
Retinal Glutamate Neurotransmission: From Physiology to Pathophysiological Mechanisms of Retinal Ganglion Cell Degeneration



Abstract
:1. Retinal Network Integration Requires Glutamate Neurotransmission
2. Distribution of Glutamate Receptors and Transporters in the Retinas
2.1. Ionotropic Receptors
2.2. Metabotropic Receptors
2.3. Glutamate Transporters
3. Vulnerability of the Retina and RGCs to Disease
4. Glutamate Excitotoxicity in the Retina
4.1. Impairment in Glutamate Clearance and the Role of Müller Glia
4.2. Glutamate Receptor-Induced RGC Degeneration
4.3. Factors Affecting NMDA Receptor-Mediated Degeneration
5. Cellular Mechanisms of NMDA Receptor-Induced Excitotoxicity in RGCs
5.1. Intracellular Calcium Dysregulation
5.2. Imbalance of Signalling Pathways and Gene Expression
5.3. Mitochondrial Dysfunction
5.4. Endoplasmic Reticular Stress
6. Final Remarks and Therapeutic Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Masland, R.H. The fundamental plan of the retina. Nat. Neurosci. 2001, 4, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia-neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, E.R.; Esguerra, M.; Kim, P.M.; Newman, E.A.; Snyder, S.H.; Zahs, K.R.; Miller, R.F. d-serine and serine racemase are present in the vertebrate retina and contribute to the physiological activation of NMDA receptors. Proc. Natl. Acad. Sci. USA 2003, 100, 6789–6794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, E.A.; Zahs, K.R. Modulation of neuronal activity by glial cells in the retina. J. Neurosci. 1998, 18, 4022–4028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bringmann, A.; Grosche, A.; Pannicke, T.; Reichenbach, A. GABA and Glutamate Uptake and Metabolism in Retinal Glial (Müller) Cells. Front. Endocrinol. 2013, 4, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, E.; Reichenbach, A. The Müller cell: A functional element of the retina. Trends Neurosci. 1996, 19, 307–312. [Google Scholar] [CrossRef]
- Higgs, M.H.; Lukasiewicz, P.D. Glutamate Uptake Limits Synaptic Excitation of Retinal Ganglion Cells. J. Neurosci. 1999, 19, 3691–3700. [Google Scholar] [CrossRef]
- Bui, B.V.; Hu, R.G.; Acosta, M.L.; Donaldson, P.; Vingrys, A.J.; Kalloniatis, M. Glutamate metabolic pathways and retinal function. J. Neurochem. 2009, 111, 589–599. [Google Scholar] [CrossRef]
- Chaya, T.; Matsumoto, A.; Sugita, Y.; Watanabe, S.; Kuwahara, R.; Tachibana, M.; Furukawa, T. Versatile functional roles of horizontal cells in the retinal circuit. Sci. Rep. 2017, 7, 5540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, H. Amacrine cells of the mammalian retina: Neurocircuitry and functional roles. Eye 1997, 11, 904–923. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.; Vaney, D.I. Amacrine Cells. In The Senses: A Comprehensive Reference; Academic Press: Cambridge, MA, USA, 2008; Volume 1, pp. 361–367. [Google Scholar]
- Bloomfield, S.A.; Dacheux, R.F. Rod Vision: Pathways and Processing in the Mammalian Retina. Prog. Retin. Eye Res. 2001, 20, 351–384. [Google Scholar] [CrossRef]
- Ravi, S.; Ahn, D.; Greschner, M.; Chichilnisky, E.J.; Field, G.D. Pathway-Specific Asymmetries between ON and OFF Visual Signals. J. Neurosci. 2018, 38, 9728–9740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witkovsky, P.; Thoreson, W.; Tranchina, D. Chapter 9 Transmission at the photoreceptor synapse. Prog. Brain Res. 2001, 131, 145–159. [Google Scholar] [CrossRef]
- Morigiwa, K.; Vardi, N. Differential expression of ionotropic glutamate receptor subunits in the outer retina. J. Comp. Neurol. 1999, 405, 173–184. [Google Scholar] [CrossRef]
- Popova, E. ON-OFF Interactions in the Retina: Role of Glycine and GABA. Curr. Neuropharmacol. 2015, 12, 509–526. [Google Scholar] [CrossRef] [Green Version]
- Vardi, N.; Duvoisin, R.; Wu, G.; Sterling, P. Localization of mGluR6 to dendrites of ON bipolar cells in primate retina. J. Comp. Neurol. 2000, 423, 402–412. [Google Scholar] [CrossRef]
- Kolb, H. Neurotransmitters in the retina. In Webvision: The Organization of the Retina and Visual System; John A. Moran Eye Center: Salt Lake City, UT, USA, 2009; pp. 407–427. [Google Scholar]
- Dieck, S.T.; Brandstätter, J.H. Ribbon synapses of the retina. Cell Tissue Res. 2006, 326, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Matthews, G.; Fuchs, P. The diverse roles of ribbon synapses in sensory neurotransmission. Nat. Rev. Neurosci. 2010, 11, 812–822. [Google Scholar] [CrossRef]
- Moser, T.; Grabner, C.P.; Schmitz, F. Sensory Processing at Ribbon Synapses in the Retina and the Cochlea. Physiol. Rev. 2020, 100, 103–144. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.-W.; Blackstone, C.; Huganir, R.; Yau, K.-W. Distribution of glutamate receptor subtypes in the vertebrate retina. Neuroscience 1995, 66, 483–497. [Google Scholar] [CrossRef]
- Brandstätter, J.H.; Koulen, P.; Wässle, H. Diversity of glutamate receptors in the mammalian retina. Vis. Res. 1998, 38, 1385–1397. [Google Scholar] [CrossRef] [Green Version]
- Harada, T.; Harada, C.; Watanabe, M.; Inoue, Y.; Sakagawa, T.; Nakayama, N.; Sasaki, S.; Okuyama, S.; Watase, K.; Wada, K.; et al. Functions of the two glutamate transporters GLAST and GLT-1 in the retina. Proc. Natl. Acad. Sci. USA 1998, 95, 4663–4666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauen, T. Diversity of glutamate transporter expression and function in the mammalian retina. Amino Acids 2000, 19, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Pircher, B.; Pircher, T.; Feigenspan, A. Ionotropic glutamate receptors in the retina-a bioinformatical meta-analysis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Dhingra, A.; Vardi, N. mGlu receptors in the retina. Wiley Interdiscip. Rev. Membr. Transp. Signal. 2012, 1, 641–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haumann, I.; Junghans, D.; Anstötz, M.; Frotscher, M. Presynaptic localization of GluK5 in rod photoreceptors suggests a novel function of high affinity glutamate receptors in the mammalian retina. PLoS ONE 2017, 12, e0172967. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, E.L.; Hack, I.; Brandstätter, J.H.; Wässle, H. Synaptic localization of NMDA receptor subunits in the rat retina. J. Comp. Neurol. 2000, 420, 98–112. [Google Scholar] [CrossRef]
- Kalloniatis, M.; Sun, D.; Foster, L.; Haverkamp, S.; Wässle, H. Localization of NMDA receptor subunits and mapping NMDA drive within the mammalian retina. Vis. Neurosci. 2004, 21, 587–597. [Google Scholar] [CrossRef]
- Koulen, P.; Kuhn, R.; Wässle, H.; Brandstätter, J.H. Modulation of the intracellular calcium concentration in photoreceptor terminals by a presynaptic metabotropic glutamate receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 9909–9914. [Google Scholar] [CrossRef] [Green Version]
- Rauen, T.; Rothstein, J.D. Differential expression of three glutamate transporter subtypes in the rat retina. Cell Tissue Res. 1996, 286, 325–336. [Google Scholar] [CrossRef]
- Pow, D.V.; Barnett, N. Developmental expression of excitatory amino acid transporter 5: A photoreceptor and bipolar cell glutamate transporter in rat retina. Neurosci. Lett. 2000, 280, 21–24. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, X.-L.; Yang, X.-L. N-Methyl-d-Aspartate Receptors in the Retina. Mol. Neurobiol. 2006, 34, 163–180. [Google Scholar] [CrossRef]
- Kugler, P.; Beyer, A. Expression of glutamate transporters in human and rat retina and rat optic nerve. Histochem. Cell Biol. 2003, 120, 199–212. [Google Scholar] [CrossRef]
- Puthussery, T.; Percival, K.A.; Venkataramani, S.; Gayet-Primo, J.; Grünert, U.; Taylor, W. Kainate Receptors Mediate Synaptic Input to Transient and Sustained OFF Visual Pathways in Primate Retina. J. Neurosci. 2014, 34, 7611–7621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandstätter, J.H.; Hartveit, E.; Sassoe-Pognetto, M.; Wässle, H. Expression of NMDA and High-affinity Kainate Receptor Subunit mRNAs in the Adult Rat Retina. Eur. J. Neurosci. 1994, 6, 1100–1112. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, A.; Benke, D.; Mohler, H.; Fritschy, J.-M. N-methyl-d-aspartate receptors containing the NR2D subunit in the retina are selectively expressed in rod bipolar cells. Neuroscience 1997, 78, 1105–1112. [Google Scholar] [CrossRef]
- Quraishi, S.; Reed, B.; Duvoisin, R.; Taylor, W. Selective activation of mGluR8 receptors modulates retinal ganglion cell light responses. Neuroscience 2010, 166, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Quraishi, S.; Gayet, J.; Morgans, C.W.; Duvoisin, R.M. Distribution of group-III metabotropic glutamate receptors in the retina. J. Comp. Neurol. 2007, 501, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Brandstätter, J.H.; Koulen, P.; Kuhn, R.; Van Der Putten, H.; Wässle, H. Compartmental Localization of a Metabotropic Glutamate Receptor (mGluR7): Two Different Active Sites at a Retinal Synapse. J. Neurosci. 1996, 16, 4749–4756. [Google Scholar] [CrossRef]
- Ghosh, K.K.; Haverkamp, S.; Wässle, H. Glutamate Receptors in the Rod Pathway of the Mammalian Retina. J. Neurosci. 2001, 21, 8636–8647. [Google Scholar] [CrossRef]
- Jensen, R.J. Activation of group II metabotropic glutamate receptors reduces directional selectivity in retinal ganglion cells. Brain Res. 2006, 1122, 86–92. [Google Scholar] [CrossRef]
- Hoffpauir, B.K.; Gleason, E.L. Modulation of Synaptic Function in Retinal Amacrine Cells. Integr. Comp. Biol. 2005, 45, 658–664. [Google Scholar] [CrossRef]
- Sucher, N.J.; Kohler, K.; Tenneti, L.; Wong, H.-K.; Gründer, T.; Fauser, S.; Wheeler-Schilling, T.; Nakanishi, N.; Lipton, S.A.; Guenther, E. N-Methyl-d-Aspartate Receptor Subunit NR3A in the Retina: Developmental Expression, Cellular Localization, and Functional Aspects. Investig. Opthalmol. Vis. Sci. 2003, 44, 4451–4456. [Google Scholar] [CrossRef] [Green Version]
- López-Colomé, A.M.; López, E.; Mendez-Flores, O.G.; Ortega, A. Glutamate Receptor Stimulation Up-Regulates Glutamate Uptake in Human Müller Glia Cells. Neurochem. Res. 2016, 41, 1797–1805. [Google Scholar] [CrossRef]
- Cervantes-Villagrana, A.R.; Garcia-Román, J.; González-Espinosa, C.; Lamas, M. Pharmacological Inhibition of N-Methyl-d-Aspartate Receptor Promotes Secretion of Vascular Endothelial Growth Factor in Müller Cells: Effects of Hyperglycemia and Hypoxia. Curr. Eye Res. 2010, 35, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Lehre, K.P.; Davanger, S.; Danbolt, N. Localization of the glutamate transporter protein GLAST in rat retina. Brain Res. 1997, 744, 129–137. [Google Scholar] [CrossRef]
- Ward, M.M.; Puthussery, T.; Foster, L.E.; Fletcher, E.L.; Jobling, A.I. Localization and expression of the glutamate transporter, excitatory amino acid transporter 4, within astrocytes of the rat retina. Cell Tissue Res. 2004, 315, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Ishita, S.; Sugawara, K.; Mawatari, K. Cystine/glutamate antiporter expression in retinal müller glial cells: Implications fordl-alpha-aminoadipate toxicity. Neuroscience 1993, 57, 473–482. [Google Scholar] [CrossRef]
