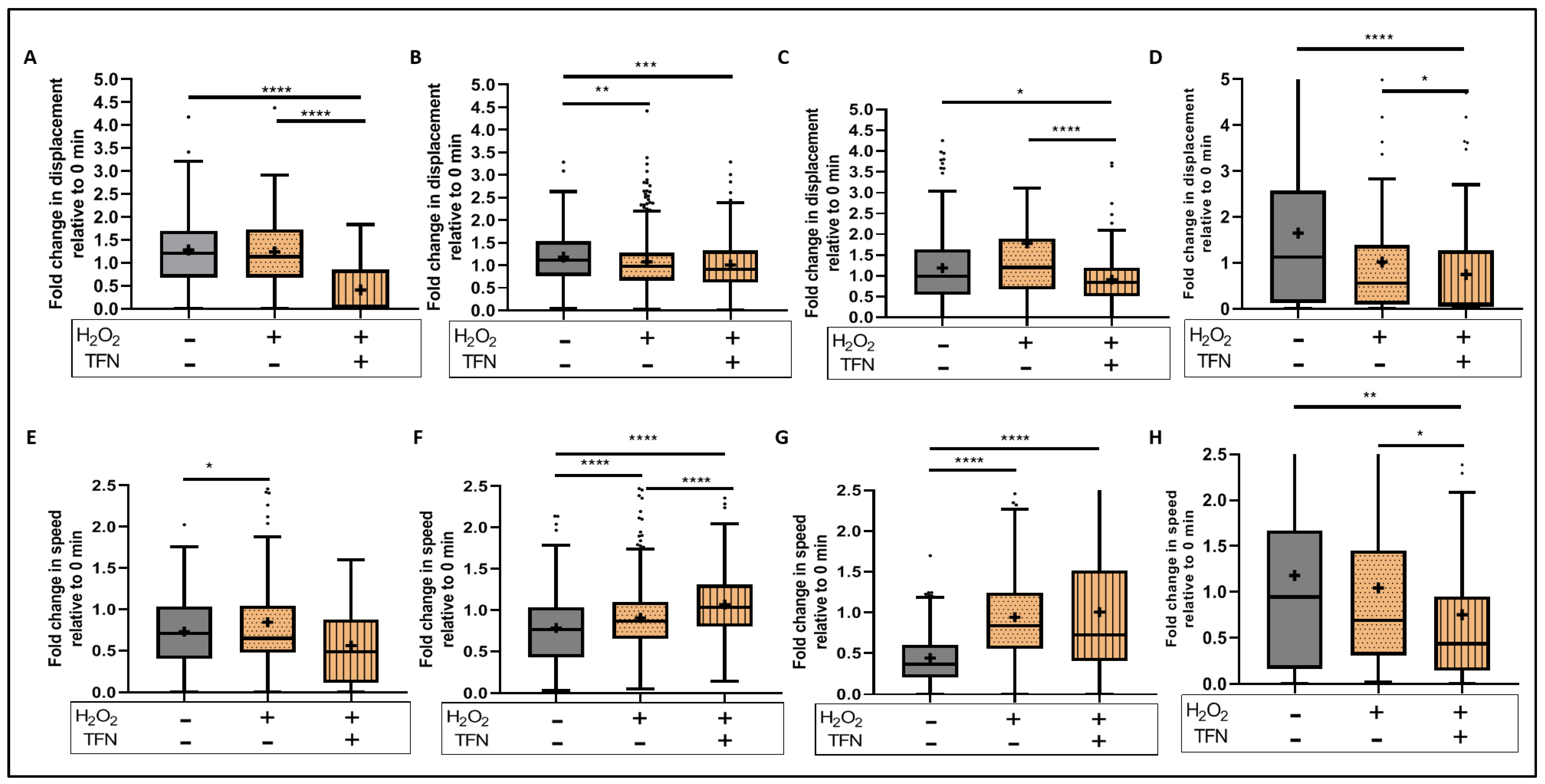
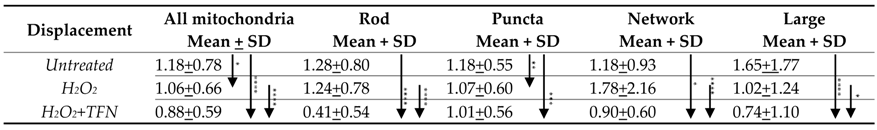
2.7. TFN Enhanced Mitochondrial Speed in Puncta-Shaped Mitochondria in Acute Hippocampal Slices Exposed to Oxidative Stress
3. Discussion
4. Materials and Methods
4.1. Experimental Design and Settings
4.2. Experimental Animals and Ethics Statement
4.3. Preparation of Acute Hippocampal Sections
4.4. Simultaneous Electrophysiology and Oxygen Partial Pressure (pO2) Recordings in Hippocampal Slices
4.5. Adenosine Triphosphate (ATP) Assay
4.6. Mitochondrial Imaging Using Two-Photon Laser Scanning Microscope in Acute Hippocampus Sections
4.7. Image Analysis of Mitochondrial Morphology and Dynamics in Acute Hippocampal Slices
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassmann, H. Multiple Sclerosis Pathology. Cold Spring Harb. Perspect. Med. 2018, 8, a028936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Sospedra, M.; Martin, R. Immunology of Multiple Sclerosis. Semin. Neurol. 2016, 36, 115–127. [Google Scholar] [CrossRef]
- Mossakowski, A.A.; Pohlan, J.; Bremer, D.; Lindquist, R.; Millward, J.M.; Bock, M.; Pollok, K.; Mothes, R.; Viohl, L.; Radbruch, M.; et al. Tracking CNS and systemic sources of oxidative stress during the course of chronic neuroinflammation. Acta Neuropathol. 2015, 130, 799–814. [Google Scholar] [CrossRef] [Green Version]
- Barsukova, A.G.; Forte, M.; Bourdette, D. Focal Increases of Axoplasmic Ca2+, Aggregation of Sodium-Calcium Exchanger, N-type Ca2+ Channel, and Actin Define the Sites of Spheroids in Axons Undergoing Oxidative Stress. J. Neurosci. 2012, 32, 12028–12037. [Google Scholar] [CrossRef] [Green Version]
- Lassmann, H. Targets of therapy in progressive MS. Mult. Scler. J. 2017, 23, 1593–1599. [Google Scholar] [CrossRef] [Green Version]
- De Barcelos, I.P.; Troxell, R.M.; Graves, J.S. Mitochondrial Dysfunction and Multiple Sclerosis. Biology 2019, 8, 37. [Google Scholar] [CrossRef] [Green Version]
- Federico, A.; Cardaioli, E.; Da Pozzo, P.; Formichi, P.; Gallus, G.N.; Radi, E. Mitochondria, oxidative stress and neurodegeneration. J. Neurol. Sci. 2012, 322, 254–262. [Google Scholar] [CrossRef]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Nikic, I.; Merkler, D.; Sorbara, C.; Brinkoetter, M.; Kreutzfeldt, M.; Bareyre, F.M.; Brück, W.; Bishop, D.; Misgeld, T.; Kerschensteiner, M. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 2011, 17, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.; Ziabreva, I.; Lassmann, H.; Turnbull, D. Mitochondrial defects in acute multiple sclerosis lesions. Brain 2008, 131, 1722–1735. [Google Scholar] [CrossRef] [PubMed]
