
Molecular Systems Architecture of Interactome in the Acute Myeloid Leukemia Microenvironment


Abstract
:Simple Summary
Abstract
1. Introduction
2. Literature Review
- creating a list of Medical Subject Headings (MeSH) keywords to optimize the recall and precision of peer-reviewed articles (listed in Table 2);
- searching and retrieving the relevant peer-reviewed articles published between January 1980 to June 2021 from PubMed, Medline, and Google Scholar, which were stored as an “Initial Set” repository;
- screening the titles and abstracts of the articles in the Initial Set repository to identify the most relevant articles based on our inclusion criteria, which were stored as the “Final Set” repository; and
- performing a full-length review of the Final Set repository using the domain experts.
The Inclusion Criteria
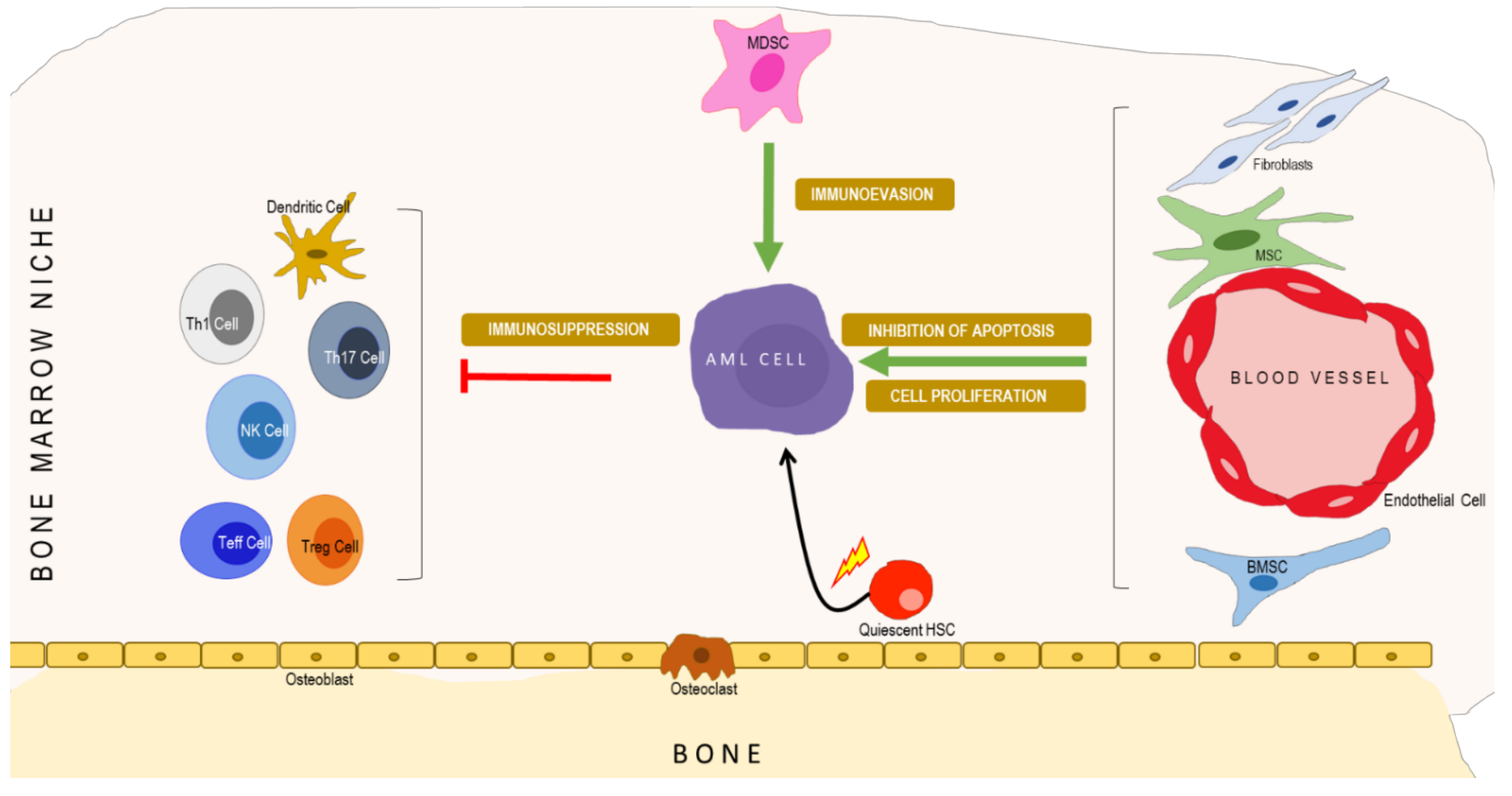
3. Molecular Systems Architecture of AML
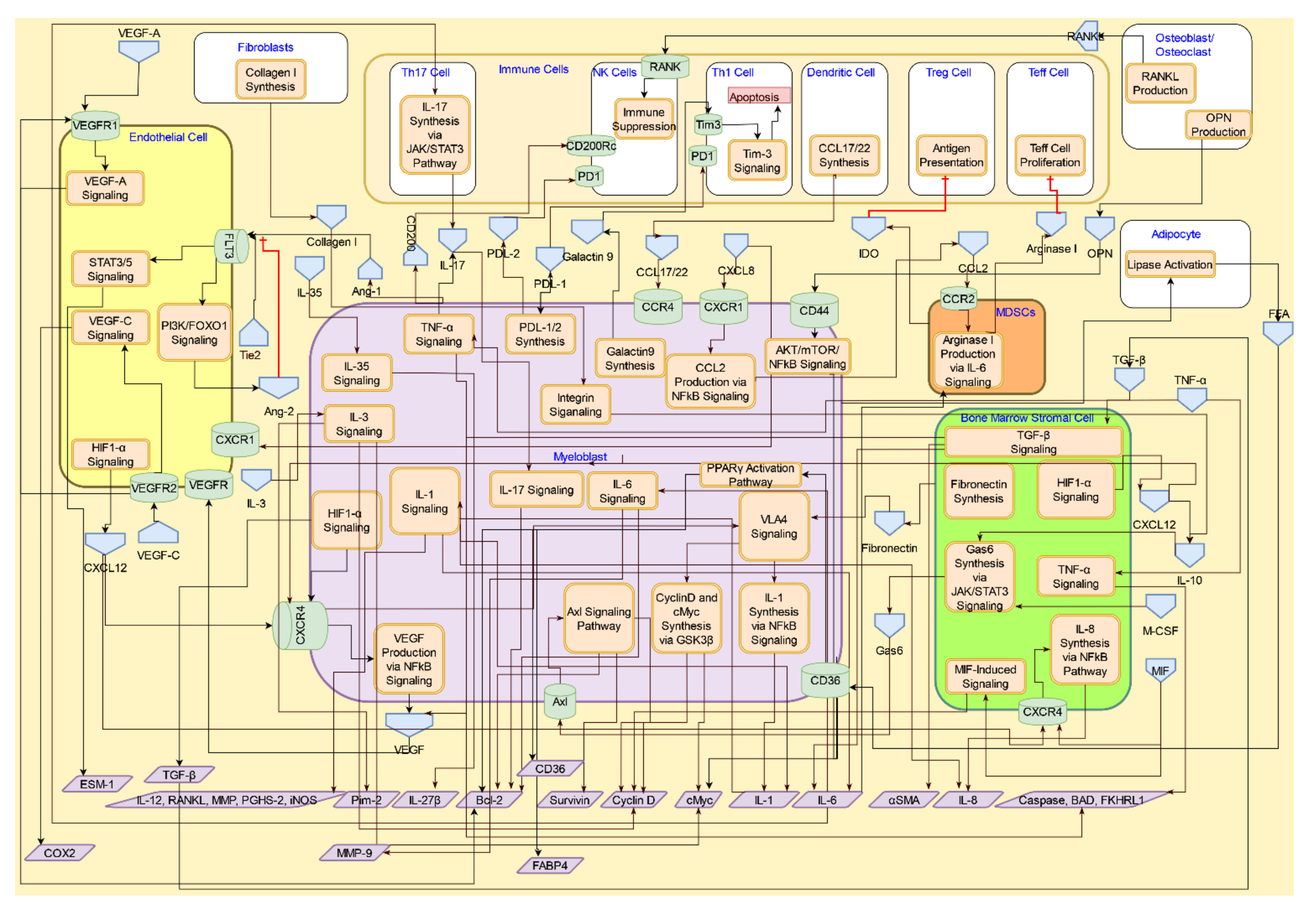
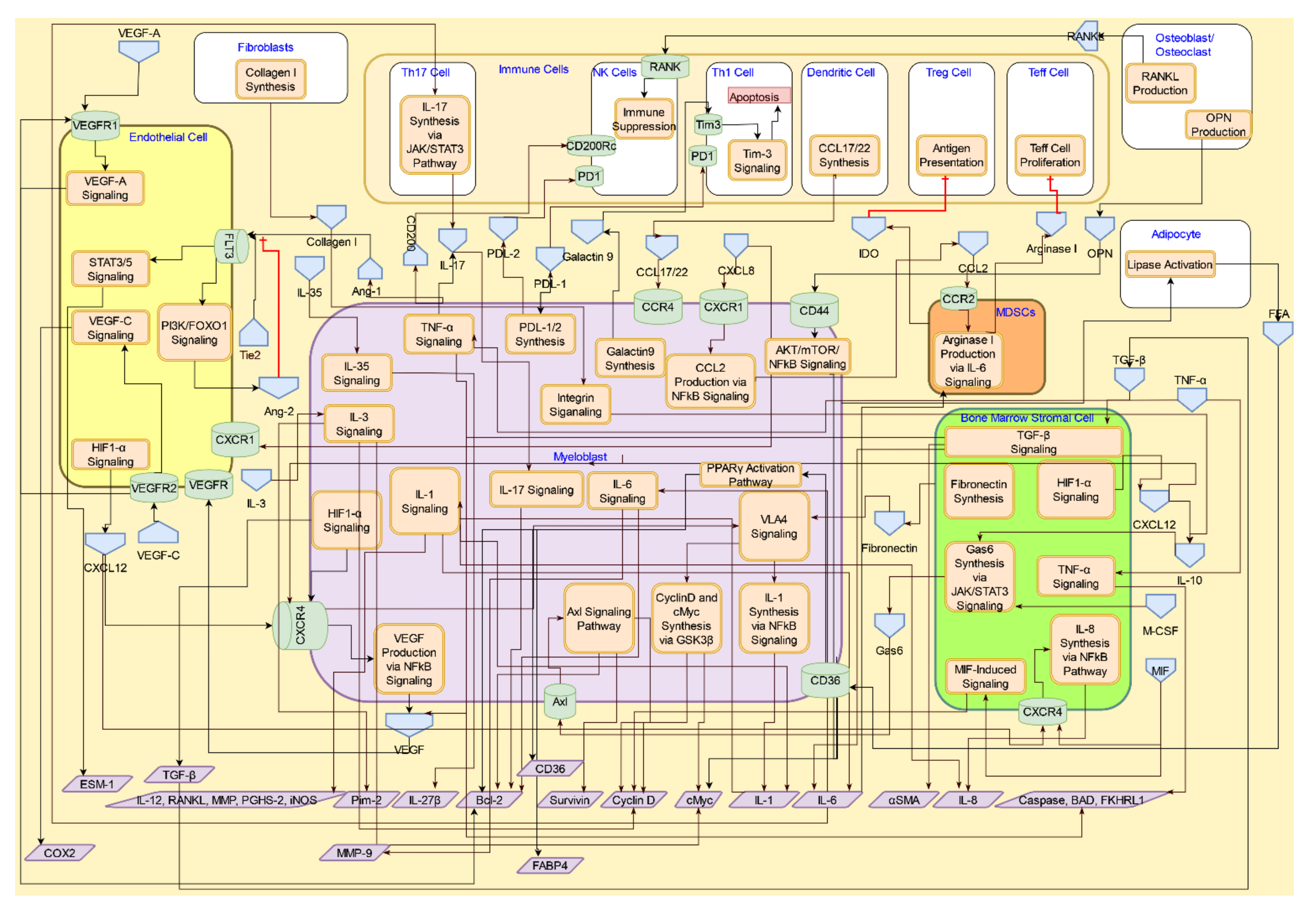
4. Interactive Signaling in the AML Microenvironment
4.1. Interactive Crosstalk between AML Cells, Bone Marrow Stromal Cells, Endothelial Cells, Osteoblasts, and Adipocytes
4.1.1. CXCR4/CXCL12 Signaling
4.1.2. TGF-β Signaling
4.1.3. RANK/RANKL and Osteopontin Signaling in Osteoblasts/Osteoclasts
4.2. Interactive Crosstalk Signaling between AML Cells and Endothelial Cells via Adhesion Molecules
4.3. Interactive Signaling with Myeloid-derived Suppressor Cells (MDSCs) in AML
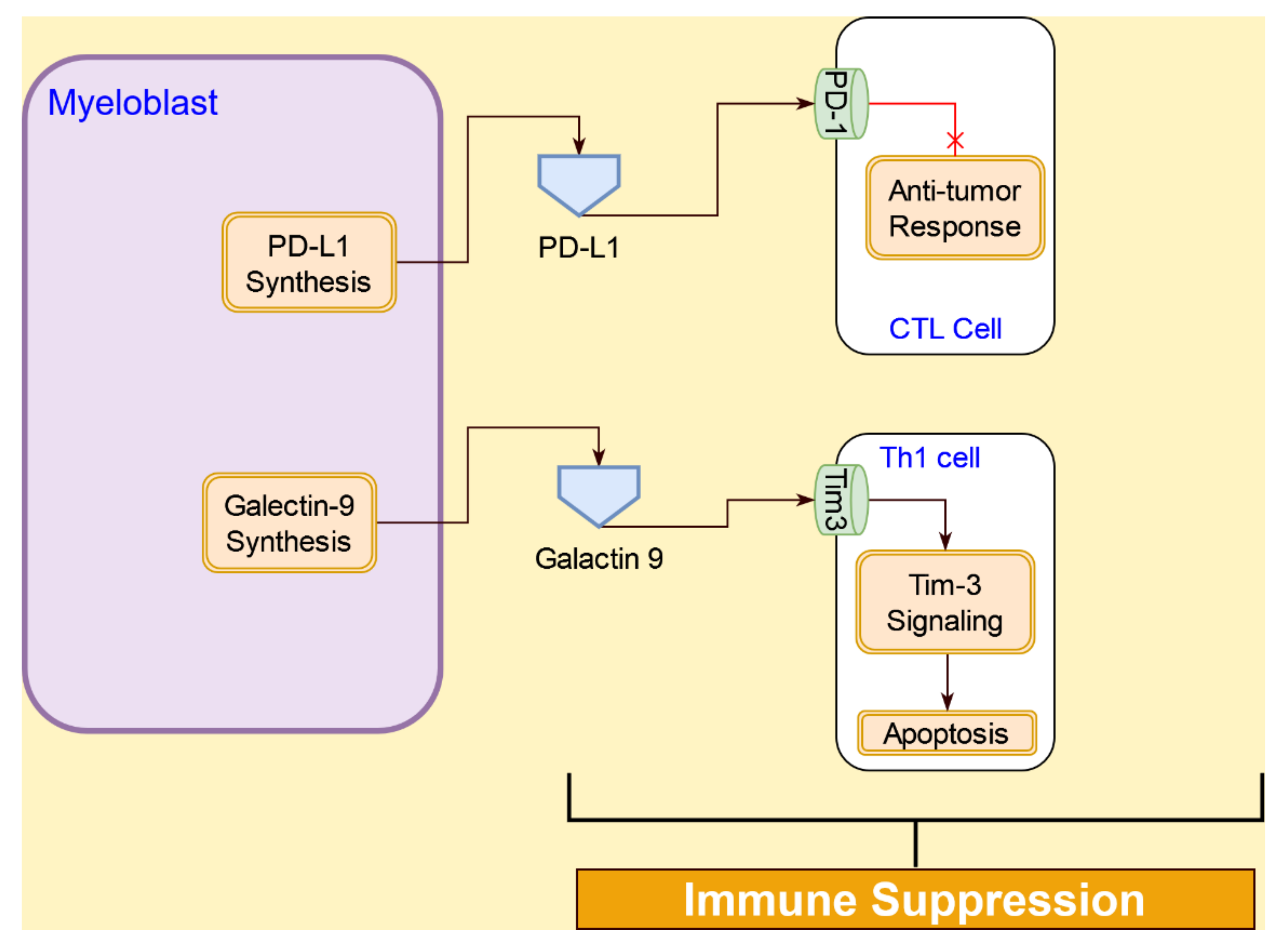
4.4. Interactive Signaling AML Cells and Immune Cells
4.4.1. Interferon α Signaling
4.4.2. Immunosuppressive Interactions of Tregs in AML Tumor Microenvironment
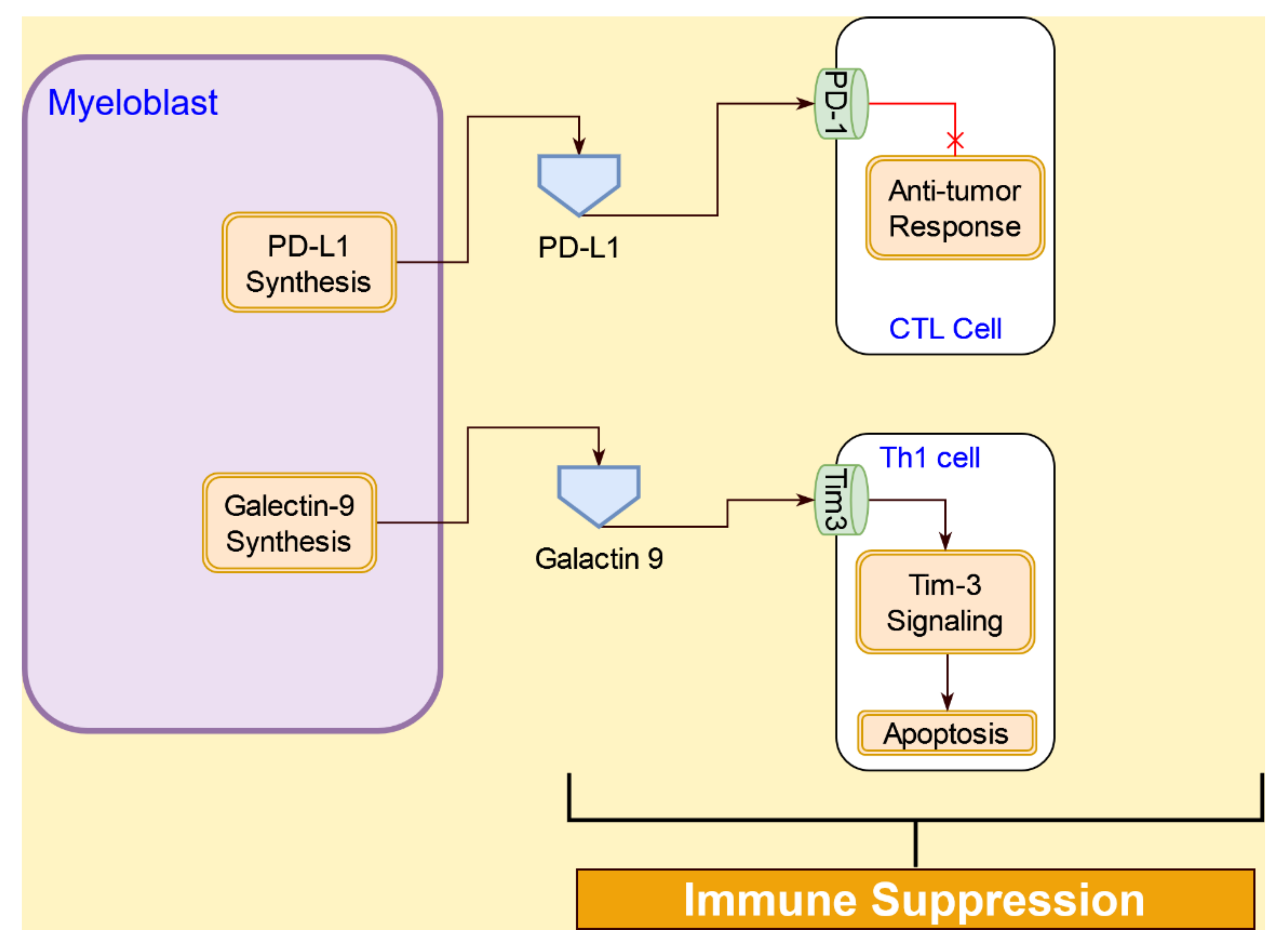
4.4.3. Immunosuppression Interactions of AML Cells with T Cells
4.4.4. Interactive Signaling with Natural Killer (NK) Cells in AML
5. Discussion
6. Future Directions
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Scholl, C.; Gilliland, D.G.; Fröhling, S. Deregulation of signaling pathways in acute myeloid leukemia. Semin. Oncol. 2008, 35, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.M.; Grant, S.; Saleiro, D.; Crispino, J.D.; Hijiya, N.; Giles, F.; Platanias, L.; Eklund, E.A. Targeting novel signaling pathways for resistant acute myeloid leukemia. Mol. Genet. Metab. 2015, 114, 397–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abboud, C.N. Another nail in the AML coffin. Blood 2009, 113, 6045–6046. [Google Scholar] [CrossRef] [PubMed]
- Steffen, B.; Müller-Tidow, C.; Schwäble, J.; Berdel, W.E.; Serve, H. The molecular pathogenesis of acute myeloid leukemia. Crit. Rev. Oncol. Hematol. 2005, 56, 195–221. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Majeti, R. Biology and relevance of human acute myeloid leukemia stem cells. Blood 2017, 129, 1577–1585. [Google Scholar] [CrossRef]
- Hanekamp, D.; Cloos, J.; Schuurhuis, G.J. Leukemic stem cells: Identification and clinical application. Int. J. Hematol. 2017, 105, 549–557. [Google Scholar] [CrossRef]
- Schonherz, A.A.; Bødker, J.S.; Schmitz, A.; Brøndum, R.F.; Jakobsen, L.H.; Roug, A.S.; Severinsen, M.T.; El-Galaly, T.C.; Jensen, P.; Johnsen, H.E.; et al. Normal myeloid progenitor cell subset-associated gene signatures for acute myeloid leukaemia subtyping with prognostic impact. PLoS ONE 2020, 15, e0229593. [Google Scholar] [CrossRef]
- Boulais, P.E.; Frenette, P.S. Making sense of hematopoietic stem cell niches. Blood 2015, 125, 2621–2629. [Google Scholar] [CrossRef] [Green Version]
- Miraki-Moud, F.; Anjos-Afonso, F.; Hodby, K.A.; Griessinger, E.; Rosignoli, G.; Lillington, D.; Jia, L.; Davies, J.K.; Cavenagh, J.; Smith, M.; et al. Acute myeloid leukemia does not deplete normal hematopoietic stem cells but induces cytopenias by impeding their differentiation. Proc. Natl. Acad. Sci. USA 2013, 110, 13576–13581. [Google Scholar] [CrossRef] [Green Version]
- Hérault, A.; Binnewies, M.; Leong, S.; Calero-Nieto, F.J.; Zhang, S.Y.; Kang, Y.A.; Wang, X.; Pietras, E.M.; Chu, S.H.; Barry-Holson, K.; et al. Myeloid progenitor cluster formation drives emergency and leukaemic myelopoiesis. Nature 2017, 544, 53–58. [Google Scholar] [CrossRef]