- Stroebel, D.; Casado, M.; Paoletti, P. Triheteromeric NMDA receptors: From structure to synaptic physiology. Curr. Opin. Physiol. 2018, 2, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hansen, K.B.; Yi, F.; Perszyk, R.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef]
- Paoletti, P. Molecular basis of NMDA receptor functional diversity. Eur. J. Neurosci. 2011, 33, 1351–1365. [Google Scholar] [CrossRef]
- Pérez-Otaño, I.; Larsen, R.S.; Wesseling, J.F. Emerging roles of GluN3-containing NMDA receptors in the CNS. Nat. Rev. Neurosci. 2016, 17, 623–635. [Google Scholar] [CrossRef]
- Bye, C.M.; McDonald, R.J. A Specific Role of Hippocampal NMDA Receptors and Arc Protein in Rapid Encoding of Novel Environmental Representations and a More General Long-Term Consolidation Function. Front. Behav. Neurosci. 2019, 13, 8. [Google Scholar] [CrossRef]
- Stafford, B.K.; Manookin, M.B.; Singer, J.H.; Demb, J.B. NMDA and AMPA receptors contribute similarly to temporal processing in mammalian retinal ganglion cells. J. Physiol. 2014, 592, 4877–4889. [Google Scholar] [CrossRef] [Green Version]
- Greger, I.H.; Watson, J.; Cull-Candy, S.G. Structural and Functional Architecture of AMPA-Type Glutamate Receptors and Their Auxiliary Proteins. Neuron 2017, 94, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Kielland, A.; Heggelund, P. AMPA and NMDA currents show different short-term depression in the dorsal lateral geniculate nucleus of the rat. J. Physiol. 2002, 542, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.F.; Ho, H.; Greger, I.H. Synaptic transmission and plasticity require AMPA receptor anchoring via its N-terminal domain. eLife 2017, 6, e23024. [Google Scholar] [CrossRef]
- Blankenship, A.G.; Ford, K.J.; Johnson, J.; Seal, R.P.; Edwards, R.H.; Copenhagen, D.R.; Feller, M.B. Synaptic and Extrasynaptic Factors Governing Glutamatergic Retinal Waves. Neuron 2009, 62, 230–241. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Ma, Y.-Y. Calcium Permeable-AMPA Receptors and Excitotoxicity in Neurological Disorders. Front. Neural Circuits 2021, 15, 82. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.; Vissel, B. The essential role of AMPA receptor GluR2 subunit RNA editing in the normal and diseased brain. Front. Mol. Neurosci. 2012, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-H.; von Gersdorff, H. Postsynaptic Plasticity Triggered by Ca2+-Permeable AMPA Receptor Activation in Retinal Amacrine Cells. Neuron 2016, 89, 507–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casimiro, T.M.; Nawy, S.; Carroll, R.C. Molecular mechanisms underlying activity-dependent AMPA receptor cycling in retinal ganglion cells. Mol. Cell. Neurosci. 2013, 56, 384–392. [Google Scholar] [CrossRef] [Green Version]
- Diamond, J.S. Calcium-Permeable AMPA Receptors in the Retina. Front. Mol. Neurosci. 2011, 4, 27. [Google Scholar] [CrossRef] [Green Version]
- Jane, D.E.; Lodge, D.; Collingridge, G.L. Kainate receptors: Pharmacology, function and therapeutic potential. Neuropharmacology 2009, 56, 90–113. [Google Scholar] [CrossRef] [PubMed]
- Yuzaki, M.; Aricescu, A.R. A GluD Coming-of-age story. Trends Neurosci. 2017, 40, 138–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohda, K.; Wang, Y.; Yuzaki, M. Mutation of a glutamate receptor motif reveals its role in gating and δ2 receptor channel properties. Nat. Neurosci. 2000, 3, 315–322. [Google Scholar] [CrossRef]
- Yang, J.-H.; Maple, B.; Gao, F.; Maguire, G.; Wu, S.M. Postsynaptic responses of horizontal cells in the tiger salamander retina are mediated by AMPA-preferring receptors. Brain Res. 1998, 797, 125–134. [Google Scholar] [CrossRef]
- Hartveit, E. Functional Organization of Cone Bipolar Cells in the Rat Retina. J. Neurophysiol. 1997, 77, 1716–1730. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Laboulaye, M.A.; Tran, N.M.; Whitney, I.E.; Benhar, I.; Sanes, J.R. Mouse Retinal Cell Atlas: Molecular Identification of over Sixty Amacrine Cell Types. J. Neurosci. 2020, 40, 5177–5195. [Google Scholar] [CrossRef]
- Veruki, M.L.; Zhou, Y.; Castilho, Á.; Morgans, C.W.; Hartveit, E. Extrasynaptic NMDA Receptors on Rod Pathway Amacrine Cells: Molecular Composition, Activation, and Signaling. J. Neurosci. 2019, 39, 627–650. [Google Scholar] [CrossRef]
- Taylor, W.R.; Chen, E.; Copenhagen, D.R. Characterization of spontaneous excitatory synaptic currents in salamander retinal ganglion cells. J. Physiol. 1995, 486, 207–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, K.; Hosoi, N.; Tachibana, M. Excitatory Synaptic Transmission in the Inner Retina: Paired Recordings of Bipolar Cells and Neurons of the Ganglion Cell Layer. J. Neurosci. 1998, 18, 4500–4510. [Google Scholar] [CrossRef] [Green Version]
- Taschenberger, H.; Engert, F.; Grantyn, R. Synaptic current kinetics in a solely AMPA-receptor-operated glutamatergic synapse formed by rat retinal ganglion neurons. J. Neurophysiol. 1995, 74, 1123–1136. [Google Scholar] [CrossRef]
- Chen, S.; Diamond, J.S. Synaptically Released Glutamate Activates Extrasynaptic NMDA Receptors on Cells in the Ganglion Cell Layer of Rat Retina. J. Neurosci. 2002, 22, 2165–2173. [Google Scholar] [CrossRef]
- Zhang, J.; Diamond, J.S. Distinct perisynaptic and synaptic localization of NMDA and AMPA receptors on ganglion cells in rat retina. J. Comp. Neurol. 2006, 498, 810–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottesman, J.; Miller, R.F. N-methyl-d-aspartate receptors contribute to the baseline noise of retinal ganglion cells. Vis. Neurosci. 2003, 20, 329–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Diamond, J.S. Subunit- and Pathway-Specific Localization of NMDA Receptors and Scaffolding Proteins at Ganglion Cell Synapses in Rat Retina. J. Neurosci. 2009, 29, 4274–4286. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.-W.; Du, W.-J.; Prober, D.A.; Du, J.-L. Müller glial Cells Participate in Retinal Waves via Glutamate Transporters and AMPA Receptors. Cell Rep. 2019, 27, 2871–2880.e2. [Google Scholar] [CrossRef] [Green Version]
- Niswender, C.M.; Conn, P.J. Metabotropic Glutamate Receptors: Physiology, Pharmacology, and Disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgs, M.; Romano, C.; Lukasiewicz, P. Presynaptic effects of group III metabotropic glutamate receptors on excitatory synaptic transmission in the retina. Neuroscience 2002, 115, 163–172. [Google Scholar] [CrossRef]
- Gleason, E. The influences of metabotropic receptor activation on cellular signaling and synaptic function in amacrine cells. Vis. Neurosci. 2011, 29, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, A.; Wheeler-Schilling, T.H.; Guenther, E. Coexpression patterns of mGLuR mRNAs in rat retinal ganglion cells: A single-cell RT-PCR study. Investig. Ophthalmol. Vis. Sci. 2000, 41, 314–319. [Google Scholar]
- Zhao, X.; Reifler, A.N.; Schroeder, M.M.; Jaeckel, E.R.; Chervenak, A.P.; Wong, K.Y. Mechanisms creating transient and sustained photoresponses in mammalian retinal ganglion cells. J. Gen. Physiol. 2017, 149, 335–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Danbolt, N.C. GABA and Glutamate Transporters in Brain. Front. Endocrinol. 2013, 4, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, T.; Akanuma, S.-I.; Usui, T.; Kubo, Y.; Tachikawa, M.; Hosoya, K.-I. Excitatory Amino Acid Transporter 1-Mediated L-Glutamate Transport at the Inner Blood–Retinal Barrier: Possible Role in L-Glutamate Elimination from the Retina. Biol. Pharm. Bull. 2015, 38, 1087–1091. [Google Scholar] [CrossRef] [Green Version]
- Vorwerk, C.K.; Naskar, R.; Schuettauf, F.; Quinto, K.; Zurakowski, D.; Gochenauer, G.; Robinson, M.B.; Mackler, A.S.; Dreyer, E.B. Depression of retinal glutamate transporter function leads to elevated intravitreal glutamate levels and ganglion cell death. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3615–3621. [Google Scholar]
- Sarthy, V.P.; Pignataro, L.; Pannicke, T.; Weick, M.; Reichenbach, A.; Harada, T.; Tanaka, K.; Marc, R. Glutamate transport by retinal Müller cells in glutamate/aspartate transporter-knockout mice. Glia 2005, 49, 184–196. [Google Scholar] [CrossRef]
- Grewer, C.; Rauen, T. Electrogenic Glutamate Transporters in the CNS: Molecular Mechanism, Pre-steady-state Kinetics, and their Impact on Synaptic Signaling. J. Membr. Biol. 2005, 203, 1–20. [Google Scholar] [CrossRef]
- Wadiche, J.; Amara, S.; Kavanaugh, M.P. Ion fluxes associated with excitatory amino acid transport. Neuron 1995, 15, 721–728. [Google Scholar] [CrossRef] [Green Version]
- Mim, C.; Balani, P.; Rauen, T.; Grewer, C. The Glutamate Transporter Subtypes EAAT4 and EAATs 1-3 Transport Glutamate with Dramatically Different Kinetics and Voltage Dependence but Share a Common Uptake Mechanism. J. Gen. Physiol. 2005, 126, 571–589. [Google Scholar] [CrossRef] [Green Version]
- Veruki, M.; Mørkve, S.H.; Hartveit, E. Activation of a presynaptic glutamate transporter regulates synaptic transmission through electrical signaling. Nat. Neurosci. 2006, 9, 1388–1396. [Google Scholar] [CrossRef] [PubMed]
- Arriza, J.L.; Eliasof, S.; Kavanaugh, M.P.; Amara, S.G. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc. Natl. Acad. Sci. USA 1997, 94, 4155–4160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukasiewcz, P.D.; Bligard, G.W.; DeBrecht, J.D. EAAT5 Glutamate Transporter-Mediated Inhibition in the Vertebrate Retina. Front. Cell. Neurosci. 2021, 15, 662859. [Google Scholar] [CrossRef]
- Bligard, G.W.; DeBrecht, J.; Smith, R.G.; Lukasiewicz, P.D. Light-Evoked Glutamate Transporter EAAT5 Activation Coordinates with Conventional Feedback Inhibition to Control Rod Bipolar Cell Output. J. Neurophysiol. 2020, 123, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- Gehlen, J.; Aretzweiler, C.; Mataruga, A.; Fahlke, C.; Müller, F. Excitatory Amino Acid Transporter EAAT5 Improves Temporal Resolution in the Retina. Eneuro 2021, 8, ENEURO.0406-21.2021. [Google Scholar] [CrossRef]
- Kovermann, P.; Engels, M.; Müller, F.; Fahlke, C. Cellular Physiology and Pathophysiology of EAAT Anion Channels. Front. Cell. Neurosci. 2022, 15, 815279. [Google Scholar] [CrossRef] [PubMed]
- Vorwerk, C.K.; Gorla, M.S.; Dreyer, E.B. An Experimental Basis for Implicating Excitotoxicity in Glaucomatous Optic Neuropathy. Surv. Ophthalmol. 1999, 43, S142–S150. [Google Scholar] [CrossRef]
- Fu, C.T.; Sretavan, D.W. Ectopic Vesicular Glutamate Release at the Optic Nerve Head and Axon Loss in Mouse Experimental Glaucoma. J. Neurosci. 2012, 32, 15859–15876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.B. Diabetic Retinopathy and the NMDA Receptor. Drug News Perspect. 2002, 15, 226–232. [Google Scholar] [CrossRef] [Green Version]