- Stys, P.K.; Zamponi, G.W.; Van Minnen, J.; Geurts, J.J.G. Will the real multiple sclerosis please stand up? Nat. Rev. Neurosci. 2012, 13, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Faissner, S.; Plemel, J.R.; Gold, R.; Yong, V.W. Progressive multiple sclerosis: From pathophysiology to therapeutic strategies. Nat. Rev. Drug Discov. 2019, 18, 905–922. [Google Scholar] [CrossRef]
- Campbell, G.; Licht-Mayer, S.; Mahad, D. Targeting mitochondria to protect axons in progressive MS. Neurosci. Lett. 2019, 710, 134258. [Google Scholar] [CrossRef]
- Bargiela, D.; Chinnery, P.F. Mitochondria in neuroinflammation—Multiple sclerosis (MS), leber hereditary optic neuropathy (LHON) and LHON-MS. Neurosci. Lett. 2019, 710, 132932. [Google Scholar] [CrossRef]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.E. Oral teriflunomide in the treatment of relapsing forms of multiple sclerosis: Clinical evidence and long-term experience. Ther. Adv. Neurol. Disord. 2017, 10, 381–396. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, P.; Comi, G.; Freedman, M.S.; Miller, A.E.; Kappos, L.; Bouchard, J.-P.; Lebrun-Frenay, C.; Mares, J.; Benamor, M.; Thangavelu, K.; et al. Long-term safety and efficacy of teriflunomide: Nine-year follow-up of the randomized TEMSO study. Neurology 2016, 86, 920–930. [Google Scholar] [CrossRef] [Green Version]
- Keen, H.I.; Conaghan, P.; Tett, S.E. Safety evaluation of leflunomide in rheumatoid arthritis. Expert Opin. Drug Saf. 2013, 12, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Bar-Or, A. Teriflunomide (Aubagio®) for the treatment of multiple sclerosis. Exp. Neurol. 2014, 262 Pt A, 57–65. [Google Scholar] [CrossRef]
- Boukalova, S.; Hubackova, S.; Milosevic, M.; Ezrova, Z.; Neuzil, J.; Rohlena, J. Dihydroorotate dehydrogenase in oxidative phosphorylation and cancer. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165759. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, B.; Pellkofer, H.; Weber, M.S. The Use of Oral Disease-Modifying Therapies in Multiple Sclerosis. Curr. Neurol. Neurosci. Rep. 2016, 16, 38. [Google Scholar] [CrossRef] [PubMed]
- Kieseier, B.C.; Warnke, C.; zu Horste, G.M.; Hartung, H.-P.; Stüve, O. Review of teriflunomide and its potential in the treatment of multiple sclerosis. Neuropsychiatr. Dis. Treat. 2009, 5, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Friese, M.A.; Schattling, B.; Fugger, L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat. Rev. Neurol. 2014, 10, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Vécsei, L. Monitoring the Redox Status in Multiple Sclerosis. Biomedicines 2020, 8, 406. [Google Scholar] [CrossRef] [PubMed]