- Cuartero, S.; Innes, A.J.; Merkenschlager, M. Towards a better understanding of cohesin mutations in AML. Front. Oncol. 2019, 9, 867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renneville, A.; Roumier, C.; Biggio, V.; Nibourel, O.; Boissel, N.; Fenaux, P.; Preudhomme, C. Cooperating gene mutations in acute myeloid leukemia: A review of the literature. Leukemia 2008, 22, 915–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway O’Brien, E.; Prideaux, S.; Chevassut, T. The epigenetic landscape of acute myeloid leukemia. Adv. Hematol. 2014, 2014, 103175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombret, H. Gene mutation and AML pathogenesis. Blood 2011, 118, 5366–5367. [Google Scholar] [CrossRef]
- Kavianpour, M.; Ahmadzadeh, A.; Shahrabi, S.; Saki, N. Significance of oncogenes and tumor suppressor genes in AML prognosis. Tumor Biol. 2016, 37, 10041–10052. [Google Scholar] [CrossRef]
- Staudt, D.; Murray, H.C.; McLachlan, T.; Alvaro, F.; Enjeti, A.K.; Verrills, N.M.; Dun, M.D. Targeting oncogenic signaling in mutant FLT3 acute myeloid leukemia: The path to least resistance. Int. J. Mol. Sci. 2018, 19, 3198. [Google Scholar] [CrossRef] [Green Version]
- Walasek, A. The new perspectives of targeted therapy in acute myeloid leukemia. Adv. Clin. Exp. Med. 2019, 28, 271–276. [Google Scholar] [CrossRef]
- Ayyadurai, V.A.S.; Deonikar, P. Bioactive compounds in green tea may improve transplant tolerance: A computational systems biology analysis. Clin. Nutr. ESPEN 2021, 46, 439–452. [Google Scholar] [CrossRef]
- Oketch-Rabah, H.A.; Hardy, M.L.; Patton, A.P.; Chung, M.; Sarma, N.D.; Yoe, C.; Ayyadurai, V.A.S.; Fox, M.A.; Jordan, S.A.; Mwamburi, M.; et al. Multi-Criteria decision analysis model for assessing the risk from multi-ingredient dietary supplements (MIDS). J. Diet. Suppl. 2021, 18, 293–315. [Google Scholar] [CrossRef] [Green Version]
- Ayyadurai, V.A.S.; Dewey, C.F. CytoSolve: A scalable computational method for dynamic integration of multiple molecular pathway models. Cell. Mol. Bioeng. 2011, 4, 28–45. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.D.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Koo, A.; Nordsletten, D.; Umeton, R.; Yankama, B.; Ayyadurai, S.; García-Cardeña, G.; Dewey, C.F. In silico modeling of shear-stress-induced nitric oxide production in endothelial cells through systems biology. Biophys. J. 2013, 104, 2295–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordsletten, D.A.; Yankama, B.; Umeton, R.; Ayyadurai, V.A.S.; Dewey, C.F. Multiscale mathematical modeling to support drug development. IEEE Trans. Biomed. Eng. 2011, 58, 3508–3512. [Google Scholar] [CrossRef] [PubMed]
- Al-Lazikani, B.; Banerji, U.; Workman, P. Combinatorial drug therapy for cancer in the post-genomic era. Nat. Biotechnol. 2012, 30, 679–692. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. BMJ 2009, 339, 332–336. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Defects in Signaling Pathways Can Lead to Cancer and Other Diseases. In Biochemistry; W H Freeman: New York, NY, USA, 2002. [Google Scholar]
- Schepers, K.; Campbell, T.B.; Passegué, E. Normal and leukemic stem cell niches: Insights and therapeutic opportunities. Cell Stem Cell 2015, 16, 254–267. [Google Scholar] [CrossRef] [Green Version]
- Cogle, C.R.; Saki, N.; Khodadi, E.; Li, J.; Shahjahani, M.; Azizidoost, S. Bone marrow niche in the myelodysplastic syndromes. Leuk. Res. 2015, 39, 1020–1027. [Google Scholar] [CrossRef]
- Rashidi, A.; DiPersio, J.F. Targeting the leukemia–stroma interaction in acute myeloid leukemia: Rationale and latest evidence. Ther. Adv. Hematol. 2016, 7, 40–51. [Google Scholar] [CrossRef]
- Su, Y.C.; Li, S.C.; Wu, Y.C.; Wang, L.M.; Chao, K.S.C.; Liao, H.F. Resveratrol downregulates interleukin-6-stimulated sonic hedgehog signaling in human acute myeloid leukemia. Evid.-Based Complement. Altern. Med. 2013, 2013, 547430. [Google Scholar] [CrossRef]