- Gowda, K.; Zinnanti, W.J.; LaNoue, K.F. The influence of diabetes on glutamate metabolism in retinas. J. Neurochem. 2011, 117, 309–320. [Google Scholar] [CrossRef]
- Kaur, C.; Foulds, W.S.; Ling, E.-A. Hypoxia-ischemia and retinal ganglion cell damage. Clin. Ophthalmol. 2008, 2, 879–889. [Google Scholar] [CrossRef] [Green Version]
- Syc-Mazurek, S.; Saidha, S.; Newsome, S.D.; Ratchford, J.N.; Levy, M.; Ford, E.; Crainiceanu, C.M.; Durbin, M.K.; Oakley, J.D.; Meyer, S.A.; et al. Optical coherence tomography segmentation reveals ganglion cell layer pathology after optic neuritis. Brain 2011, 135, 521–533. [Google Scholar] [CrossRef]
- Bojcevski, J.; Stojic, A.; Hoffmann, D.B.; Williams, S.K.; Müller, A.; Diem, R.; Fairless, R. Influence of retinal NMDA receptor activity during autoimmune optic neuritis. J. Neurochem. 2020, 153, 693–709. [Google Scholar] [CrossRef]
- Kale, N. Optic neuritis as an early sign of multiple sclerosis. Eye Brain 2016, 8, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Hirotaka, Y.; Toshiyuki, O.; Masayasu, K.; Baba, T.; Shuichi, Y. Retinal sensitivity reduced in patients with neuromyelitis optica spectrum disorder with no history of optic neuritis. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5116. [Google Scholar]
- Ziccardi, L.; Barbano, L.; Boffa, L.; Albanese, M.; Grzybowski, A.; Centonze, D.; Parisi, V. Morphological Outer Retina Findings in Multiple Sclerosis Patients with or without Optic Neuritis. Front. Neurol. 2020, 11, 858. [Google Scholar] [CrossRef]
- Grimaldi, A.; Brighi, C.; Peruzzi, G.; Ragozzino, D.; Bonanni, V.; Limatola, C.; Ruocco, G.; Di Angelantonio, S. Inflammation, neurodegeneration and protein aggregation in the retina as ocular biomarkers for Alzheimer’s disease in the 3xTg-AD mouse model. Cell Death Dis. 2018, 9, 685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asanad, S.; Felix, C.M.; Fantini, M.; Harrington, M.G.; Sadun, A.A.; Karanjia, R. Retinal ganglion cell dysfunction in preclinical Alzheimer’s disease: An electrophysiologic biomarker signature. Sci. Rep. 2021, 11, 6344. [Google Scholar] [CrossRef]
- Cuenca, N. The retina as a biomarker of Parkinson disease. Investig. Ophthalmol. Vis. Sci. 2019, 60, 8. [Google Scholar]
- Normando, E.M.; Davis, B.; De Groef, L.; Nizari, S.; Turner, L.A.; Ravindran, N.; Pahlitzsch, M.; Brenton, J.; Malaguarnera, G.; Guo, L.; et al. The retina as an early biomarker of neurodegeneration in a rotenone-induced model of Parkinson’s disease: Evidence for a neuroprotective effect of rosiglitazone in the eye and brain. Acta Neuropathol. Commun. 2016, 4, 86. [Google Scholar] [CrossRef] [Green Version]
- London, A.; Benhar, I.; Schwartz, M. The retina as a window to the brain—from eye research to CNS disorders. Nat. Rev. Neurol. 2013, 9, 44–53. [Google Scholar] [CrossRef]
- Liu, X.; Lau, A.; Hou, H.; Moghimi, S.; Proudfoot, J.A.; Chan, E.; Do, J.; Camp, A.; Welsbie, D.; de Moraes, C.G.; et al. Progressive Thinning of Retinal Nerve Fiber Layer and Ganglion Cell–Inner Plexiform Layer in Glaucoma Eyes with Disc Hemorrhage. Ophthalmol. Glaucoma 2021, 4, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, E.B.; Urias, M.G.; Penha, F.M.; Badaró, E.; Novais, E.; Meirelles, R.; Farah, M.E. Diabetes induces changes in neuroretina before retinal vessels: A spectral-domain optical coherence tomography study. Int. J. Retin. Vitr. 2015, 1, 4. [Google Scholar] [CrossRef] [Green Version]
- Akbari, M.; Abdi, P.; Fard, M.A.; Afzali, M.; Ameri, A.; Yazdani-Abyaneh, A.; Mohammadi, M.; Moghimi, S. Retinal Ganglion Cell Loss Precedes Retinal Nerve Fiber Thinning in Nonarteritic Anterior Ischemic Optic Neuropathy. J. Neuro-Ophthalmol. 2016, 36, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Al-Mujaini, A.S.; Al-Mujaini, M.S.; Sabt, B.I. Retinal nerve fiber layer thickness in multiple sclerosis with and without optic neuritis: A four-year follow-up study from Oman. BMC Ophthalmol. 2021, 21, 391. [Google Scholar] [CrossRef] [PubMed]
- López-De-Eguileta, A.; Lage, C.; López-García, S.; Pozueta, A.; García-Martínez, M.; Kazimierczak, M.; Bravo, M.; De Arcocha-Torres, M.; Banzo, I.; Jimenez-Bonilla, J.; et al. Ganglion cell layer thinning in prodromal Alzheimer’s disease defined by amyloid PET. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 570–578. [Google Scholar] [CrossRef]
- Cunha, J.P.; Proença, R.P.; Dias-Santos, A.; Almeida, R.; Águas, H.; Alves, M.; Papoila, A.L.; Louro, C.; Castanheira-Dinis, A. OCT in Alzheimer’s disease: Thinning of the RNFL and superior hemiretina. Graefe’s Arch. Clin. Exp. Ophthalmol. 2017, 255, 1827–1835. [Google Scholar] [CrossRef]
- Sung, M.S.; Choi, S.-M.; Kim, J.; Ha, J.Y.; Kim, B.-C.; Heo, H.; Park, S.W. Inner retinal thinning as a biomarker for cognitive impairment in de novo Parkinson’s disease. Sci. Rep. 2019, 9, 11832. [Google Scholar] [CrossRef] [Green Version]
- Hajee, M.E.; March, W.F.; Lazzaro, D.R.; Wolintz, A.H.; Shrier, E.M.; Glazman, S.; Bodis-Wollner, I.G. Inner Retinal Layer Thinning in Parkinson Disease. Arch. Ophthalmol. 2009, 127, 737–741. [Google Scholar] [CrossRef] [Green Version]
- Downs, J.C. Optic nerve head biomechanics in aging and disease. Exp. Eye Res. 2015, 133, 19–29. [Google Scholar] [CrossRef] [Green Version]
- McAllister, A.S. A review of the vascular anatomy of the optic nerve head and its clinical implications. Cureus 2013, 5, 98. [Google Scholar] [CrossRef] [Green Version]
- Hofman, P.; Hoyng, P.; VanderWerf, F.; Vrensen, G.F.; Schlingemann, O.R. Lack of blood-brain barrier properties in microvessels of the prelaminar optic nerve head. Investig. Ophthalmol. Vis. Sci. 2001, 42, 895–901. [Google Scholar]
- Muench, N.; Patel, S.; Maes, M.; Donahue, R.; Ikeda, A.; Nickells, R. The Influence of Mitochondrial Dynamics and Function on Retinal Ganglion Cell Susceptibility in Optic Nerve Disease. Cells 2021, 10, 1593. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.J.; Attwell, D. The Energetics of CNS White Matter. J. Neurosci. 2012, 32, 356–371. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef]
- Sanes, J.R.; Masland, R.H. The Types of Retinal Ganglion Cells: Current Status and Implications for Neuronal Classification. Annu. Rev. Neurosci. 2015, 38, 221–246. [Google Scholar] [CrossRef]
- Christensen, I.; Lu, B.; Yang, N.; Huang, K.; Wang, P.; Tian, N. The Susceptibility of Retinal Ganglion Cells to Glutamatergic Excitotoxicity Is Type-Specific. Front. Neurosci. 2019, 13, 219. [Google Scholar] [CrossRef] [Green Version]
- Milla-Navarro, S.; Diaz-Tahoces, A.; Ortuño-Lizarán, I.; Fernández, E.; Cuenca, N.; Germain, F.; de la Villa, P. Visual Disfunction due to the Selective Effect of Glutamate Agonists on Retinal Cells. Int. J. Mol. Sci. 2021, 22, 6245. [Google Scholar] [CrossRef]
- El-Danaf, R.N.; Huberman, A.D. Characteristic Patterns of Dendritic Remodeling in Early-Stage Glaucoma: Evidence from Genetically Identified Retinal Ganglion Cell Types. J. Neurosci. 2015, 35, 2329–2343. [Google Scholar] [CrossRef] [Green Version]
- Della Santina, L.; Inman, D.; Lupien, C.B.; Horner, P.J.; Wong, R.O.L. Differential Progression of Structural and Functional Alterations in Distinct Retinal Ganglion Cell Types in a Mouse Model of Glaucoma. J. Neurosci. 2013, 33, 17444–17457. [Google Scholar] [CrossRef]
- Ou, Y.; Jo, R.E.; Ullian, E.M.; Wong, R.O.; Della Santina, L. Selective Vulnerability of Specific Retinal Ganglion Cell Types and Synapses after Transient Ocular Hypertension. J. Neurosci. 2016, 36, 9240–9252. [Google Scholar] [CrossRef] [PubMed]
- VanderWall, K.B.; Lu, B.; Alfaro, J.S.; Allsop, A.R.; Carr, A.S.; Wang, S.; Meyer, J.S. Differential susceptibility of retinal ganglion cell subtypes in acute and chronic models of injury and disease. Sci. Rep. 2020, 10, 17359. [Google Scholar] [CrossRef] [PubMed]
- Daniel, S.; Clark, A.F.; McDowell, C.M. Subtype-specific response of retinal ganglion cells to optic nerve crush. Cell Death Discov. 2018, 4, 67. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.C.S.; Cloherty, S.L.; Ibbotson, M.R.; O’brien, B.J.; O’brien, B.J. Intrinsic physiological properties of rat retinal ganglion cells with a comparative analysis. J. Neurophysiol. 2012, 108, 2008–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreyer, E.B.; Pan, Z.-H.; Storm, S.; Lipton, S.A. Greater sensitivity of larger retinal ganglion cells to NMDA-mediated cell death. Neuroreport 1994, 5, 629–631. [Google Scholar] [CrossRef]
- Vorwerk, C.; Kreutz, M.; Böckers, T.; Brosz, M.; Dreyer, E.; Sabel, B. Susceptibility of retinal ganglion cells to excitotoxicity depends on soma size and retinal eccentricity. Curr. Eye Res. 1999, 19, 59–65. [Google Scholar] [CrossRef]
- Duan, X.; Qiao, M.; Bei, F.; Kim, I.-J.; He, Z.; Sanes, J.R. Subtype-Specific Regeneration of Retinal Ganglion Cells following Axotomy: Effects of Osteopontin and mTOR Signaling. Neuron 2015, 85, 1244–1256. [Google Scholar] [CrossRef] [Green Version]
- Mayer, C.; Bruehl, C.; Salt, E.L.; Diem, R.; Draguhn, A.; Fairless, R. Selective Vulnerability of αOFF Retinal Ganglion Cells during Onset of Autoimmune Optic Neuritis. Neuroscience 2018, 393, 258–272. [Google Scholar] [CrossRef]
- Margolis, D.J.; Gartland, A.J.; Euler, T.; Detwiler, P.B. Dendritic Calcium Signaling in ON and OFF Mouse Retinal Ganglion Cells. J. Neurosci. 2010, 30, 7127–7138. [Google Scholar] [CrossRef] [Green Version]
- Margolis, D.J.; Detwiler, P. Different Mechanisms Generate Maintained Activity in ON and OFF Retinal Ganglion Cells. J. Neurosci. 2007, 27, 5994–6005. [Google Scholar] [CrossRef]
- Wen, X.; Cahill, A.L.; Barta, C.; Thoreson, W.B.; Nawy, S. Elevated Pressure Increases Ca2+ Influx Through AMPA Receptors in Select Populations of Retinal Ganglion Cells. Front. Cell. Neurosci. 2018, 12, 162. [Google Scholar] [CrossRef] [Green Version]
- Vargas, J.L.C.; Osswald, I.K.; Unsain, N.; Aurousseau, M.R.; Barker, P.A.; Bowie, D.; Di Polo, A. Soluble Tumor Necrosis Factor Alpha Promotes Retinal Ganglion Cell Death in Glaucoma via Calcium-Permeable AMPA Receptor Activation. J. Neurosci. 2015, 35, 12088–12102. [Google Scholar] [CrossRef] [Green Version]
- Cuthbertson, A.R.; Mandel, E.T. Anatomy of the mouse retina. Capillary basement membrane thickness. Investig. Ophthalmol. Vis. Sci. 1986, 27, 1653–1658. [Google Scholar]
- Chen, W.-Y.; Han, X.; Cui, L.-J.; Yu, C.-X.; Sheng, W.-L.; Yu, J.; Yuan, F.; Zhong, Y.-M.; Yang, X.-L.; Weng, S.-J. Cell-Subtype-Specific Remodeling of Intrinsically Photosensitive Retinal Ganglion Cells in Streptozotocin-Induced Diabetic Mice. Diabetes 2021, 70, 1157–1169. [Google Scholar] [CrossRef] [PubMed]
- Catalani, E.; Cervia, D. Diabetic retinopathy: A matter of retinal ganglion cell homeostasis. Neural Regen. Res. 2020, 15, 1253–1254. [Google Scholar] [CrossRef]
- Fairless, R.; Bading, H.; Diem, R. Pathophysiological Ionotropic Glutamate Signalling in Neuroinflammatory Disease as a Therapeutic Target. Front. Neurosci. 2021, 15, 741280. [Google Scholar] [CrossRef]