- Malla, B.; Cotten, S.; Ulshoefer, R.; Paul, F.; Hauser, A.E.; Niesner, R.; Bros, H.; Infante-Duarte, C. Teriflunomide preserves peripheral nerve mitochondria from oxidative stress-mediated alterations. Ther. Adv. Chronic Dis. 2020, 11, 2040622320944773. [Google Scholar] [CrossRef]
- Nitsch, R.; Pohl, E.E.; Smorodchenko, A.; Infante-Duarte, C.; Aktas, O.; Zipp, F. Direct Impact of T Cells on Neurons Revealed by Two-Photon Microscopy in Living Brain Tissue. J. Neurosci. 2004, 24, 2458–2464. [Google Scholar] [CrossRef] [Green Version]
- Bros, H.; Millward, J.M.; Paul, F.; Niesner, R.; Infante-Duarte, C. Oxidative damage to mitochondria at the nodes of Ranvier precedes axon degeneration in ex vivo transected axons. Exp. Neurol. 2014, 261, 127–135. [Google Scholar] [CrossRef]
- Ohashi, M.; Hirano, T.; Watanabe, K.; Shoji, H.; Ohashi, N.; Baba, H.; Endo, N.; Kohno, T. Hydrogen peroxide modulates neuronal excitability and membrane properties in ventral horn neurons of the rat spinal cord. Neuroscience 2016, 331, 206–220. [Google Scholar] [CrossRef]
- Scialò, F.; Fernández-Ayala, D.J.; Sanz, A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front. Physiol. 2017, 8, 428. [Google Scholar] [CrossRef] [PubMed]
- Löffler, M.; Carrey, E.A.; Zameitat, E. New perspectives on the roles of pyrimidines in the central nervous system. Nucleosides Nucleotides Nucleic Acids 2018, 37, 290–306. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.A.; Cash, N.J.; Ouyang, Y.; Morton, J.; Chvanov, M.; Latawiec, D.; Awais, M.; Tepikin, A.; Sutton, R.; Criddle, D.N. Oxidative stress alters mitochondrial bioenergetics and modifies pancreatic cell death independently of cyclophilin D, resulting in an apoptosis-to-necrosis shift. J. Biol. Chem. 2018, 293, 8032–8047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Bliek, A.M. Fussy mitochondria fuse in response to stress. EMBO J. 2009, 28, 1533–1534. [Google Scholar] [CrossRef] [PubMed]
- Aryaman, J.; Johnston, I.G.; Jones, N.S. Mitochondrial Heterogeneity. Front. Genet. 2019, 9, 718. [Google Scholar] [CrossRef] [Green Version]
- Campello, S.; Scorrano, L. Mitochondrial shape changes: Orchestrating cell pathophysiology. EMBO Rep. 2010, 11, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Hoitzing, H.; Johnston, I.G.; Jones, N.S. What is the function of mitochondrial networks? A theoretical assessment of hypotheses and proposal for future research. BioEssays 2015, 37, 687–700. [Google Scholar] [CrossRef] [Green Version]