- Chang, Y.-T.; Hernandez, D.; Alonso, S.; Gao, M.; Su, M.; Ghiaur, G.; Levis, M.J.; Jones, R.J. Role of CYP3A4 in bone marrow microenvironment–mediated protection of FLT3/ITD AML from tyrosine kinase inhibitors. Blood Adv. 2019, 3, 908. [Google Scholar] [CrossRef]
- Chen, T.; Zhang, G.; Kong, L.; Xu, S.; Wang, Y.; Dong, M. Leukemia-Derived exosomes induced IL-8 production in bone marrow stromal cells to protect the leukemia cells against chemotherapy. Life Sci. 2019, 221, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M.; Campbell, P. FLT3 inhibitors in acute myeloid leukemia: Current and future. J. Oncol. Pharm. Pract. 2019, 25, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Tohumeken, S.; Baur, R.; Böttcher, M.; Stoll, A.; Loschinski, R.; Panagiotidis, K.; Braun, M.; Saul, D.; Völkl, S.; Baur, A.S.; et al. Palmitoylated proteins on AML-derived extracellular vesicles promote myeloid-derived suppressor cell differentiation via TLR2/Akt/mTOR signaling. Cancer Res. 2020, 80, 3663–3676. [Google Scholar] [CrossRef] [PubMed]
- Leisch, M.; Greil, R.; Pleyer, L. IDO in MDS/AML disease progression and its role in resistance to azacitidine: A potential new drug target? Br. J. Haematol. 2020, 190, 314. [Google Scholar] [CrossRef]
- Zhang, Y.; Dépond, M.; He, L.; Foudi, A.; Kwarteng, E.O.; Lauret, E.; Plo, I.; Desterke, C.; Dessen, P.; Fujii, N.; et al. CXCR4/CXCL12 axis counteracts hematopoietic stem cell exhaustion through selective protection against oxidative stress. Sci. Rep. 2016, 6, 37827. [Google Scholar] [CrossRef] [Green Version]
- Sison, E.A.R.; Brown, P. The bone marrow microenvironment and leukemia: Biology and therapeutic targeting. Expert Rev. Hematol. 2011, 4, 271–283. [Google Scholar] [CrossRef] [Green Version]
- Schioppa, T.; Uranchimeg, B.; Saccani, A.; Biswas, S.K.; Doni, A.; Rapisarda, A.; Bernasconi, S.; Saccani, S.; Nebuloni, M.; Vago, L.; et al. Regulation of the chemokine receptor CXCR4 by hypoxia. J. Exp. Med. 2003, 198, 1391–1402. [Google Scholar] [CrossRef] [Green Version]
- Burger, J.A.; Kipps, T.J. CXCR4: A key receptor in the crosstalk between tumor cells and their microenvironment. Blood 2006, 107, 1761–1767. [Google Scholar] [CrossRef]
- Peng, G.; Liu, Y. Hypoxia-Inducible factors in cancer stem cells and inflammation. Trends Pharmacol. Sci. 2015, 36, 374. [Google Scholar] [CrossRef] [Green Version]
- Deynoux, M.; Sunter, N.; Hérault, O.; Mazurier, F. Hypoxia and hypoxia-inducible factors in leukemias. Front. Oncol. 2016, 6, 41. [Google Scholar] [CrossRef] [Green Version]
- Yazdani, Z.; Mousavi, Z.; Moradabadi, A.; Hassanshahi, G. Significance of CXCL12/CXCR4 Ligand/Receptor axis in various aspects of acute myeloid leukemia. Cancer Manag. Res. 2020, 12, 2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schajnovitz, A.; Itkin, T.; D’Uva, G.; Kalinkovich, A.; Golan, K.; Ludin, A.; Cohen, D.; Shulman, Z.; Avigdor, A.; Nagler, A.; et al. CXCL12 secretion by bone marrow stromal cells is dependent on cell contact and mediated by connexin-43 and connexin-45 gap junctions. Nat. Immunol. 2011, 12, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Meng, F.; Lu, Q. Expression profile screening and bioinformatics analysis of circRNA, LncRNA, and mRNA in acute myeloid leukemia drug-resistant cells. Turkish J. Hematol. 2020, 37, 104. [Google Scholar]
- Zhou, W.; Guo, S.; Liu, M.; Burow, M.E.; Wang, G. Targeting CXCL12/CXCR4 axis in tumor immunotherapy. Curr. Med. Chem. 2019, 26, 3026. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, R.; Panneerselvam, J.; Chen, A.; Zhao, Y.D.; Munshi, A.; Ramesh, R. HuR-targeted nanotherapy in combination with AMD3100 suppresses CXCR4 expression, cell growth, migration and invasion in lung cancer. Cancer Gene Ther. 2015, 22, 581–590. [Google Scholar] [CrossRef] [Green Version]
- De Clercq, E. AMD3100/CXCR4 Inhibitor. Front. Immunol. 2015, 6, 276. [Google Scholar] [CrossRef]
- Kuhne, M.R.; Mulvey, T.; Belanger, B.; Chen, S.; Pan, C.; Chong, C.; Cao, F.; Niekro, W.; Kempe, T.; Henning, K.A.; et al. BMS-936564/MDX-1338: A fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin. Cancer Res. 2013, 19, 357–366. [Google Scholar] [CrossRef] [Green Version]