- Elewerenz, J.; Emaher, P. Chronic Glutamate Toxicity in Neurodegenerative Diseases—What is the Evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef]
- Lucas, D.R.; Newhouse, J.P. The Toxic Effect of Sodium L-Glutamate on the Inner Layers of the Retina. Arch. Ophthalmol. 1957, 58, 193–201. [Google Scholar] [CrossRef]
- Sisk, D.R.; Kuwabara, T. Histologic changes in the inner retina of albino rats following intravitreal injection of mono-sodiuml-glutamate. Graefe’s Arch. Clin. Exp. Ophthalmol. 1985, 223, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Lotery, A.J. Glutamate excitotoxicity in glaucoma: Truth or fiction? Eye 2005, 19, 369–370. [Google Scholar] [CrossRef] [Green Version]
- Izumi, Y.; Shimamoto, K.; Benz, A.M.; Hammerman, S.B.; Olney, J.W.; Zorumski, C.F. Glutamate transporters and retinal excitotoxicity. Glia 2002, 39, 58–68. [Google Scholar] [CrossRef] [PubMed]
- Siliprandi, R.; Canella, R.; Carmignoto, G.; Schiavo, N.; Zanellato, A.; Zanoni, R.; Vantini, G. N-methyl-d-aspartate-induced neurotoxicity in the adult rat retina. Vis. Neurosci. 1992, 8, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Lambuk, L.; Jafri, A.J.A.; Iezhitsa, I.; Agarwal, R.; Bakar, N.S.; Agarwal, P.; Abdullah, A.; Ismail, N.M. Dose-dependent effects of NMDA on retinal and optic nerve morphology in rats. Int. J. Ophthalmol. 2019, 12, 746–753. [Google Scholar] [CrossRef]
- Mizutani, M.; Gerhardinger, C.; Lorenzi, M. Müller cell changes in human diabetic retinopathy. Diabetes 1998, 47, 445–449. [Google Scholar] [CrossRef]
- Li, Q.; Puro, D.G. Diabetes-induced dysfunction of the glutamate transporter in retinal Müller cells. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3109–3116. [Google Scholar]
- Yanagisawa, M.; Namekata, K.; Aida, T.; Katou, S.; Takeda, T.; Harada, T.; Fuse, N.; The Glaucoma Gene Research Group; Tanaka, K. EAAT1 variants associated with glaucoma. Biochem. Biophys. Res. Commun. 2020, 529, 943–949. [Google Scholar] [CrossRef]
- Naskar, R.; Vorwerk, C.K.; Dreyer, E.B. Concurrent downregulation of a glutamate transporter and receptor in glaucoma. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1940–1944. [Google Scholar]
- Russo, R.; Cavaliere, F.; Varano, G.P.; Milanese, M.; Adornetto, A.; Nucci, C.; Bonanno, G.; Morrone, L.A.; Corasaniti, M.T.; Bagetta, G. Impairment of Neuronal Glutamate Uptake and Modulation of the Glutamate Transporter GLT-1 Induced by Retinal Ischemia. PLoS ONE 2013, 8, e69250. [Google Scholar] [CrossRef] [Green Version]
- Vallejo-Illarramendi, A.; Domercq, M.; Cerda, F.P.; Ravid, R.; Matute, C. Increased expression and function of glutamate transporters in multiple sclerosis. Neurobiol. Dis. 2006, 21, 154–164. [Google Scholar] [CrossRef]
- Pampliega, O.; Domercq, M.; Soria, F.N.; Villoslada, P.; Rodríguez-Antigüedad, A.; Matute, C. Increased expression of cystine/glutamate antiporter in multiple sclerosis. J. Neuroinflamm. 2011, 8, 63. [Google Scholar] [CrossRef] [Green Version]
- Sulkowski, G.; Dąbrowska-Bouta, B.; Salińska, E.; Strużyńska, L. Modulation of Glutamate Transport and Receptor Binding by Glutamate Receptor Antagonists in EAE Rat Brain. PLoS ONE 2014, 9, e113954. [Google Scholar] [CrossRef] [Green Version]
- Alba-Arbalat, S.; Andorra, M.; Sanchez-Dalmau, B.; Camos-Carreras, A.; Dotti-Boada, M.; Pulido-Valdeolivas, I.; Llufriu, S.; Blanco, Y.; Sepulveda, M.; Saiz, A.; et al. In Vivo Molecular Changes in the Retina of Patients with Multiple Sclerosis. Investig. Opthalmology Vis. Sci. 2021, 62, 11. [Google Scholar] [CrossRef]
- Shen, W.; Fruttiger, M.; Zhu, L.; Chung, S.H.; Barnett, N.L.; Kirk, J.K.; Lee, S.R.; Coorey, N.J.; Killingsworth, M.C.; Sherman, L.S.; et al. Conditional Muller Cell Ablation Causes Independent Neuronal and Vascular Pathologies in a Novel Transgenic Model. J. Neurosci. 2012, 32, 15715–15727. [Google Scholar] [CrossRef]
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.N.; Osborne, N.N.; Reichenbach, A. Müller cells in the healthy and diseased retina. Prog. Retin. Eye Res. 2006, 25, 397–424. [Google Scholar] [CrossRef]
- García-Bermúdez, M.Y.; Freude, K.K.; Mouhammad, Z.A.; van Wijngaarden, P.; Martin, K.K.; Kolko, M. Glial Cells in Glaucoma: Friends, Foes, and Potential Therapeutic Targets. Front. Neurol. 2021, 12, 624983. [Google Scholar] [CrossRef]
- Slezak, M.; Grosche, A.; Niemiec, A.; Tanimoto, N.; Pannicke, T.; Münch, T.A.; Crocker, B.; Isope, P.; Härtig, W.; Beck, S.C.; et al. Relevance of Exocytotic Glutamate Release from Retinal Glia. Neuron 2012, 74, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Jeremy, S.M.; Livne-Bar, I. Quiescent Retinal Glia are Protective, but their Activation Increases Vulnerability to Acute RGC Injury In Vivo. ARVO Annu. Meet. Abstr. 2014, 55, 13. [Google Scholar]
- Ju, W.-K.; Lindsey, J.D.; Angert, M.; Patel, A.; Weinreb, R.N. Glutamate receptor activation triggers OPA1 release and induces apoptotic cell death in ischemic rat retina. Mol. Vis. 2008, 14, 2629–2638. [Google Scholar]
- Passlick, S.; Rose, C.R.; Petzold, G.C.; Henneberger, C. Disruption of Glutamate Transport and Homeostasis by Acute Metabolic Stress. Front. Cell. Neurosci. 2021, 15, 522–529. [Google Scholar] [CrossRef]
- Vohra, R.; Kolko, M. Lactate: More Than Merely a Metabolic Waste Product in the Inner Retina. Mol. Neurobiol. 2020, 57, 2021–2037. [Google Scholar] [CrossRef]
- Toft-Kehler, A.K.; Gurubaran, I.S.; Desler, C.; Rasmussen, L.J.; Skytt, D.M.; Kolko, M. Oxidative Stress-Induced Dysfunction of Müller Cells During Starvation. Investig. Opthalmol. Vis. Sci. 2016, 57, 2721–2728. [Google Scholar] [CrossRef]
- Mysona, B.; Dun, Y.; Duplantier, J.; Ganapathy, V.; Smith, S.B. Effects of hyperglycemia and oxidative stress on the glutamate transporters GLAST and system xc−In mouse retinal Müller glial cells. Cell Tissue Res. 2009, 335, 477–488. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.J.; Jun, H.O.; Kim, J.H.; Kim, J.H. Astrocytic cystine/glutamate antiporter is a key regulator of erythropoietin expression in the ischemic retina. FASEB J. 2019, 33, 6045–6054. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The Cystine/Glutamate Antiporter System xc−In Health and Disease: From Molecular Mechanisms to Novel Therapeutic Opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef] [Green Version]
- Newman, E.A. Glial cell regulation of neuronal activity and blood flow in the retina by release of gliotransmitters. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140195. [Google Scholar] [CrossRef]
- Coughlin, B.A.; Feenstra, D.J.; Mohr, S. Müller cells and diabetic retinopathy. Vis. Res. 2017, 139, 93–100. [Google Scholar] [CrossRef]
- Ishida, S.; Usui, T.; Yamashiro, K.; Kaji, Y.; Ahmed, E.; Carrasquillo, K.G.; Amano, S.; Hida, T.; Oguchi, Y.; Adamis, A.P. VEGF164 Is Proinflammatory in the Diabetic Retina. Investig. Opthalmol. Vis. Sci. 2003, 44, 2155–2162. [Google Scholar] [CrossRef] [Green Version]
- Abhary, S.; Burdon, K.P.; Gupta, A.; Lake, S.; Selva, D.; Petrovsky, N.; Craig, J.E. Common Sequence Variation in the VEGFA Gene Predicts Risk of Diabetic Retinopathy. Investig. Opthalmol. Vis. Sci. 2009, 50, 5552–5558. [Google Scholar] [CrossRef] [Green Version]
- Kida, T.; Oku, H.; Osuka, S.; Horie, T.; Ikeda, T. Hyperglycemia-induced VEGF and ROS production in retinal cells is inhibited by the mTOR inhibitor, rapamycin. Sci. Rep. 2021, 11, 1885. [Google Scholar] [CrossRef]
- Schlüter, A.; Aksan, B.; Diem, R.; Fairless, R.; Mauceri, D. VEGFD Protects Retinal Ganglion Cells and, consequently, Capillaries against Excitotoxic Injury. Mol. Ther. Methods Clin. Dev. 2020, 17, 281–299. [Google Scholar] [CrossRef] [Green Version]
- Mac Nair, E.C.; Fernandes, A.K.; Schlamp, C.L.; Libby, R.T.; Nickells, R.W. Tumor necrosis factor alpha has an early protective effect on retinal ganglion cells after optic nerve crush. J. Neuroinflamm. 2014, 11, 194. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Cheng, Y.; Zhang, S.; Sun, X.; Wu, J. TRPV4-induced Müller cell gliosis and TNF-α elevation-mediated retinal ganglion cell apoptosis in glaucomatous rats via JAK2/STAT3/NF-κB pathway. J. Neuroinflamm. 2021, 18, 271. [Google Scholar] [CrossRef] [PubMed]
- Dyer, M.A.; Cepko, C.L. Control of Müller glial cell proliferation and activation following retinal injury. Nat. Neurosci. 2000, 3, 873–880. [Google Scholar] [CrossRef]
- Yang, L.-P.; Sun, H.-L.; Wu, L.-M.; Guo, X.-J.; Dou, H.-L.; Tso, M.O.M.; Zhao, L.; Li, S.-M. Baicalein Reduces Inflammatory Process in a Rodent Model of Diabetic Retinopathy. Investig. Opthalmol. Vis. Sci. 2009, 50, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Abcouwer, S.F. Angiogenic Factors and Cytokines in Diabetic Retinopathy. J. Clin. Cell. Immunol. 2011, 1, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Pereiro, X.; Ruzafa, N.; Acera, A.; Fonollosa, A.; Rodriguez, F.D.; Vecino, E. Dexamethasone protects retinal ganglion cells but not Müller glia against hyperglycemia in vitro. PLoS ONE 2018, 13, e0207913. [Google Scholar] [CrossRef]
- Lieth, E.; Barber, A.J.; Xu, B.; Dice, C.; Ratz, M.J.; Tanase, D.; Strother, J.M. Glial reactivity and impaired glutamate metabolism in short-term experimental diabetic retinopathy. Penn State Retina Research Group. Diabetes 1998, 47, 815–820. [Google Scholar] [CrossRef]
- Kaur, C.; Sivakumar, V.; Foulds, W.S. Early Response of Neurons and Glial Cells to Hypoxia in the Retina. Investig. Opthalmol. Vis. Sci. 2006, 47, 1126–1141. [Google Scholar] [CrossRef]
- Chen, Q.; Olney, J.W.; Price, M.T.; Romano, C. Biochemical and morphological analysis of non-NMDA receptor mediated excitotoxicity in chick embryo retina. Vis. Neurosci. 1999, 16, 131–139. [Google Scholar] [CrossRef]
- Lebrun-Julien, F.; Duplan, L.; Pernet, V.; Osswald, I.; Sapieha, P.; Bourgeois, P.; Dickson, K.; Bowie, D.; Barker, P.A.; Di Polo, A. Excitotoxic Death of Retinal Neurons In Vivo Occurs via a Non-Cell-Autonomous Mechanism. J. Neurosci. 2009, 29, 5536–5545. [Google Scholar] [CrossRef] [Green Version]
- Liberatore, F.; Bucci, D.; Mascio, G.; Madonna, M.; Di Pietro, P.; Beneventano, M.; Puliti, A.M.; Battaglia, G.; Bruno, V.; Nicoletti, F.; et al. Permissive role for mGlu1 metabotropic glutamate receptors in excitotoxic retinal degeneration. Neuroscience 2017, 363, 142–149. [Google Scholar] [CrossRef]
- Kuehn, M.; Fingert, J.; Kwon, Y. Retinal Ganglion Cell Death in Glaucoma: Mechanisms and Neuroprotective Strategies. Ophthalmol. Clin. N. Am. 2005, 18, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Vargas, J.L.C.; Di Polo, A. The molecular basis of retinal ganglion cell death in glaucoma. Prog. Retin. Eye Res. 2012, 31, 152–181. [Google Scholar] [CrossRef] [PubMed]
- Evangelho, K.; Mogilevskaya, M.; Losada-Barragan, M.; Vargas-Sanchez, J.K. Pathophysiology of primary open-angle glaucoma from a neuroinflammatory and neurotoxicity perspective: A review of the literature. Int. Ophthalmol. 2017, 39, 259–271. [Google Scholar] [CrossRef]