- Palmer, C.S.; Osellame, L.D.; Stojanovski, D.; Ryan, M.T. The regulation of mitochondrial morphology: Intricate mechanisms and dynamic machinery. Cell. Signal. 2011, 23, 1534–1545. [Google Scholar] [CrossRef]
- Rafelski, S.M. Mitochondrial network morphology: Building an integrative, geometrical view. BMC Biol. 2013, 11, 71. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Vorobjev, I.A.; Popkov, V.A.; Babenko, V.A.; Zorova, L.D.; Pevzner, I.B.; Silachev, D.N.; Zorov, S.D.; Andrianova, N.V.; Plotnikov, E.Y. Lessons from the Discovery of Mitochondrial Fragmentation (Fission): A Review and Update. Cells 2019, 8, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009, 28, 1589–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casals, L.M.; Sebastián, D.; Brea, J.; Rico-Leo, E.M.; Palacín, M.; Fernández-Salguero, P.M.; Loza, M.I.; Albericio, F.; Zorzano, A. Identification of New Activators of Mitochondrial Fusion Reveals a Link between Mitochondrial Morphology and Pyrimidine Metabolism. Cell Chem. Biol. 2018, 25, 268–278.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Li, Z.; Zhang, Q.; Chen, L.; Huang, X.; Zhang, Y.; Liu, X.; Liu, W.; Li, W. Mechanisms Underlying H2O2-Evoked Carbonyl Modification of Cytoskeletal Protein and Axon Injury in PC-12 Cells. Cell. Physiol. Biochem. 2018, 48, 1088–1098. [Google Scholar] [CrossRef] [PubMed]
- Löffler, M.; Carrey, E.A.; Knecht, W. The pathway to pyrimidines: The essential focus on dihydroorotate dehydrogenase, the mitochondrial enzyme coupled to the respiratory chain. Nucleosides Nucleotides Nucleic Acids 2020, 39, 1281–1305. [Google Scholar] [CrossRef] [PubMed]
- Gülden, M.; Jess, A.; Kammann, J.; Maser, E.; Seibert, H. Cytotoxic potency of H2O2 in cell cultures: Impact of cell concentration and exposure time. Free Radic. Biol. Med. 2010, 49, 1298–1305. [Google Scholar] [CrossRef]
- Iwakami, S.; Misu, H.; Takeda, T.; Sugimori, M.; Matsugo, S.; Kaneko, S.; Takamura, T. Concentration-dependent Dual Effects of Hydrogen Peroxide on Insulin Signal Transduction in H4IIEC Hepatocytes. PLoS ONE 2011, 6, e27401. [Google Scholar] [CrossRef]
- Berndt, N.; Rösner, J.; Haq, R.U.; Kann, O.; Kovács, R.; Holzhütter, H.-G.; Spies, C.; Liotta, A. Possible neurotoxicity of the anesthetic propofol: Evidence for the inhibition of complex II of the respiratory chain in area CA3 of rat hippocampal slices. Arch. Toxicol. 2018, 92, 3191–3205. [Google Scholar] [CrossRef]