- Epstein, J.; Barlogie, B.; Johnson, C.L.; Yaccoby, S.; Pearse, R.N.; Choi, Y. Myeloma interacts with the bone marrow microenvironment to induce osteoclastogenesis and is dependent on osteoclast activity. Br. J. Haematol. 2002, 116, 278–290. [Google Scholar]
- Le, P.M.; Andreeff, M.; Lokesh Battula, V. Osteogenic niche in the regulation of normal hematopoiesis and leukemogenesis. Haematologica 2018, 103, 1945. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zheng, L.; Yuan, Q.; Zhen, G.; Crane, J.L.; Zhou, X.; Cao, X. Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res. 2018, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Heldin, C.H.; Moustakas, A. Signaling receptors for TGF-β family members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Lettirio, J. Transforming growth factor-β signaling in normal and malignant hematopoiesis. Leukemia 2003, 17, 1731–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korn, C.; Méndez-Ferrer, S. Myeloid malignancies and the microenvironment. Blood 2017, 129, 811–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabe, Y.; Shi, Y.X.; Zeng, Z.; Jin, L.; Shikami, M.; Hatanaka, Y.; Miida, T.; Hsu, F.J.; Andreeff, M.; Konopleva, M. TGF-β-Neutralizing antibody 1D11 enhances cytarabine-induced apoptosis in AML cells in the bone marrow microenvironment. PLoS ONE 2013, 8, 62785. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Hideshima, T.; Nguyen, A.N.; Munoz, O.; Podar, K.; Hamasaki, M.; Ishitsuka, K.; Yasui, H.; Richardson, P.; Chakravarty, S.; et al. Transforming growth factor β receptor I kinase inhibitor down-regulates cytokine secretion and multiple myeloma cell growth in the bone marrow microenvironment. Clin. Cancer Res. 2004, 10, 7540–7546. [Google Scholar] [CrossRef] [Green Version]
- Selnø, A.T.H.; Schlichtner, S.; Yasinska, I.M.; Sakhnevych, S.S.; Fiedler, W.; Wellbrock, J.; Klenova, E.; Pavlova, L.; Gibbs, B.F.; Degen, M.; et al. Transforming growth factor beta type 1 (TGF-β) and hypoxia-inducible factor 1 (HIF-1) transcription complex as master regulators of the immunosuppressive protein galectin-9 expression in human cancer and embryonic cells. Aging 2020, 12, 23478. [Google Scholar] [CrossRef]
- Yuan, B.; Dana, F.E.; Ly, S.; Yan, Y.; Ruvolo, V.; Shpall, E.J.; Konopleva, M.; Andreeff, M.; Battula, V.L. Bone marrow stromal cells induce an ALDH+ stem cell-like phenotype and enhance therapy resistance in AML through a TGF-β-p38-ALDH2 pathway. PLoS ONE 2020, 15, e0242809. [Google Scholar] [CrossRef]
- Passaro, D.; Di Tullio, A.; Abarrategi, A.; Rouault-Pierre, K.; Foster, K.; Ariza-McNaughton, L.; Montaner, B.; Chakravarty, P.; Bhaw, L.; Diana, G.; et al. Increased vascular permeability in the bone marrow microenvironment contributes to disease progression and drug response in acute myeloid leukemia. Cancer Cell 2017, 32, 324–341.e6. [Google Scholar] [CrossRef] [Green Version]
- Schmiedel, B.J.; Grosse-Hovest, L.; Salih, H.R. A “vicious cycle” of NK-cell immune evasion in acute myeloid leukemia mediated by RANKL? Oncoimmunology 2013, 2, e23850. [Google Scholar] [CrossRef] [Green Version]
- Roodman, G.D. Pathogenesis of myeloma bone disease. Leukemia 2009, 23, 435–441. [Google Scholar] [CrossRef]
- Mohammadi, S.; Ghaffari, S.H.; Shaiegan, M.; Zarif, M.N.; Nikbakht, M.; Akbari Birgani, S.; Alimoghadam, K.; Ghavamzadeh, A. Acquired expression of osteopontin selectively promotes enrichment of leukemia stem cells through AKT/mTOR/PTEN/β-catenin pathways in AML cells. Life Sci. 2016, 152, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Pievani, A.; Biondi, M.; Tomasoni, C.; Biondi, A.; Serafini, M. Location first: Targeting acute myeloid leukemia within its niche. J. Clin. Med. 2020, 9, 1513. [Google Scholar] [CrossRef]
- Han, J.; Koh, Y.J.; Moon, H.R.; Ryoo, H.G.; Cho, C.H.; Kim, I.; Koh, G.Y. Adipose tissue is an extramedullary reservoir for functional hematopoietic stem and progenitor cells. Blood 2010, 115, 957–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafat, M.S.; Oellerich, T.; Mohr, S.; Robinson, S.D.; Edwards, D.R.; Marlein, C.R.; Piddock, R.E.; Fenech, M.; Zaitseva, L.; Abdul-Aziz, A.; et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017, 129, 1320–1332. [Google Scholar] [CrossRef]