- Adachi, K.; Fujita, Y.; Morizane, C.; Akaike, A.; Ueda, M.; Satoh, M.; Masai, H.; Kashii, S.; Honda, Y. Inhibition of NMDA receptors and nitric oxide synthase reduces ischemic injury of the retina. Eur. J. Pharmacol. 1998, 350, 53–57. [Google Scholar] [CrossRef]
- Osborne, N.; Ugarte, M.; Chao, M.; Chidlow, G.; Bae, J.; Wood, J.; Nash, M. Neuroprotection in Relation to Retinal Ischemia and Relevance to Glaucoma. Surv. Ophthalmol. 1999, 43, S102–S128. [Google Scholar] [CrossRef]
- Ng, Y.-K.; Zeng, X.-X.; Ling, E.-A. Expression of glutamate receptors and calcium-binding proteins in the retina of streptozotocin-induced diabetic rats. Brain Res. 2004, 1018, 66–72. [Google Scholar] [CrossRef]
- Santiago, A.R.; Hughes, J.M.; Kamphuis, W.; Schlingemann, R.O.; Ambrósio, A.F. Diabetes changes ionotropic glutamate receptor subunit expression level in the human retina. Brain Res. 2008, 1198, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Bengtson, C.P.; Buchthal, B.; Hagenston, A.M.; Bading, H. Coupling of NMDA receptors and TRPM4 guides discovery of unconventional neuroprotectants. Science 2020, 370, 6513. [Google Scholar] [CrossRef] [PubMed]
- Papouin, T.; Oliet, S.H.R. Organization, control and function of extrasynaptic NMDA receptors. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130601. [Google Scholar] [CrossRef] [PubMed]
- Franchini, L.; Carrano, N.; Di Luca, M.; Gardoni, F. Synaptic GluN2A-Containing NMDA Receptors: From Physiology to Pathological Synaptic Plasticity. Int. J. Mol. Sci. 2020, 21, 1538. [Google Scholar] [CrossRef] [Green Version]
- Hardingham, G.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Bai, N.; Aida, T.; Yanagisawa, M.; Katou, S.; Sakimura, K.; Mishina, M.; Tanaka, K. NMDA receptor subunits have different roles in NMDA-induced neurotoxicity in the retina. Mol. Brain 2013, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.-D.; Chen, J.; Li, F.; Gao, F.; Wu, J.; Miao, Y.; Wang, Z. Enhanced Expression of NR2B Subunits of NMDA Receptors in the Inherited Glaucomatous DBA/2J Mouse Retina. Neural Plast. 2013, 2013, 670254. [Google Scholar] [CrossRef]
- Ishikawa, M. Abnormalities in Glutamate Metabolism and Excitotoxicity in the Retinal Diseases. Science 2013, 2013, 528940. [Google Scholar] [CrossRef] [Green Version]
- Sucher, N.J.; Lipton, S.A.; Dreyer, E.B. Molecular basis of glutamate toxicity in retinal ganglion cells. Vis. Res. 1997, 37, 3483–3493. [Google Scholar] [CrossRef] [Green Version]
- Hare, W.A.; Wheeler, L. Experimental Glutamatergic Excitotoxicity in Rabbit Retinal Ganglion Cells: Block by Memantine. Investig. Opthalmol. Vis. Sci. 2009, 50, 2940–2948. [Google Scholar] [CrossRef]
- Macrez, R.; Ortega, M.C.; Bardou, I.; Mehra, A.; Fournier, A.; Van der Pol, S.M.A.; Haelewyn, B.; Maubert, E.; Lesept, F.; Chevilley, A.; et al. Neuroendothelial NMDA receptors as therapeutic targets in experimental autoimmune encephalomyelitis. Brain 2016, 139, 2406–2419. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wong, W.T. Microglia-Müller Cell Interactions in the Retina. Adv. Exp. Med. Biol. 2014, 801, 333–338. [Google Scholar] [CrossRef] [Green Version]
- Tsoka, P.; Barbisan, P.R.; Kataoka, K.; Chen, X.N.; Tian, B.; Bouzika, P.; Miller, J.W.; Paschalis, E.I.; Vavvas, D.G. NLRP3 inflammasome in NMDA-induced retinal excitotoxicity. Exp. Eye Res. 2019, 181, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Bading, H. Therapeutic targeting of the pathological triad of extrasynaptic NMDA receptor signaling in neurodegenerations. J. Exp. Med. 2017, 214, 569–578. [Google Scholar] [CrossRef] [PubMed]
- VanDongen, A.; Blanke, M. Activation Mechanisms of the NMDA Receptor. In Biology of the NMDA Receptor; Informa UK Limited: London, UK, 2008; pp. 283–312. [Google Scholar]
- Sargoy, A.; Sun, X.; Barnes, S.; Brecha, N.C. Differential Calcium Signaling Mediated by Voltage-Gated Calcium Channels in Rat Retinal Ganglion Cells and Their Unmyelinated Axons. PLoS ONE 2014, 9, e84507. [Google Scholar] [CrossRef]
- Schubert, T.; Akopian, A. Actin filaments regulate voltage-gated ion channels in salamander retinal ganglion cells. Neuroscience 2004, 125, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Sivakumar, V.; Foulds, W.S.; Luu, C.D.; Ling, E.-A. Hypoxia-Induced Activation of N-methyl-d-aspartate Receptors Causes Retinal Ganglion Cell Death in the Neonatal Retina. J. Neuropathol. Exp. Neurol. 2012, 71, 330–347. [Google Scholar] [CrossRef] [Green Version]
- Kocsis, J.; Rand, M.; Chen, B.; Waxman, S.; Pourcho, R. Kainate elicits elevated nuclear calcium signals in retinal neurons via calcium-induced calcium release. Brain Res. 1993, 616, 273–282. [Google Scholar] [CrossRef]
- Leinders-Zufall, T.; Rand, M.N.; Waxman, S.G.; Kocsis, J.D. Differential Role of Two Ca2+-Permeable Non-NMDA Glutamate Channels in Rat Retinal Ganglion Cells: Kainate-Induced Cytoplasmic and Nuclear Ca2+ Signals. J. Neurophysiol. 1994, 72, 2503–2516. [Google Scholar] [CrossRef]
- Hartwick, A.T.E.; Hamilton, C.M.; Baldridge, W.H. Glutamatergic calcium dynamics and deregulation of rat retinal ganglion cells. J. Physiol. 2008, 586, 3425–3446. [Google Scholar] [CrossRef]
- Brittain, M.K.; Brustovetsky, T.; Sheets, P.L.; Brittain, J.M.; Khanna, R.; Cummins, T.R.; Brustovetsky, N. Delayed calcium dysregulation in neurons requires both the NMDA receptor and the reverse Na+/Ca2+ exchanger. Neurobiol. Dis. 2012, 46, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Brustovetsky, T.; Bolshakov, A.; Brustovetsky, N. Calpain activation and Na+/Ca2+ exchanger degradation occur downstream of calcium deregulation in hippocampal neurons exposed to excitotoxic glutamate. J. Neurosci. Res. 2009, 88, 1317–1328. [Google Scholar] [CrossRef] [Green Version]
- Inokuchi, Y.; Shimazawa, M.; Nakajima, Y.; Komuro, I.; Matsuda, T.; Baba, A.; Araie, M.; Kita, S.; Iwamoto, T.; Hara, H. A Na+/Ca2+ exchanger isoform, NCX1, is involved in retinal cell death after N-methyl-d-aspartate injection and ischemia-reperfusion. J. Neurosci. Res. 2009, 87, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.Z.; Zhang, D.; Abele, A.E.; Lipton, S.A. Blockade of NMDA receptor-mediated mobilization of intracellular Ca2+ prevents neurotoxicity. Brain Res. 1992, 598, 196–202. [Google Scholar] [CrossRef]
- Karagas, N.E.; Venkatachalam, K. Roles for the Endoplasmic Reticulum in Regulation of Neuronal Calcium Homeostasis. Cells 2019, 8, 1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruiz, A.; Matute, C.; Alberdi, E. Endoplasmic reticulum Ca2+ release through ryanodine and IP3 receptors contributes to neuronal excitotoxicity. Cell Calcium 2009, 46, 273–281. [Google Scholar] [CrossRef]
- Duchen, M.R. Mitochondria and calcium: From cell signalling to cell death. J. Physiol. 2000, 529, 57–68. [Google Scholar] [CrossRef]
- Hajnóczky, G.; Csordás, G.; Muniswamy, M.; Pacher, P. The machinery of local Ca2+ signalling between sarco-endo-plasmic reticulum and mitochondria. J. Physiol. 2000, 529, 69–81. [Google Scholar] [CrossRef]
- Duncan, R.S.; Goad, D.L.; Grillo, M.A.; Kaja, S.; Payne, A.J.; Koulen, P. Control of Intracellular Calcium Signaling as a Neuroprotective Strategy. Molecules 2010, 15, 1168–1195. [Google Scholar] [CrossRef] [Green Version]
- Möckel, V.; Fischer, G. Vulnerability to excitotoxic stimuli of cultured rat hippocampal neurons containing the calcium-binding proteins calretinin and calbindin D28K. Brain Res. 1994, 648, 109–120. [Google Scholar] [CrossRef]
- Fei, Z.; Fei, F.; Wu, X.-Q.; Su, N. Homer signaling pathways as effective therapeutic targets for ischemic and traumatic brain injuries and retinal lesions. Neural Regen. Res. 2022, 17, 1454. [Google Scholar] [CrossRef]
- Brandstätter, J.H.; Dick, O.; Boeckers, T.M. The postsynaptic scaffold proteins ProSAP1/Shank2 and Homer1 are associated with glutamate receptor complexes at rat retinal synapses. J. Comp. Neurol. 2004, 475, 551–563. [Google Scholar] [CrossRef]
- Bertaso, F.; Roussignol, G.; Worley, P.; Bockaert, J.; Fagni, L.; Ango, F. Homer1a-Dependent Crosstalk Between NMDA and Metabotropic Glutamate Receptors in Mouse Neurons. PLoS ONE 2010, 5, e9755. [Google Scholar] [CrossRef] [Green Version]
- Fei, F.; Li, J.; Rao, W.; Liu, W.; Chen, X.; Su, N.; Wang, Y.; Fei, Z. Upregulation of Homer1a Promoted Retinal Ganglion Cell Survival After Retinal Ischemia and Reperfusion via Interacting with Erk Pathway. Cell. Mol. Neurobiol. 2015, 35, 1039–1048. [Google Scholar] [CrossRef]
- Kaja, S.; Naumchuk, Y.; Grillo, S.L.; Borden, P.K.; Koulen, P. Differential up-regulation of Vesl-1/Homer 1 protein isoforms associated with decline in visual performance in a preclinical glaucoma model. Vis. Res. 2013, 94, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Inoue, A.; Okabe, S. The dynamic organization of postsynaptic proteins: Translocating molecules regulate synaptic function. Curr. Opin. Neurobiol. 2003, 13, 332–340. [Google Scholar] [CrossRef]
- Chen, M.; Lu, T.-J.; Chen, X.-J.; Zhou, Y.; Chen, Q.; Feng, X.-Y.; Xu, L.; Duan, W.-H.; Xiong, Z.-Q. Differential Roles of NMDA Receptor Subtypes in Ischemic Neuronal Cell Death and Ischemic Tolerance. Stroke 2008, 39, 3042–3048. [Google Scholar] [CrossRef]
- Liu, Y.; Wong, T.P.; Aarts, M.; Rooyakkers, A.; Liu, L.; Lai, T.W.; Wu, D.C.; Lu, J.; Tymianski, M.; Craig, A.M.; et al. NMDA Receptor Subunits Have Differential Roles in Mediating Excitotoxic Neuronal Death Both In Vitro and In Vivo. J. Neurosci. 2007, 27, 2846–2857. [Google Scholar] [CrossRef] [Green Version]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Okamoto, K.-I.; Hayashi, Y.; Sheng, M. The Importance of Dendritic Mitochondria in the Morphogenesis and Plasticity of Spines and Synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef] [Green Version]
- Schwaller, B. Cytosolic Ca2+ Buffers. Cold Spring Harb. Perspect. Biol. 2010, 2, a004051. [Google Scholar] [CrossRef] [PubMed]
- Kovács-Öller, T.; Szarka, G.; Ganczer, A.; Tengölics, Á.; Balogh, B.; Völgyi, B. Expression of Ca2+-Binding Buffer Proteins in the Human and Mouse Retinal Neurons. Int. J. Mol. Sci. 2019, 20, 2229. [Google Scholar] [CrossRef] [Green Version]
- Fairless, R.; Williams, S.K.; Diem, R. Calcium-Binding Proteins as Determinants of Central Nervous System Neuronal Vulnerability to Disease. Int. J. Mol. Sci. 2019, 20, 2146. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.J.H.; Siddiqui, A.M.; Sabljic, T.F.; Ball, A.K. Changes in parvalbumin immunoreactive retinal ganglion cells and amacrine cells after optic nerve injury. Exp. Eye Res. 2016, 145, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Kwon, O.J.; Kim, J.Y.; Kim, S.Y.; Jeon, C.J. Alterations in the localization of calbindin D28K-, calretinin-, and par-valbumin-immunoreactive neurons of rabbit retinal ganglion cell layer from ischemia and reperfusion. Mol. Cells 2005, 19, 382–390. [Google Scholar]