- Schindelin, J.; Rueden, C.T.; Hiner, M.C.; Eliceiri, K.W. The ImageJ ecosystem: An open platform for biomedical image analysis. Mol. Reprod. Dev. 2015, 82, 518–529. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Thevenaz, P.; Ruttimann, U.; Unser, M. A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. 1998, 7, 27–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arganda-Carreras, I.; Kaynig, V.; Rueden, C.; Eliceiri, K.W.; Schindelin, J.; Cardona, A.; Seung, H.S. Trainable Weka Segmentation: A machine learning tool for microscopy pixel classification. Bioinformatics 2017, 33, 2424–2426. [Google Scholar] [CrossRef] [PubMed]
- Jaqaman, K.; Loerke, D.; Mettlen, M.; Kuwata, H.; Grinstein, S.; Schmid, S.L.; Danuser, G. Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods 2008, 5, 695–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawls, J.; Knecht, W.; Diekert, K.; Lill, R.; Löffler, M. Requirements for the mitochondrial import and localization of dihydroorotate dehydrogenase. Eur. J. Biochem. 2000, 267, 2079–2087. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 |
 |
 |
 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malla, B.; Liotta, A.; Bros, H.; Ulshöfer, R.; Paul, F.; Hauser, A.E.; Niesner, R.; Infante-Duarte, C. Teriflunomide Preserves Neuronal Activity and Protects Mitochondria in Brain Slices Exposed to Oxidative Stress. Int. J. Mol. Sci. 2022, 23, 1538. https://doi.org/10.3390/ijms23031538
Malla B, Liotta A, Bros H, Ulshöfer R, Paul F, Hauser AE, Niesner R, Infante-Duarte C. Teriflunomide Preserves Neuronal Activity and Protects Mitochondria in Brain Slices Exposed to Oxidative Stress. International Journal of Molecular Sciences. 2022; 23(3):1538. https://doi.org/10.3390/ijms23031538
Chicago/Turabian StyleMalla, Bimala, Agustin Liotta, Helena Bros, Rebecca Ulshöfer, Friedemann Paul, Anja E. Hauser, Raluca Niesner, and Carmen Infante-Duarte. 2022. "Teriflunomide Preserves Neuronal Activity and Protects Mitochondria in Brain Slices Exposed to Oxidative Stress" International Journal of Molecular Sciences 23, no. 3: 1538. https://doi.org/10.3390/ijms23031538
APA StyleMalla, B., Liotta, A., Bros, H., Ulshöfer, R., Paul, F., Hauser, A. E., Niesner, R., & Infante-Duarte, C. (2022). Teriflunomide Preserves Neuronal Activity and Protects Mitochondria in Brain Slices Exposed to Oxidative Stress. International Journal of Molecular Sciences, 23(3), 1538. https://doi.org/10.3390/ijms23031538