- Sengenès, C.; Bouloumié, A.; Hauner, H.; Berlan, M.; Busse, R.; Lafontan, M.; Galitzky, J. Involvement of a cGMP-dependent pathway in the natriuretic peptide-mediated hormone-sensitive lipase phosphorylation in human adipocytes. J. Biol. Chem. 2003, 278, 48617–48626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabe, Y.; Yamamoto, S.; Saitoh, K.; Sekihara, K.; Monma, N.; Ikeo, K.; Mogushi, K.; Shikami, M.; Ruvolo, V.; Ishizawa, J.; et al. Bone marrow adipocytes facilitate fatty acid oxidation activating ampk and a transcriptional network supporting survival of acute monocytic leukemia cells. Cancer Res. 2017, 77, 1453–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levesque, J.P.; Winkler, I.G. Cell adhesion molecules in normal and malignant hematopoiesis: From bench to bedside. Curr. Stem Cell Reports 2016, 2, 356–367. [Google Scholar] [CrossRef] [Green Version]
- Becker, P.S.; Kopecky, K.J.; Wilks, A.N.; Chien, S.; Harlan, J.M.; Willman, C.L.; Petersdorf, S.H.; Stirewalt, D.L.; Papayannopoulou, T.; Appelbaum, F.R. Very late antigen-4 function of myeloblasts correlates with improved overall survival for patients with acute myeloid leukemia. Blood 2009, 113, 866–874. [Google Scholar] [CrossRef]
- Jacamo, R.; Chen, Y.; Wang, Z.; Wencai, M.; Zhang, M.; Spaeth, E.L.; Wang, Y.; Battula, V.L.; Mak, P.Y.; Schallmoser, K.; et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood 2014, 123, 2691–2702. [Google Scholar] [CrossRef]
- Ganghammer, S.; Hutterer, E.; Hinterseer, E.; Brachtl, G.; Asslaber, D.; Krenn, P.W.; Girbl, T.; Berghammer, P.; Geisberger, R.; Egle, A.; et al. CXCL12-induced VLA-4 activation is impaired in trisomy 12 chronic lymphocytic leukemia cells: A role for CCL21. Oncotarget 2015, 6, 12048–12060. [Google Scholar] [CrossRef] [Green Version]
- Al-Hussaini, M.; Dipersio, J.F. Small molecule inhibitors in acute myeloid leukemia: From the bench to the clinic. Expert Rev. Hematol. 2014, 7, 439–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Dedhar, S. Integrin-Linked kinase (ILK) and its interactors: A new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J. Cell Biol. 2001, 155, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Shishido, S.; Bönig, H.; Kim, Y.M. Role of integrin alpha4 in drug resistance of leukemia. Front. Oncol. 2014, 4, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, I.G.; Barbier, V.; Nowlan, B.; Jacobsen, R.N.; Forristal, C.E.; Patton, J.T.; Magnani, J.L.; Lévesque, J.P. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat. Med. 2012, 18, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Barbier, V.; Erbani, J.; Fiveash, C.; Davies, J.M.; Tay, J.; Tallack, M.R.; Lowe, J.; Magnani, J.L.; Pattabiraman, D.R.; Perkins, A.C.; et al. Endothelial E-selectin inhibition improves acute myeloid leukaemia therapy by disrupting vascular niche-mediated chemoresistance. Nat. Commun. 2020, 11, 2042. [Google Scholar] [CrossRef]
- Erbani, J.; Tay, J.; Barbier, V.; Levesque, J.P.; Winkler, I.G. Acute myeloid leukemia chemo-resistance is mediated by e-selectin receptor CD162 in bone marrow niches. Front. Cell Dev. Biol. 2020, 8, 688. [Google Scholar] [CrossRef]
- Chien, S.; Haq, S.U.; Pawlus, M.; Moon, R.T.; Estey, E.H.; Appelbaum, F.R.; Othus, M.; Magnani, J.L.; Becker, P.S. Adhesion of acute myeloid leukemia blasts to e-selectin in the vascular niche enhances their survival by mechanisms such as wnt activation. Blood 2013, 122, 61. [Google Scholar] [CrossRef]
- Pyzer, A.R.; Stroopinsky, D.; Rajabi, H.; Washington, A.; Tagde, A.; Coll, M.; Fung, J.; Bryant, M.P.; Cole, L.; Palmer, K.; et al. MUC1-mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood 2017, 129, 1791–1801. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-Derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, R.W.; Rodriguez, P.C.; Toque, H.A.; Priya Narayanan, S.; Caldwell, R.B. Arginase: A multifaceted enzyme important in health and disease. Physiol. Rev. 2018, 98, 641–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mussai, F.; Egan, S.; Higginbotham-Jones, J.; Perry, T.; Beggs, A.; Odintsova, E.; Loke, J.; Pratt, G.; U, K.P.; Lo, A.; et al. Arginine dependence of acute myeloid leukemia blast proliferation: A novel therapeutic target. Blood 2015, 125, 2386–2396. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; Prendergast, G. Indoleamine 2,3-Dioxygenase in immune suppression and cancer. Curr. Cancer Drug Targets 2007, 7, 31–40. [Google Scholar] [CrossRef] [PubMed]