- Park, H.-S.; Park, S.-J.; Park, S.-H.; Chun, M.-H.; Oh, S.-J. Shifting of parvalbumin expression in the rat retina in experimentally induced diabetes. Acta Neuropathol. 2008, 115, 241–248. [Google Scholar] [CrossRef]
- Gunn, D.J.; Gole, A.G.; Barnett, N.L. Specific amacrine cell changes in an induced mouse model of glaucoma. Clin. Exp. Ophthalmol. 2011, 39, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Krieger, B.; Qiao, M.; Rousso, D.L.; Sanes, J.R.; Meister, M. Four alpha ganglion cell types in mouse retina: Function, structure, and molecular signatures. PLoS ONE 2017, 12, e0180091. [Google Scholar] [CrossRef] [Green Version]
- Kántor, O.; Mezey, S.; Adeghate, J.; Naumann, A.; Nitschke, R.; Énzsöly, A.; Szabó, A.; Lukáts, Á.; Németh, J.; Somogyvári, Z.; et al. Calcium buffer proteins are specific markers of human retinal neurons. Cell Tissue Res. 2016, 365, 29–50. [Google Scholar] [CrossRef]
- Fan, W.; Li, X.; Cooper, N.G.F. CaMKIIαB Mediates a Survival Response in Retinal Ganglion Cells Subjected to a Glutamate Stimulus. Investig. Opthalmol. Vis. Sci. 2007, 48, 3854–3863. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Kitaoka, Y.; Hayashi, Y.; Kumai, T.; Munemasa, Y.; Fujino, H.; Kobayashi, S.; Ueno, S. Calcium/calmodulin-dependent protein kinase II regulates the phosphorylation of CREB in NMDA-induced retinal neurotoxicity. Brain Res. 2007, 1184, 306–315. [Google Scholar] [CrossRef]
- Saura, C.A.; Valero, J. The role of CREB signaling in Alzheimer’s disease and other cognitive disorders. Rev. Neurosci. 2011, 22, 153–169. [Google Scholar] [CrossRef]
- Fudalej, E.; Justyniarska, M.; Kasarełło, K.; Dziedziak, J.; Szaflik, J.P.; Cudnoch-Jędrzejewska, A. Neuroprotective Factors of the Retina and Their Role in Promoting Survival of Retinal Ganglion Cells: A Review. Ophthalmic Res. 2021, 64, 345–355. [Google Scholar] [CrossRef]
- Kimura, A.; Namekata, K.; Guo, X.; Harada, C.; Harada, T. Neuroprotection, Growth Factors and BDNF-TrkB Signalling in Retinal Degeneration. Int. J. Mol. Sci. 2016, 17, 1584. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Chen, H.; Yi, J.; Troy, J.B.; Zhang, H.F.; Liu, X. Long-Term Protection of Retinal Ganglion Cells and Visual Function by Brain-Derived Neurotrophic Factor in Mice with Ocular Hypertension. Investig. Opthalmol. Vis. Sci. 2016, 57, 3793–3802. [Google Scholar] [CrossRef] [Green Version]
- Domenici, L.; Origlia, N.; Falsini, B.; Cerri, E.; Barloscio, D.; Fabiani, C.; Sansò, M.; Giovannini, L. Rescue of Retinal Function by BDNF in a Mouse Model of Glaucoma. PLoS ONE 2014, 9, e115579. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.; You, Y.; Li, J.; Gupta, V.; Golzan, M.; Klistorner, A.; van den Buuse, M.; Graham, S. BDNF impairment is associated with age-related changes in the inner retina and exacerbates experimental glaucoma. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1567–1578. [Google Scholar] [CrossRef]
- Rocha, M.; Martins, R.A.; Linden, R. Activation of NMDA receptors protects against glutamate neurotoxicity in the retina: Evidence for the involvement of neurotrophins. Brain Res. 1999, 827, 79–92. [Google Scholar] [CrossRef]
- Dai, M.; Xia, X.-B.; Xiong, S.-Q. BDNF regulates GLAST and glutamine synthetase in mouse retinal Müller cells. J. Cell. Physiol. 2012, 227, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Harada, C.; Guo, X.; Namekata, K.; Kimura, A.; Nakamura, K.; Tanaka, K.; Parada, L.F.; Harada, T. Glia- and neuron-specific functions of TrkB signalling during retinal degeneration and regeneration. Nat. Commun. 2011, 2, 189. [Google Scholar] [CrossRef] [Green Version]
- Afarid, M.; Namvar, E.; Sanie-Jahromi, F. Diabetic Retinopathy and BDNF: A Review on Its Molecular Basis and Clinical Applications. J. Ophthalmol. 2020, 2020, 1602739. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, S.-Z.; Wang, P.-P.; Yu, S.-P.; Zheng, Z.; Xu, X. Calcium mediates high glucose-induced HIF-1α and VEGF expression in cultured rat retinal Müller cells through CaMKII-CREB pathway. Acta Pharmacol. Sin. 2012, 33, 1030–1036. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Lv, F.-L.; Wang, G.-H. Effects of HIF-1α on diabetic retinopathy angiogenesis and VEGF expression. Eur. Rev. Med Pharmacol. Sci. 2018, 22, 5071–5076. [Google Scholar]
- Wang, X.; Wang, G.; Wang, Y. Intravitreous Vascular Endothelial Growth Factor and Hypoxia-Inducible Factor 1a in Patients with Proliferative Diabetic Retinopathy. Am. J. Ophthalmol. 2009, 148, 883–889. [Google Scholar] [CrossRef]
- Ambati, J. Elevated γ-Aminobutyric Acid, Glutamate, and Vascular Endothelial Growth Factor Levels in the Vitreous of Patients with Proliferative Diabetic Retinopathy. Arch. Ophthalmol. 1997, 115, 1161–1166. [Google Scholar] [CrossRef]
- Manabe, S.-I.; Lipton, S.A. Divergent NMDA signals leading to proapoptotic and antiapoptotic pathways in the rat retina. Investig. Opthalmol. Vis. Sci. 2003, 44, 385–392. [Google Scholar] [CrossRef] [Green Version]
- Munemasa, Y.; Ohtani-Kaneko, R.; Kitaoka, Y.; Kuribayashi, K.; Isenoumi, K.; Kogo, J.; Yamashita, K.; Kumai, T.; Kobayashi, S.; Hirata, K.; et al. Contribution of mitogen-activated protein kinases to NMDA-induced neurotoxicity in the rat retina. Brain Res. 2005, 1044, 227–240. [Google Scholar] [CrossRef]
- Russo, R.; Cavaliere, F.; Berliocchi, L.; Nucci, C.; Gliozzi, M.; Mazzei, C.; Tassorelli, C.; Corasaniti, M.T.; Rotiroti, D.; Bagetta, G.; et al. Modulation of pro-survival and death-associated pathways under retinal ischemia/reperfusion: Effects of NMDA receptor blockade. J. Neurochem. 2008, 107, 1347–1357. [Google Scholar] [CrossRef]
- Du, H.-Y.; Wang, R.; Li, J.-L.; Luo, H.; Xie, X.-Y.; Yan, R.; Jian, Y.-L.; Cai, J.-Y. Ligustrazine induces viability, suppresses apoptosis and autophagy of retinal ganglion cells with ischemia/reperfusion injury through the PI3K/Akt/mTOR signaling pathway. Bioengineered 2021, 12, 507–515. [Google Scholar] [CrossRef]
- Li, R.; Jin, Y.; Li, Q.; Sun, X.; Zhu, H.; Cui, H. MiR-93-5p targeting PTEN regulates the NMDA-induced autophagy of retinal ganglion cells via AKT/mTOR pathway in glaucoma. Biomed. Pharmacother. 2018, 100, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Agostinone, J.; Alarcon-Martinez, L.; Gamlin, C.; Yu, W.-Q.; Wong, R.O.L.; Di Polo, A. Insulin signalling promotes dendrite and synapse regeneration and restores circuit function after axonal injury. Brain 2018, 141, 1963–1980. [Google Scholar] [CrossRef] [PubMed]
- Teotia, P.; Van Hook, M.J.; Fischer, D.; Ahmad, I. Human retinal ganglion cell axon regeneration by recapitulating developmental mechanisms: Effects of recruitment of the mTOR pathway. Development 2019, 146, dev178012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madrakhimov, S.B.; Yang, J.Y.; Kim, J.H.; Han, J.W.; Park, T.K. mTOR-dependent dysregulation of autophagy contributes to the retinal ganglion cell loss in streptozotocin-induced diabetic retinopathy. Cell Commun. Signal. 2021, 19, 29. [Google Scholar] [CrossRef]
- Yu, D.-Y.; Cringle, S.J.; Balaratnasingam, C.; Morgan, W.H.; Yu, P.; Su, E.-N. Retinal ganglion cells: Energetics, compartmentation, axonal transport, cytoskeletons and vulnerability. Prog. Retin. Eye Res. 2013, 36, 217–246. [Google Scholar] [CrossRef] [PubMed]
- Seager, R.; Lee, L.; Henley, J.M.; Wilkinson, K.A. Mechanisms and roles of mitochondrial localisation and dynamics in neuronal function. Neuronal Signal. 2020, 4, NS20200008. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.M.; Griffiths, P.G.; Johnson, A.M.; Turnbull, D.M. Histochemical localisation of mitochondrial enzyme activity in human optic nerve and retina. Br. J. Ophthalmol. 1999, 83, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Ito, Y.A.; Di Polo, A. Mitochondrial dynamics, transport, and quality control: A bottleneck for retinal ganglion cell viability in optic neuropathies. Mitochondrion 2017, 36, 186–192. [Google Scholar] [CrossRef]
- Man, C.Y.W.; Chinnery, P.; Griffiths, P. Optic neuropathies – Importance of spatial distribution of mitochondria as well as function. Med Hypotheses 2005, 65, 1038–1042. [Google Scholar] [CrossRef]
- Lee, S.; Van Bergen, N.J.; Kong, G.Y.; Chrysostomou, V.; Waugh, H.S.; O’Neill, E.C.; Crowston, J.G.; Trounce, I.A. Mitochondrial dysfunction in glaucoma and emerging bioenergetic therapies. Exp. Eye Res. 2011, 93, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G. Oxidative stress in glaucomatous neurodegeneration: Mechanisms and consequences. Prog. Retin. Eye Res. 2006, 25, 490–513. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Pang, Y.; Zhang, Z.; Li, X.; Wang, C.; Lei, Y.; Li, A.; Yu, L.; Ye, J. Mitochondria-targeted antioxidant peptide SS-31 mediates neuroprotection in a rat experimental glaucoma model. Acta Biochim. et Biophys. Sin. 2019, 51, 411–421. [Google Scholar] [CrossRef]
- Lee, D.; Kim, K.-Y.; Shim, M.S.; Kim, S.Y.; Ellisman, M.H.; Weinreb, R.N.; Ju, W.-K. Coenzyme Q10 ameliorates oxidative stress and prevents mitochondrial alteration in ischemic retinal injury. Apoptosis 2014, 19, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.; Kim, K.-Y.; Noh, Y.H.; Chai, S.; Lindsey, J.D.; Ellisman, M.H.; Weinreb, R.N.; Ju, W.-K. Brimonidine Blocks Glutamate Excitotoxicity-Induced Oxidative Stress and Preserves Mitochondrial Transcription Factor A in Ischemic Retinal Injury. PLoS ONE 2012, 7, e47098. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A.; Abbas, S.N. Diabetes-Induced Mitochondrial Dysfunction in the Retina. Investig. Opthalmol. Vis. Sci. 2003, 44, 5327–5334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef]
- Gleichmann, M.; Mattson, M.P. Neuronal Calcium Homeostasis and Dysregulation. Antioxid. Redox Signal. 2011, 14, 1261–1273. [Google Scholar] [CrossRef] [Green Version]
- Mira, R.G.; Cerpa, W. Building a Bridge Between NMDAR-Mediated Excitotoxicity and Mitochondrial Dysfunction in Chronic and Acute Diseases. Cell. Mol. Neurobiol. 2021, 41, 1413–1430. [Google Scholar] [CrossRef]
- Winkler, B.S. Glycolytic and oxidative metabolism in relation to retinal function. J. Gen. Physiol. 1981, 77, 667–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashii, S.; Mandai, M.; Kikuchi, M.; Honda, Y.; Tamura, Y.; Kaneda, K.; Akaike, A. Dual actions of nitric oxide inN-methyl-d-aspartate receptor-mediated neurotoxicity in cultured retinal neurons. Brain Res. 1996, 711, 93–101. [Google Scholar] [CrossRef]
- Lei, S.Z.; Pan, Z.-H.; Aggarwal, S.K.; Chen, H.-S.V.; Hartman, J.; Sucher, N.; Lipton, S.A. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron 1992, 8, 1087–1099. [Google Scholar] [CrossRef]