- De Veirman, K.; Van Valckenborgh, E.; Lahmar, Q.; Geeraerts, X.; De Bruyne, E.; Menu, E.; Van Riet, I.; Vanderkerken, K.; Van Ginderachter, J.A. Myeloid-Derived suppressor cells as therapeutic target in hematological malignancies. Front. Oncol. 2014, 4, 349. [Google Scholar] [CrossRef]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-Derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Sun, Y.; Niu, J.; Jarugumilli, G.K.; Wu, X. Protein lipidation in cell signaling and diseases: Function, regulation, and therapeutic opportunities. Cell Chem. Biol. 2018, 25, 817–831. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Li, Y.; Zhu, B. T-Cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- Anguille, S.; Lion, E.; Willemen, Y.; Van Tendeloo, V.F.I.; Berneman, Z.N.; Smits, E.L.J.M. Interferon-α in acute myeloid leukemia: An old drug revisited. Leukemia 2011, 25, 739–748. [Google Scholar] [CrossRef] [Green Version]
- Jedema, I.; Barge, R.M.Y.; Willemze, R.; Falkenburg, J.H.F. High susceptibility of human leukemic cells to Fas-induced apoptosis is restricted to G1 phase of the cell cycle and can be increased by interferon treatment. Leukemia 2003, 17, 576–584. [Google Scholar] [CrossRef] [Green Version]
- Müller, L.; Aigner, P.; Stoiber, D. Type I interferons and natural killer cell regulation in cancer. Front. Immunol. 2017, 8, 304. [Google Scholar] [CrossRef] [Green Version]
- Cuartero, S.; Weiss, F.D.; Dharmalingam, G.; Guo, Y.; Ing-Simmons, E.; Masella, S.; Robles-Rebollo, I.; Xiao, X.; Wang, Y.-F.; Barozzi, I.; et al. Control of inducible gene expression links cohesin to hematopoietic progenitor self-renewal and differentiation. Nat. Immunol. 2018, 19, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Smits, E.L.J.; Anguille, S.; Berneman, Z.N. Interferon α may be back on track to treat acute myeloid leukemia. Oncoimmunology 2013, 2, e23619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adeegbe, D.O.; Nishikawa, H. Natural and induced T regulatory cells in cancer. Front. Immunol. 2013, 4, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workman, C.J.; Szymczak-Workman, A.L.; Collison, L.W.; Pillai, M.R.; Vignali, D.A.A. The development and function of regulatory T cells. Cell. Mol. Life Sci. 2009, 66, 2603–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isidori, A.; Loscocco, F.; Ciciarello, M.; Visani, G.; Corradi, G.; Forte, D.; Lecciso, M.; Ocadlikova, D.; Parisi, S.; Salvestrini, V.; et al. Renewing the immunological approach to AML treatment: From novel pathways to innovative therapies. Cancer Res. Front. 2016, 2, 226–251. [Google Scholar] [CrossRef]
- Ustun, C.; Miller, J.S.; Munn, D.H.; Weisdorf, D.J.; Blazar, B.R. Regulatory T cells in acute myelogenous leukemia: Is it time for immunomodulation? Blood 2011, 118, 5084–5095. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Dong, Y.; Yang, Q.; Xu, W.; Jiang, S.; Yu, Z.; Yu, K.; Zhang, S. Acute myeloid leukemia cells express ICOS ligand to promote the expansion of regulatory T cells. Front. Immunol. 2018, 9, 2227. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez, L.; Jang, M.; Zhang, T.; Akhtari, M.; Alachkar, H. Midostaurin reduces regulatory T cells markers in acute myeloid leukemia. Sci. Rep. 2018, 8, 17544. [Google Scholar] [CrossRef]
- Klein, M.; Bopp, T. Cyclic AMP represents a crucial component of treg cell-mediated immune regulation. Front. Immunol. 2016, 7, 315. [Google Scholar] [CrossRef] [Green Version]
- Gondek, D.C.; Lu, L.-F.; Quezada, S.A.; Sakaguchi, S.; Noelle, R.J. Cutting edge: Contact-Mediated suppression by CD4 + CD25 + Regulatory cells involves a granzyme b-dependent, perforin-independent mechanism. J. Immunol. 2005, 174, 1783–1786. [Google Scholar] [CrossRef] [Green Version]