- Schmetterer, L.; Findl, O.; Fasching, P.; Ferber, W.; Strenn, K.; Breiteneder, H.; Adam, H.; Eichler, H.-G.; Wolzt, M. Nitric Oxide and Ocular Blood Flow in Patients with IDDM. Diabetes 1997, 46, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Engerman, R.L.; Case, G.L.; Kern, T.S. Retinal glutamate in diabetes and effect of antioxidants. Neurochem. Int. 2001, 38, 385–390. [Google Scholar] [CrossRef]
- Neufeld, A.H.; Das, S.; Vora, S.; Gachie, E.; Kawai, S.-I.; Manning, P.T.; Connor, J.R. A Prodrug of a Selective Inhibitor of Inducible Nitric Oxide Synthase is Neuroprotective in the Rat Model of Glaucoma. J. Glaucoma 2002, 11, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Sattler, R.; Xiong, Z.; Lu, W.-Y.; Hafner, M.; MacDonald, J.F.; Tymianski, M. Specific Coupling of NMDA Receptor Activation to Nitric Oxide Neurotoxicity by PSD-95 Protein. Science 1999, 284, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.; Caprioli, J.; Koseki, Y. Nitric Oxide Mediates Excitotoxic and Anoxic Damage in Rat Retinal Ganglion Cells Cocultured with Astroglia. Arch. Ophthalmol. 1999, 117, 1524–1529. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Kim, J.H.; Kim, Y.H.; Park, C.K. Expression of neuronal nitric oxide synthase in the retina of a rat model of chronic glaucoma. Vis. Res. 2007, 47, 2732–2740. [Google Scholar] [CrossRef] [Green Version]
- Aarts, M.; Liu, Y.; Liu, L.; Besshoh, S.; Arundine, M.; Gurd, J.W.; Wang, Y.-T.; Salter, M.W.; Tymianski, M. Treatment of Ischemic Brain Damage by Perturbing NMDA Receptor- PSD-95 Protein Interactions. Science 2002, 298, 846–850. [Google Scholar] [CrossRef]
- Wittmann, M.; Bengtson, C.P.; Bading, H. Extrasynaptic NMDA receptors: Mediators of excitotoxic cell death The involvement of NMDA receptors in neuron death. In Pharmacology of Cerebral Ischemia; Medpharm Scientific Publishers: Stuttgart, Germany, 2004. [Google Scholar]
- Aizenman, E.; Loring, R.H.; Reynolds, I.J.; Rosenberg, P. The Redox Biology of Excitotoxic Processes: The NMDA Receptor, TOPA Quinone, and the Oxidative Liberation of Intracellular Zinc. Front. Neurosci. 2020, 14, 778. [Google Scholar] [CrossRef] [PubMed]
- Sucher, N.J.; Lipton, S.A. Redox modulatory site of the NMDA receptor-channel complex: Regulation by oxidized glutathione. J. Neurosci. Res. 1991, 30, 582–591. [Google Scholar] [CrossRef]
- Marshall, J.; Wong, K.; Rupasinghe, C.N.; Tiwari, R.; Zhao, X.; Berberoglu, E.D.; Sinkler, C.; Liu, J.; Lee, I.; Parang, K.; et al. Inhibition of N-Methyl-d-aspartate-induced Retinal Neuronal Death by Polyarginine Peptides Is Linked to the Attenuation of Stress-induced Hyperpolarization of the Inner Mitochondrial Membrane Potential. J. Biol. Chem. 2015, 290, 22030–22048. [Google Scholar] [CrossRef] [Green Version]
- Kwong, J.M.; Lam, T. N-Methyl-d-Aspartate (NMDA) Induced Apoptosis in Adult Rabbit Retinas. Exp. Eye Res. 2000, 71, 437–444. [Google Scholar] [CrossRef]
- Hoffmann, D.B.; Williams, S.K.; Jovana, B.; Müller, A.; Stadelmann, C.; Naidoo, V.; Bahr, B.A.; Diem, R.; Fairless, R. Calcium Influx and Calpain Activation Mediate Preclinical Retinal Neurodegeneration in Autoimmune Optic Neuritis. J. Neuropathol. Exp. Neurol. 2013, 72, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Fileta, J.; Rawe, I.; Qu, J.; Grosskreutz, C.L. Calpain Activation in Experimental Glaucoma. Investig. Opthalmol. Vis. Sci. 2010, 51, 3049–3054. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Dobberfuhl, A.; Filippopoulos, T.; Ingelsson, M.; Fileta, J.B.; Poulin, N.R.; Grosskreutz, C.L. Transcriptional Up-Regulation and Activation of Initiating Caspases in Experimental Glaucoma. Am. J. Pathol. 2005, 167, 673–681. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lopez, D.; Davey, P.G.; Cameron, D.J.; Nguyen, K.; Tran, J.; Marquez, E.; Liu, Y.; Bi, X.; Baudry, M. Calpain-1 and calpain-2 play opposite roles in retinal ganglion cell degeneration induced by retinal ischemia/reperfusion injury. Neurobiol. Dis. 2016, 93, 121–128. [Google Scholar] [CrossRef]
- Nakajima, E.; David, L.L.; Bystrom, C.; Shearer, T.R.; Azuma, M. Calpain-Specific Proteolysis in Primate Retina: Contribution of Calpains in Cell Death. Investig. Opthalmol. Vis. Sci. 2006, 47, 5469–5475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crish, S.; Calkins, D. Neurodegeneration in glaucoma: Progression and calcium-dependent intracellular mechanisms. Neurosci. 2011, 176, 1–11. [Google Scholar] [CrossRef] [Green Version]
- McKernan, D.P.; Guerin, M.B.; O’Brien, C.J.; Cotter, T.G. A Key Role for Calpains in Retinal Ganglion Cell Death. Investig. Opthalmol. Vis. Sci. 2007, 48, 5420–5430. [Google Scholar] [CrossRef]
- Azuma, M.; Shearer, T. The Role of Calcium-Activated Protease Calpain in Experimental Retinal Pathology. Surv. Ophthalmol. 2008, 53, 150–163. [Google Scholar] [CrossRef] [Green Version]
- Ryu, M.; Yasuda, M.; Shi, D.; Shanab, A.Y.; Watanabe, R.; Himori, N.; Omodaka, K.; Yokoyama, Y.; Takano, J.; Saido, T.; et al. Critical role of calpain in axonal damage-induced retinal ganglion cell death. J. Neurosci. Res. 2012, 90, 802–815. [Google Scholar] [CrossRef]
- Miao, Y.; Dong, L.-D.; Chen, J.; Hu, X.-C.; Yang, X.-L.; Wang, Z. Involvement of Calpain/p35-p25/Cdk5/NMDAR Signaling Pathway in Glutamate-Induced Neurotoxicity in Cultured Rat Retinal Neurons. PLoS ONE 2012, 7, e42318. [Google Scholar] [CrossRef]
- Coughlin, L.; Morrison, R.S.; Horner, P.J.; Inman, D.M. Mitochondrial Morphology Differences and Mitophagy Deficit in Murine Glaucomatous Optic Nerve. Investig. Opthalmol. Vis. Sci. 2015, 56, 1437–1446. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Bell, C.M.; Berlinicke, C.A.; Marsh-Armstrong, N.; Zack, D.J. Programmed switch in the mitochondrial degradation pathways during human retinal ganglion cell differentiation from stem cells is critical for RGC survival. Redox Biol. 2020, 34, 101465. [Google Scholar] [CrossRef] [PubMed]
- Rezaie, T.; Child, A.; Hitchings, R.; Brice, G.; Miller, L.; Coca-Prados, M.; Héon, E.; Krupin, T.; Ritch, R.; Kreutzer, D.; et al. Adult-Onset Primary Open-Angle Glaucoma Caused by Mutations in Optineurin. Science 2002, 295, 1077–1079. [Google Scholar] [CrossRef] [PubMed]
- Hombrebueno, J.R.; Cairns, L.; Dutton, L.R.; Lyons, T.J.; Brazil, D.P.; Moynagh, P.; Curtis, T.M.; Xu, H. Uncoupled turnover disrupts mitochondrial quality control in diabetic retinopathy. JCI Insight 2019, 4, e129760. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Hu, X.; Sun, X. Overexpression of parkin protects retinal ganglion cells in experimental glaucoma. Cell Death Dis. 2018, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Dai, Y.; Sun, X. Parkin overexpression protects retinal ganglion cells against glutamate excitotoxicity. Mol. Vis. 2017, 23, 447–456. [Google Scholar]
- Hu, X.; Zhuang, D.; Zhang, R.; Sun, X.; Lu, Q.; Dai, Y. The small molecule inhibitor PR-619 protects retinal ganglion cells against glutamate excitotoxicity. NeuroReport 2020, 31, 1134–1141. [Google Scholar] [CrossRef]
- Zaninello, M.; Palikaras, K.; Sotiriou, A.; Tavernarakis, N.; Scorrano, L. Sustained intracellular calcium rise mediates neuronal mitophagy in models of autosomal dominant optic atrophy. Cell Death Differ. 2021, 29, 167–177. [Google Scholar] [CrossRef]
- Chen, H.; Chan, D.C. Critical dependence of neurons on mitochondrial dynamics. Curr. Opin. Cell Biol. 2006, 18, 453–459. [Google Scholar] [CrossRef]
- Koppers, M.; Özkan, N.; Farías, G.G. Complex Interactions Between Membrane-Bound Organelles, Biomolecular Condensates and the Cytoskeleton. Front. Cell Dev. Biol. 2020, 8, 1661. [Google Scholar] [CrossRef]
- Majander, A.; João, C.; Rider, A.T.; Henning, G.B.; Votruba, M.; Moore, A.T.; Yu-Wai-Man, P.; Stockman, A. The Pattern of Retinal Ganglion Cell Loss in OPA1-Related Autosomal Dominant Optic Atrophy Inferred From Temporal, Spatial, and Chromatic Sensitivity Losses. Investig. Opthalmol. Vis. Sci. 2017, 58, 502–516. [Google Scholar] [CrossRef] [Green Version]
- González-Menéndez, I.; Reinhard, K.; Tolivia, J.; Wissinger, B.; Münch, T.A. Influence ofOpa1Mutation on Survival and Function of Retinal Ganglion Cells. Investig. Opthalmol. Vis. Sci. 2015, 56, 4835–4845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushnareva, E.Y.; Gerencser, A.A.; Bossy, B.; Ju, W.-K.; White, A.D.; Waggoner, J.; Ellisman, M.H.; Perkins, G.; Bossy-Wetzel, E. Loss of OPA1 disturbs cellular calcium homeostasis and sensitizes for excitotoxicity. Cell Death Differ. 2012, 20, 353–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.; Alavi, M.; Kim, K.-Y.; Kang, T.; Scott, R.T.; Noh, Y.H.; Lindsey, J.D.; Wissinger, B.; Ellisman, M.H.; Weinreb, R.N.; et al. A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis. 2011, 2, e240. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.-K.; Kim, K.-Y.; Angert, M.; Duong-Polk, K.X.; Lindsey, J.D.; Ellisman, M.H.; Weinreb, R.N. Memantine Blocks Mitochondrial OPA1 and CytochromecRelease and Subsequent Apoptotic Cell Death in Glaucomatous Retina. Investig. Opthalmol. Vis. Sci. 2009, 50, 707–716. [Google Scholar] [CrossRef]
- Jassim, A.H.; Inman, D.M.; Mitchell, C.H. Crosstalk Between Dysfunctional Mitochondria and Inflammation in Glaucomatous Neurodegeneration. Front. Pharmacol. 2021, 12, 699623. [Google Scholar] [CrossRef]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.-K. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell. Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef]
- Görlach, A.; Klappa, P.; Kietzmann, T. The Endoplasmic Reticulum: Folding, Calcium Homeostasis, Signaling, and Redox Control. Antioxid. Redox Signal. 2006, 8, 1391–1418. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Paillusson, S.; Stoica, R.; Gomez-Suaga, P.; Lau, D.; Mueller, S.; Miller, T.; Miller, C.C. There’s Something Wrong with my MAM; the ER–Mitochondria Axis and Neurodegenerative Diseases. Trends Neurosci. 2016, 39, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Öztürk, Z.; O’Kane, C.J.; Pérez-Moreno, J.J. Axonal Endoplasmic Reticulum Dynamics and Its Roles in Neurodegeneration. Front. Neurosci. 2020, 14, 48. [Google Scholar] [CrossRef]
- Horak, M.; Petralia, R.S.; Kaniakova, M.; Sans, N. ER to synapse trafficking of NMDA receptors. Front. Cell. Neurosci. 2014, 8, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucharz, K.; Krogh, M.; Na Ng, A.; Toresson, H. NMDA Receptor Stimulation Induces Reversible Fission of the Neuronal Endoplasmic Reticulum. PLoS ONE 2009, 4, e5250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Na Ng, A.; Doherty, A.J.; Lombroso, P.J.; Emptage, N.J.; Collingridge, G.L. Rapid regulation of endoplasmic reticulum dynamics in dendritic spines by NMDA receptor activation. Mol. Brain 2014, 7, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doh, S.H.; Kim, J.H.; Lee, K.M.; Park, H.Y.; Park, C.K. Retinal ganglion cell death induced by endoplasmic reticulum stress in a chronic glaucoma model. Brain Res. 2010, 1308, 158–166. [Google Scholar] [CrossRef]