- Janikashvili, N.; Bonnotte, B.; Katsanis, E.; Larmonier, N. The dendritic cell-regulatory T lymphocyte crosstalk contributes to tumor-induced tolerance. Clin. Dev. Immunol. 2011, 2011, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, J.L. PD-1 signaling in primary T cells. Immunol. Rev. 2009, 229, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Yang, H.; Bueso-Ramos, C.; Dinardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef]
- Asayama, T.; Tamura, H.; Ishibashi, M.; Kuribayashi-Hamada, Y.; Onodera-Kondo, A.; Okuyama, N.; Yamada, A.; Shimizu, M.; Moriya, K.; Takahashi, H.; et al. Functional expression of Tim-3 on blasts and clinical impact of its ligand galectin-9 in myelodysplastic syndromes. Oncotarget 2017, 8, 88904–88917. [Google Scholar] [CrossRef] [Green Version]
- Freeman, G.J.; Casasnovas, J.M.; Umetsu, D.T.; Dekruyff, R.H. TIM genes: A family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol. Rev. 2010, 235, 172–189. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves Silva, I.; Yasinska, I.M.; Sakhnevych, S.S.; Fiedler, W.; Wellbrock, J.; Bardelli, M.; Varani, L.; Hussain, R.; Siligardi, G.; Ceccone, G.; et al. The Tim-3-galectin-9 secretory pathway is involved in the immune escape of human acute myeloid leukemia cells. EBioMedicine 2017, 22, 44–57. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Munger, M.E.; Veenstra, R.G.; Weigel, B.J.; Hirashima, M.; Munn, D.H.; Murphy, W.J.; Azuma, M.; Anderson, A.C.; Kuchroo, V.K.; et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood 2011, 117, 4501–4510. [Google Scholar] [CrossRef]
- Malhotra, A.; Shanker, A. NK cells: Immune cross-talk and therapeutic implications. Immunotherapy 2011, 3, 1143–1166. [Google Scholar] [CrossRef] [Green Version]
- Lima, M.; Leander, M.; Santos, M.; Santos, A.H.; Lau, C.; Queirós, M.L.; Gonçalves, M.; Fonseca, S.; Moura, J.; Teixeira, M.D.A.; et al. Chemokine receptor expression on normal blood CD56+ NK-cells elucidates cell partners that comigrate during the innate and adaptive immune responses and identifies a transitional NK-Cell population. J. Immunol. Res. 2015, 2015, 839684. [Google Scholar] [CrossRef] [Green Version]
- Kittang, A.O.; Hatfield, K.; Sand, K.; Reikvam, H.; Bruserud, Ø. The chemokine network in acute myelogenous leukemia: Molecular mechanisms involved in leukemogenesis and therapeutic implications. Curr. Top Microbiol. Immunol. 2010, 341, 149–172. [Google Scholar] [PubMed]
- Dulphy, N.; Chrétien, A.S.; Khaznadar, Z.; Fauriat, C.; Nanbakhsh, A.; Caignard, A.; Chouaib, S.; Olive, D.; Toubert, A. Underground adaptation to a hostile environment: Acute myeloid leukemia vs. natural killer cells. Front. Immunol. 2016, 7, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaznadar, Z.; Boissel, N.; Agaugué, S.; Henry, G.; Cheok, M.; Vignon, M.; Geromin, D.; Cayuela, J.-M.; Castaigne, S.; Pautas, C.; et al. Defective NK cells in acute myeloid leukemia patients at diagnosis are associated with blast transcriptional signatures of immune evasion. J. Immunol. 2015, 195, 2580–2590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martner, A.; Thorén, F.B.; Aurelius, J.; Hellstrand, K. Immunotherapeutic strategies for relapse control in acute myeloid leukemia. Blood Rev. 2013, 27, 209–216. [Google Scholar] [CrossRef]
- Hegedűs, C.; Kovács, K.; Polgár, Z.; Regdon, Z.; Szabó, É.; Robaszkiewicz, A.; Forman, H.J.; Martner, A.; Virág, L. Redox control of cancer cell destruction. Redox Biol. 2018, 16, 59–74. [Google Scholar] [CrossRef]
- Wang, Z.; Guan, W.; Wang, M.; Chen, J.; Zhang, L.; Xiao, Y.; Wang, L.; Li, Y.; Yu, L. AML1-ETO inhibits acute myeloid leukemia immune escape by CD48. Leuk. Lymphoma 2021, 62, 937–943. [Google Scholar] [CrossRef]