- Hata, N.; Oshitari, T.; Yokoyama, A.; Mitamura, Y.; Yamamoto, S. Increased expression of IRE1α and stress-related signal transduction proteins in ischemia-reperfusion injured retina. Clin. Ophthalmol. 2008, 2, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wang, J.J.; Yu, Q.; Wang, M.; Zhang, S.X. Endoplasmic reticulum stress is implicated in retinal inflammation and diabetic retinopathy. FEBS Lett. 2009, 583, 1521–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indrieri, A.; Pizzarelli, R.; Franco, B.; De Leonibus, E. Dopamine, Alpha-Synuclein, and Mitochondrial Dysfunctions in Parkinsonian Eyes. Front. Neurosci. 2020, 14, 567129. [Google Scholar] [CrossRef]
- Veys, L.; Vandenabeele, M.; Ortuño-Lizarán, I.; Baekelandt, V.; Cuenca, N.; Moons, L.; De Groef, L. Retinal α-synuclein deposits in Parkinson’s disease patients and animal models. Acta Neuropathol. 2019, 137, 379–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancino, R.; Martucci, A.; Cesareo, M.; Giannini, C.; Corasaniti, M.T.; Bagetta, G.; Nucci, C. Glaucoma and Alzheimer Disease: One Age-Related Neurodegenerative Disease of the Brain. Curr. Neuropharmacol. 2018, 16, 971–977. [Google Scholar] [CrossRef]
- Chiasseu, M.; Alarcon-Martinez, L.; Belforte, N.; Quintero, H.; Dotigny, F.; Destroismaisons, L.; Velde, C.V.; Panayi, F.; Louis, C.; Di Polo, A. Tau accumulation in the retina promotes early neuronal dysfunction and precedes brain pathology in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 58. [Google Scholar] [CrossRef]
- Leger, F.; Fernagut, P.-O.; Canron, M.-H.; Léoni, S.; Vital, C.; Tison, F.; Bezard, E.; Vital, A. Protein Aggregation in the Aging Retina. J. Neuropathol. Exp. Neurol. 2011, 70, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.X.; Sanders, E.; Fliesler, S.J.; Wang, J.J. Endoplasmic reticulum stress and the unfolded protein responses in retinal degeneration. Exp. Eye Res. 2014, 125, 30–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroeger, H.; Chiang, W.-C.; Felden, J.; Nguyen, A.; Lin, J.H. ER stress and unfolded protein response in ocular health and disease. FEBS J. 2019, 286, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi, Y.; Nakajima, Y.; Shimazawa, M.; Kurita, T.; Kubo, M.; Saito, A.; Sajiki, H.; Kudo, T.; Aihara, M.; Imaizumi, K.; et al. Effect of an Inducer of BiP, a Molecular Chaperone, on Endoplasmic Reticulum (ER) Stress-Induced Retinal Cell Death. Investig. Opthalmol. Vis. Sci. 2009, 50, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Boriushkin, E.; Wang, J.J.; Li, J.; Jing, G.; Seigel, G.M.; Zhang, S.X. Identification of p58IPK as a Novel Neuroprotective Factor for Retinal Neurons. Investig. Opthalmol. Vis. Sci. 2015, 56, 1374–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaughlin, T.; Dhimal, N.; Li, J.; Wang, J.J.; Zhang, S.X. p58IPK Is an Endogenous Neuroprotectant for Retinal Ganglion Cells. Front. Aging Neurosci. 2018, 10, 267. [Google Scholar] [CrossRef] [Green Version]
- Shimazawa, M.; Inokuchi, Y.; Ito, Y.; Murata, H.; Aihara, M.; Miura, M.; Araie, M.; Hara, H. Involvement of ER stress in retinal cell death. Mol. Vis. 2007, 13, 578–587. [Google Scholar]
- Uchibayashi, R.; Tsuruma, K.; Inokuchi, Y.; Shimazawa, M.; Hara, H. Involvement of Bid and caspase-2 in endoplasmic reticulum stress- and oxidative stress-induced retinal ganglion cell death. J. Neurosci. Res. 2011, 89, 1783–1794. [Google Scholar] [CrossRef]
- Awai, M.; Koga, T.; Inomata, Y.; Oyadomari, S.; Gotoh, T.; Mori, M.; Tanihara, H. NMDA-induced retinal injury is mediated by an endoplasmic reticulum stress-related protein, CHOP/GADD. J. Neurochem. 2006, 96, 43–52. [Google Scholar] [CrossRef]
- Kashani, A.H.; Asanad, S.; Chan, J.W.; Singer, M.B.; Zhang, J.; Sharifi, M.; Khansari, M.M.; Abdolahi, F.; Shi, Y.; Biffi, A.; et al. Past, present and future role of retinal imaging in neurodegenerative disease. Prog. Retin. Eye Res. 2021, 83, 100938. [Google Scholar] [CrossRef]
- Colligris, P.; De Lara, M.J.P.; Colligris, B.; Pintor, J. Ocular Manifestations of Alzheimer’s and Other Neurodegenerative Diseases: The Prospect of the Eye as a Tool for the Early Diagnosis of Alzheimer’s Disease. J. Ophthalmol. 2018, 2018, 8538573. [Google Scholar] [CrossRef] [PubMed]
- Askou, A.L.; Jakobsen, T.S.; Corydon, T.J. Retinal gene therapy: An eye-opener of the 21st century. Gene Ther. 2021, 28, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Thakur, S.; Sharma, P.K.; Malviya, R. A Review: Recent Strategies Involved in Brain Targeting Through Ocular Route-Patents and Application. Ann. Pharmacol. Pharm. 2017, 2, 1043. [Google Scholar]
- Opere, C.A.; Heruye, S.H.; Njie-Mbye, Y.-F.; Ohia, S.E.; Sharif, N.A. Regulation of Excitatory Amino Acid Transmission in the Retina: Studies on Neuroprotection. J. Ocul. Pharmacol. Ther. 2018, 34, 107–118. [Google Scholar] [CrossRef]
- Hare, W.; WoldeMussie, E.; Lai, R.; Ton, H.; Ruiz, G.; Feldmann, B.; Wijono, M.; Chun, T.; Wheeler, L. Efficacy and Safety of Memantine, an NMDA-Type Open-Channel Blocker, for Reduction of Retinal Injury Associated with Experimental Glaucoma in Rat and Monkey. Surv. Ophthalmol. 2001, 45, S284–S289. [Google Scholar] [CrossRef]
- Sühs, K.-W.; Fairless, R.; Williams, S.K.; Heine, K.; Cavalié, A.; Diem, R. N-Methyl-d-Aspartate Receptor Blockade Is Neuroprotective in Experimental Autoimmune Optic Neuritis. J. Neuropathol. Exp. Neurol. 2014, 73, 507–518. [Google Scholar] [CrossRef] [Green Version]
- Celiker, H.; Yuksel, N.; Solakoglu, S.; Karabas, L.; Aktar, F.; Caglar, Y. Neuroprotective Effects of Memantine in the Retina of Glaucomatous Rats: An Electron Microscopic Study. J. Ophthalmic Vis. Res. 2016, 11, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Fuwa, M.; Kageyama, M.; Ohashi, K.; Sasaoka, M.; Sato, R.; Tanaka, M.; Tashiro, K. Nafamostat and sepimostat identified as novel neuroprotective agents via NR2B N-methyl-d-aspartate receptor antagonism using a rat retinal excitotoxicity model. Sci. Rep. 2019, 9, 20409–20412. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, R.; Chen, J.; Wu, J. Neuroprotective effects of DAAO are mediated via the ERK1/2 signaling pathway in a glaucomatous animal model. Exp. Eye Res. 2020, 190, 107892. [Google Scholar] [CrossRef]
- Watanabe, K.; Asano, D.; Ushikubo, H.; Morita, A.; Mori, A.; Sakamoto, K.; Ishii, K.; Nakahara, T. Metformin Protects against NMDA-Induced Retinal Injury through the MEK/ERK Signaling Pathway in Rats. Int. J. Mol. Sci. 2021, 22, 4439. [Google Scholar] [CrossRef]
- Iezhitsa, I.; Lambuk, L.; Agarwal, R.; Agarwal, P.; Peresypkina, A.; Pobeda, A.; Ismail, N.M. Magnesium acetyltaurate prevents retinal damage and visual impairment in rats through suppression of NMDA-induced upregulation of NF-κB, p53 and AP-1 (c-Jun/c-Fos). Neural Regen. Res. 2021, 16, 2330–2344. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Sase, K.; Tsukahara, C.; Arizono, I.; Takagi, H.; Kitaoka, Y. Pemafibrate prevents retinal neuronal cell death in NMDA-induced excitotoxicity via inhibition of p-c-Jun expression. Mol. Biol. Rep. 2021, 48, 195–202. [Google Scholar] [CrossRef]
- Qi, X.; Lewin, A.S.; Sun, L.; Hauswirth, W.W.; Guy, J. Suppression of Mitochondrial Oxidative Stress Provides Long-term Neuroprotection in Experimental Optic Neuritis. Investig. Opthalmol. Vis. Sci. 2007, 48, 681–691. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Li, S.; Miao, L.; Huang, H.; Liang, F.; Teng, X.; Xu, L.; Wang, Q.; Xiao, W.; Ridder, W.H.; et al. Rescue of Glaucomatous Neurodegeneration by Differentially Modulating Neuronal Endoplasmic Reticulum Stress Molecules. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 5891–5903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, H.; Mori, M.; Harashima, M.; Hashizume, T.; Furiya, M.; Mukaigaito, C.; Takemura, E.; Yamada, M.; Mise, K.; Yuan, B.; et al. Apolipoprotein E-Containing Lipoproteins and LRP1 Protect From NMDA-Induced Excitotoxicity Associated with Reducing α2-Macroglobulin in Müller Glia. Investig. Opthalmol. Vis. Sci. 2021, 62, 23. [Google Scholar] [CrossRef] [PubMed]
- Boesl, F.; Drexler, K.; Müller, B.; Seitz, R.; Weber, G.R.; Priglinger, S.G.; Fuchshofer, R.; Tamm, E.R.; Ohlmann, A. Endogenous Wnt/β-catenin signaling in Müller cells protects retinal ganglion cells from excitotoxic damage. Mol. Vis 2020, 26, 135–149. [Google Scholar]
- Kurose, T.; Sugano, E.; Sugai, A.; Shiraiwa, R.; Kato, M.; Mitsuguchi, Y.; Takai, Y.; Tabata, K.; Honma, Y.; Tomita, H. Neuroprotective effect of a dietary supplement against glutamate-induced excitotoxicity in retina. Int. J. Ophthalmol. 2019, 12, 1231–1237. [Google Scholar] [CrossRef]
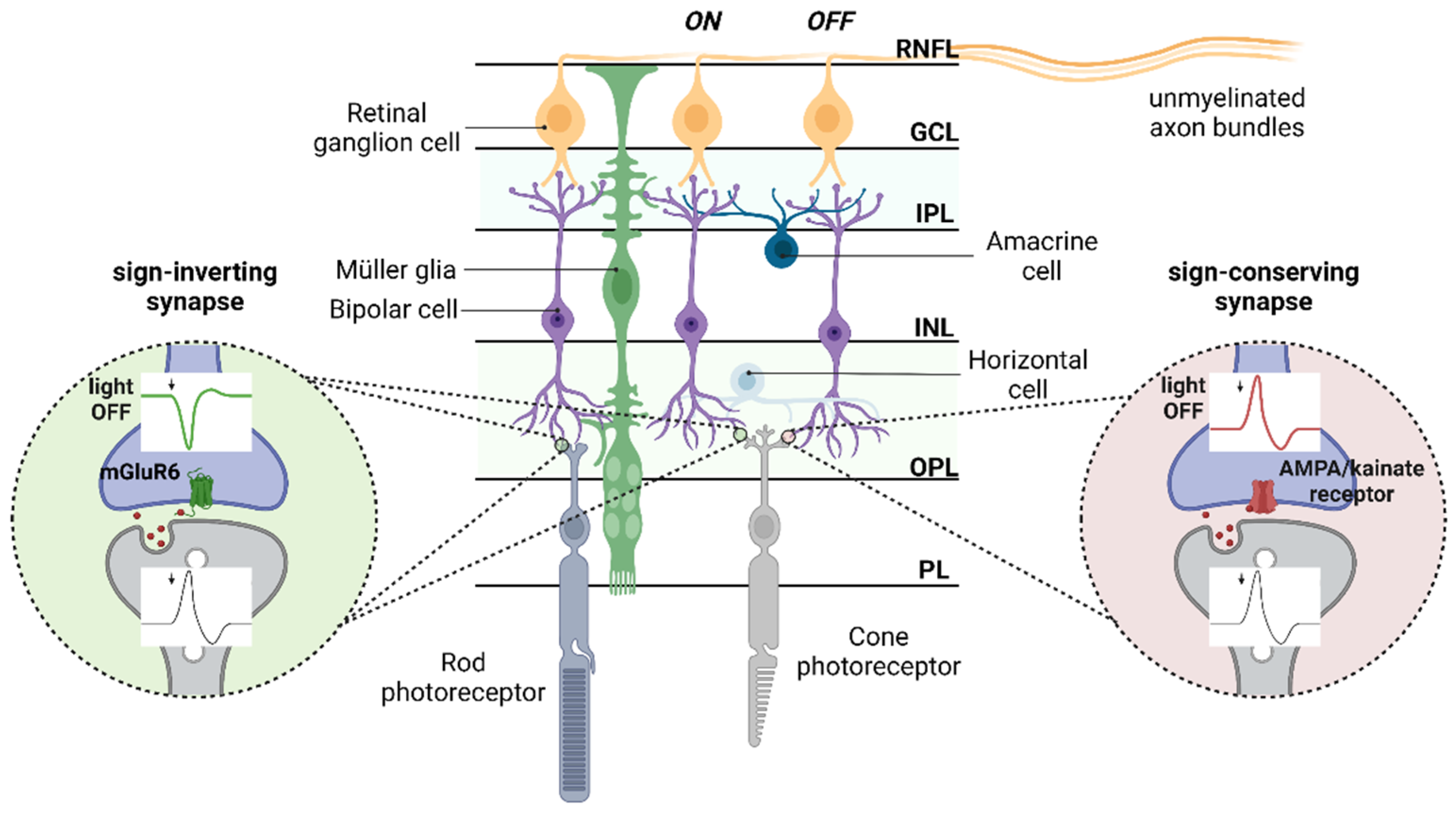
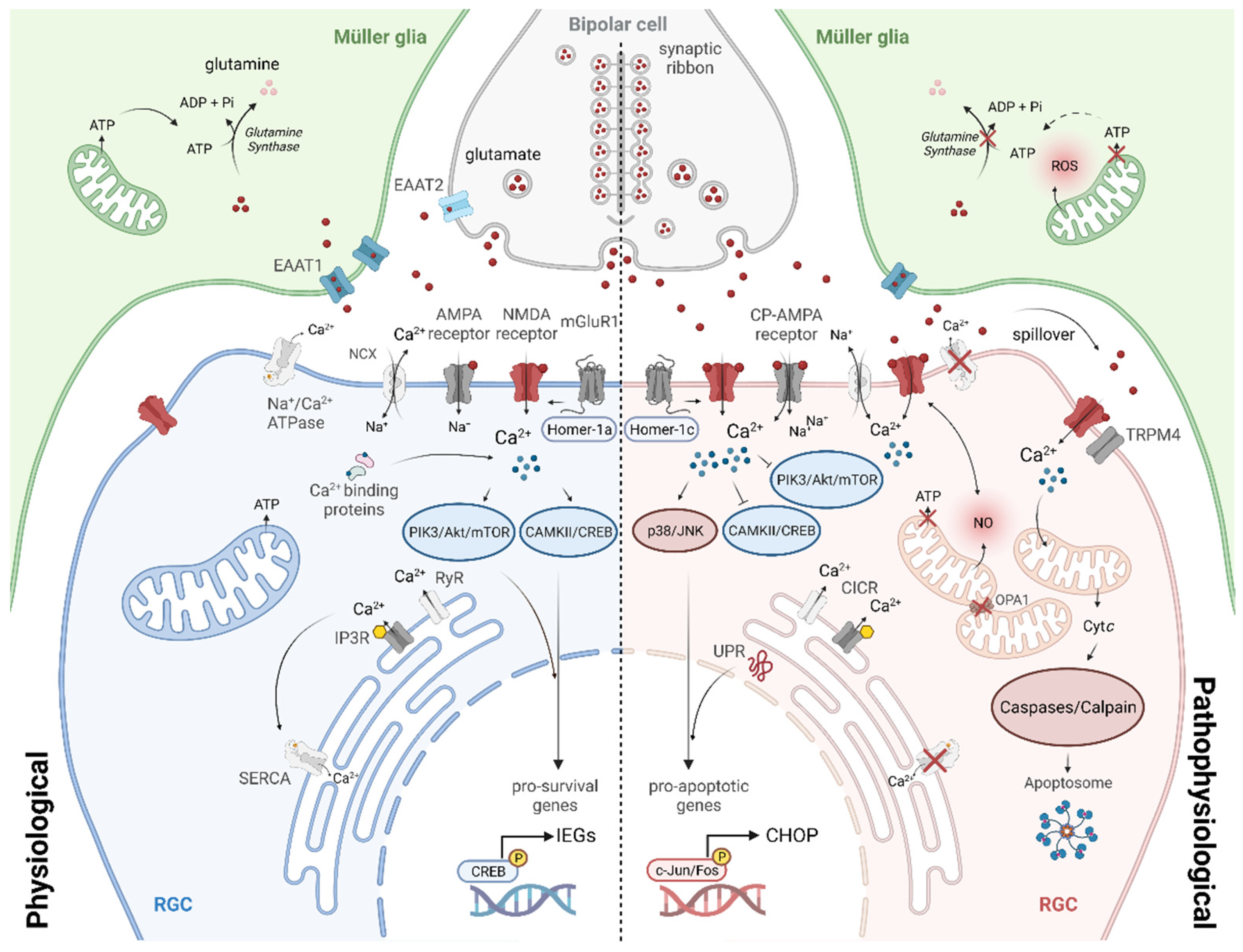

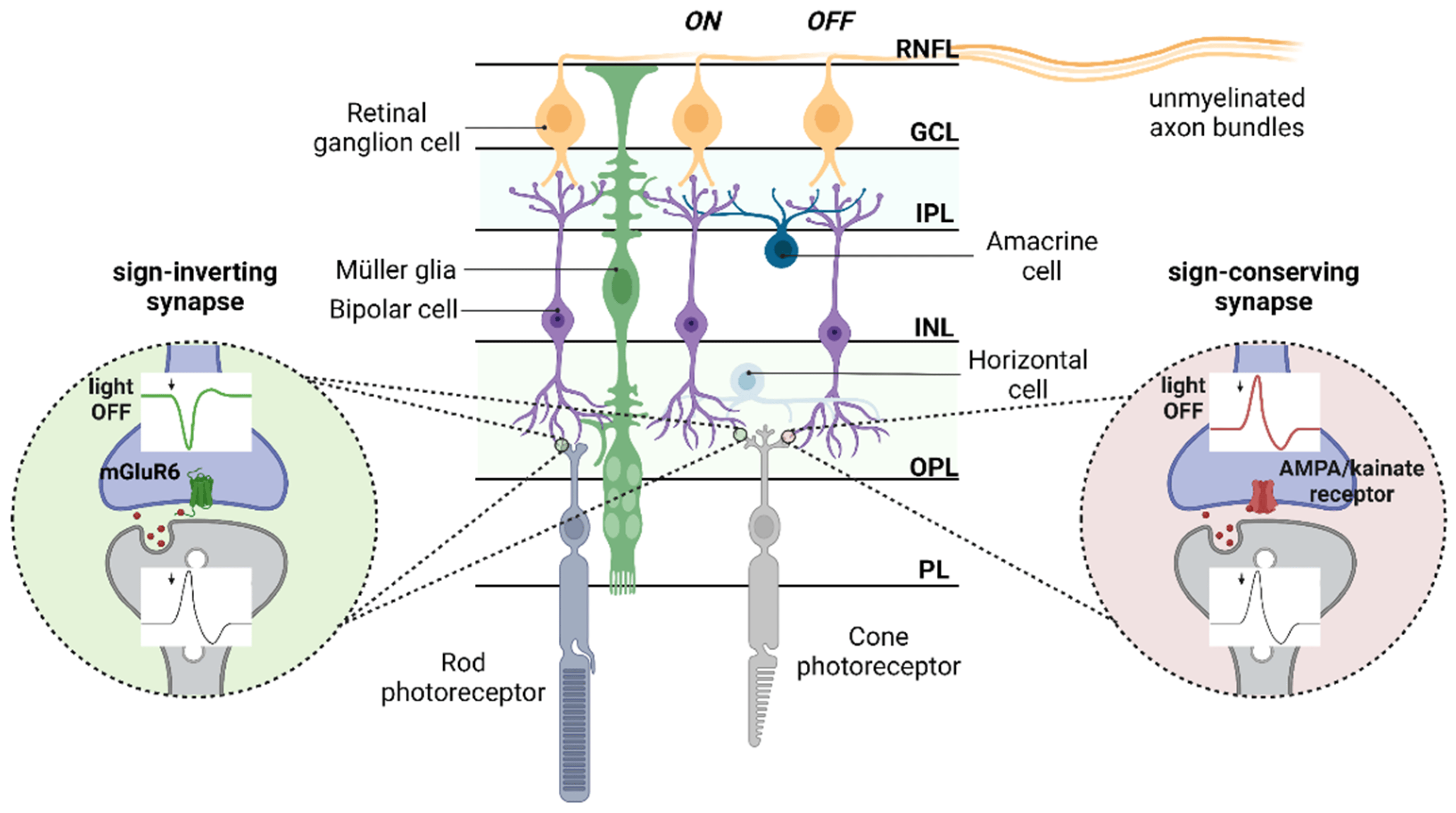{kind=link}
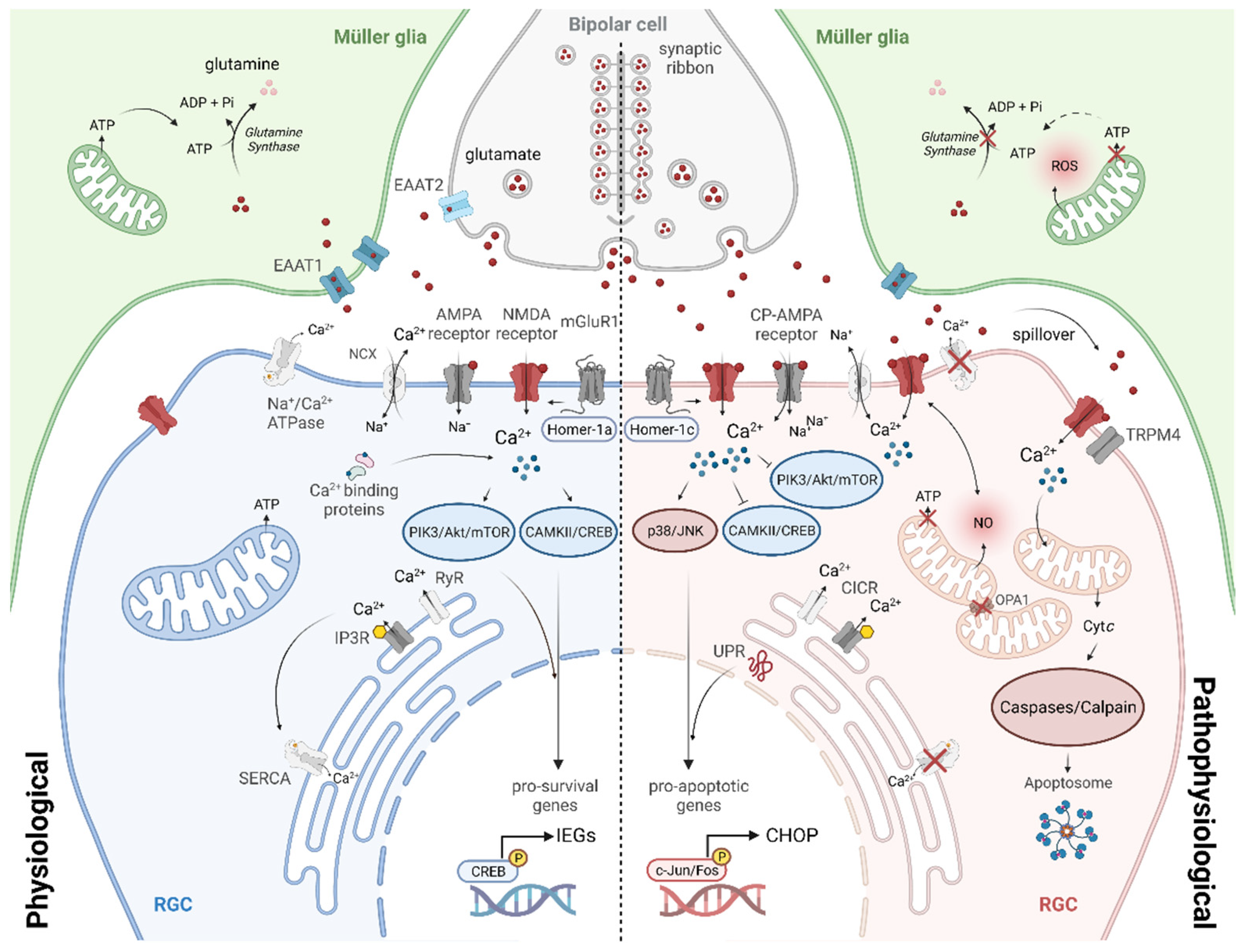{kind=link}
| Ionotropic Glutamate Receptors | Metabotropic Glutamate Receptors | Glutamate Transporters | |||
|---|---|---|---|---|---|
| AMPA Receptors (GluA1-2) | Kainate Receptors (GluK1-5) | NMDA Receptors (GluN1, 2A-D, 3A-B) | Group I-III Metabotropic Receptors (mGluR1-8) | EAAT1-5 xCT | |
| Photoreceptors | - | GluK5 only presynaptic [28] | GluN1, 2B only presynaptic [29,30] | mGluR8 [31] | EAAT2 [24,32] EAAT5 [33] |
| Horizontal cells | GluA2-4 [26] | - | GluN1 [29,34]/ additional subunits unknown | - | EAAT3 [32,35] |
| Bipolar cells | GluA1-4 [22] | GluK1 (OFF-BCs) [36] GluK1-3 [22,37] | GluN1,2C-D [23,37] GluN2D only presynaptic in rod BCs [29,38] | mGluR6 (ON-BCs) [17] mGluR8 [39,40] mGluR7 presynaptic [40,41] | EAAT2 [24,32] EAAT3 [32,35] EAAT5 [33] |
| Amacrine cells | GluA1-4 [22,26] | GluK2-5 [22,26,37] GluD1-2 [42] | GluN1,2A-C [29,37] | mGluR1 [22] mGluR2 [43] mGluR5 [44] mGluR7 postsynaptic [40,41] | EAAT2 [24,32] EAAT3 [32,35] |
| Retinal ganglion cells | GluA1-4 [22,26] | GluK2-5 [22,26,37] | GluN1,2-C [23,26,29,37] GluN3A [45] | mGluR1 [22] | EAAT3 [32,35] |
| Müllerglia | AMPA receptor (in vitro) [46] * GluA4 [22] | - | NMDA receptor (in vitro) [47,48] * | - | EAAT1 [24,32,48] EAAT4 [49] xCT [50] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boccuni, I.; Fairless, R. Retinal Glutamate Neurotransmission: From Physiology to Pathophysiological Mechanisms of Retinal Ganglion Cell Degeneration. Life 2022, 12, 638. https://doi.org/10.3390/life12050638
Boccuni I, Fairless R. Retinal Glutamate Neurotransmission: From Physiology to Pathophysiological Mechanisms of Retinal Ganglion Cell Degeneration. Life. 2022; 12(5):638. https://doi.org/10.3390/life12050638
Chicago/Turabian StyleBoccuni, Isabella, and Richard Fairless. 2022. "Retinal Glutamate Neurotransmission: From Physiology to Pathophysiological Mechanisms of Retinal Ganglion Cell Degeneration" Life 12, no. 5: 638. https://doi.org/10.3390/life12050638
APA StyleBoccuni, I., & Fairless, R. (2022). Retinal Glutamate Neurotransmission: From Physiology to Pathophysiological Mechanisms of Retinal Ganglion Cell Degeneration. Life, 12(5), 638. https://doi.org/10.3390/life12050638