- Scoville, S.D.; Nalin, A.P.; Chen, L.; Chen, L.; Zhang, M.H.; McConnell, K.; Casas, S.B.; Ernst, G.; Al-Rahman Traboulsi, A.; Hashi, N.; et al. Human AML activates the aryl hydrocarbon receptor pathway to impair NK cell development and function. Blood 2018, 132, 1792–1804. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, J.; Wang, M.; Zhang, L.; Yu, L. One stone, two birds: The roles of tim-3 in acute myeloid leukemia. Front. Immunol. 2021, 12, 1098. [Google Scholar] [CrossRef]
- Darwish, N.H.E.; Sudha, T.; Godugu, K.; Elbaz, O.; Abdelghaffar, H.A.; Hassan, E.E.A.; Mousa, S.A. Acute myeloid leukemia stem cell markers in prognosis and targeted therapy: Potential impact of BMI-1, TIM-3 and CLL-1. Oncotarget 2016, 7, 57811–57820. [Google Scholar] [CrossRef] [Green Version]
- Ayyadurai, V.A.S.; Deonikar, P.; Ali, A.; Rockel, J.; Kapoor, M. Molecular Systems Architecture of Human Knee Osteoarthritis. Available online: https://cytosolve.com/human-knee-osteoarthritis/ (accessed on 18 April 2021).












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class I Genes | Class II Genes | Class III Genes | Other Genes | |
|---|---|---|---|---|
| Signal Transduction | Differentiation | Epigenetic Regulation | Tumor Suppression | Oncogenes |
| FLT3 | RUNX1 (AML1) | TET2 | WT1 | PML-RARa |
| KIT | CBFα | IDH1/IDH2 | TP53 | FLT3-ITD |
| NRAS, KRAS | CEBPα | DNMT3A | AML-ETO | |
| JAK2 | NPM1 | ASXL1 | CBFB-MYH11 | |
| PTPN11 | PU1 | EZH2 | ||
| MLL | Cohesin | |||
| RARα | NPM1 | |||
| MeSH Keywords |
|---|
| Human acute myeloid leukemia CXCR4 CXCL12 Signaling NOT review |
| Human acute myeloid leukemia TGF-β Signaling NOT review |
| Human acute myeloid leukemia VLA-4 VCAM-1 Signaling NOT review |
| Human acute myeloid leukemia Arginase NOT review |
| Human acute myeloid leukemia IDO NOT review |
| Human acute myeloid leukemia PD-1 PD-L1 Signaling NOT review |
| Human acute myeloid leukemia NK cells NOT review |
| Human acute myeloid leukemia BMSC NOT review |
| Human acute myeloid leukemia MDSC NOT review |
| Human acute myeloid leukemia Endothelial Cell NOT review |
| Human acute myeloid leukemia Treg cells NOT review |
| Human acute myeloid leukemia MSC cells NOT review |
| Human acute myeloid leukemia Fibroblast cells NOT review |
| Human acute myeloid leukemia Th1 cells NOT review |
| Human acute myeloid leukemia Th17 cells NOT review |
| Human acute myeloid leukemia Teff cells NOT review |
| Human acute myeloid leukemia Osteoblasts/Osteoclast cells NOT review |
| Human acute myeloid leukemia Adipocytes NOT review |
| Name of Symbol | Symbol | Description |
|---|---|---|
| Double-sided Orange Rectangle |  | Molecular pathway |
| Black Arrow |  | Receptor/Ligand Binding, Signal propagation |
| Red Flat Arrow |  | Inhibition of signal propagation |
| Green Cylinder |  | Cell surface receptor |
| Purple Lozenge |  | mRNA |
| Blue Pentagram |  | Protein/small molecule |
| Physiological Effect | Cell Type | Potential Target |
|---|---|---|
| Suppression of Immune Response | AML Cell | PD-L1, IL-6, Galactin-9, CCL2, CXCR1, IDO |
| Th1-Cell | Tim-3, PD-1 | |
| NK Cell | PD-1, AHR | |
| MDSC | Arginase, CCR2 | |
| Cell Proliferation | BMSC | Fibronectin, Gas-6, CXCR4/CXCL12 |
| Osteoblast/Osteoclast | OPN, CXCR4/CXCL12 | |
| Cell Apoptosis | AML Cell | Axl, IL-17, IL-6 |
| Adipocytes | FAO |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayyadurai, V.A.S.; Deonikar, P.; McLure, K.G.; Sakamoto, K.M. Molecular Systems Architecture of Interactome in the Acute Myeloid Leukemia Microenvironment. Cancers 2022, 14, 756. https://doi.org/10.3390/cancers14030756
Ayyadurai VAS, Deonikar P, McLure KG, Sakamoto KM. Molecular Systems Architecture of Interactome in the Acute Myeloid Leukemia Microenvironment. Cancers. 2022; 14(3):756. https://doi.org/10.3390/cancers14030756
Chicago/Turabian StyleAyyadurai, V. A. Shiva, Prabhakar Deonikar, Kevin G. McLure, and Kathleen M. Sakamoto. 2022. "Molecular Systems Architecture of Interactome in the Acute Myeloid Leukemia Microenvironment" Cancers 14, no. 3: 756. https://doi.org/10.3390/cancers14030756
APA StyleAyyadurai, V. A. S., Deonikar, P., McLure, K. G., & Sakamoto, K. M. (2022). Molecular Systems Architecture of Interactome in the Acute Myeloid Leukemia Microenvironment. Cancers, 14(3), 756. https://doi.org/10.3390/cancers14030756