Imaging Review of the Lung Parenchymal Complications in Patients with IPF

 ,
,
Abstract
:1. Introduction
2. Imaging
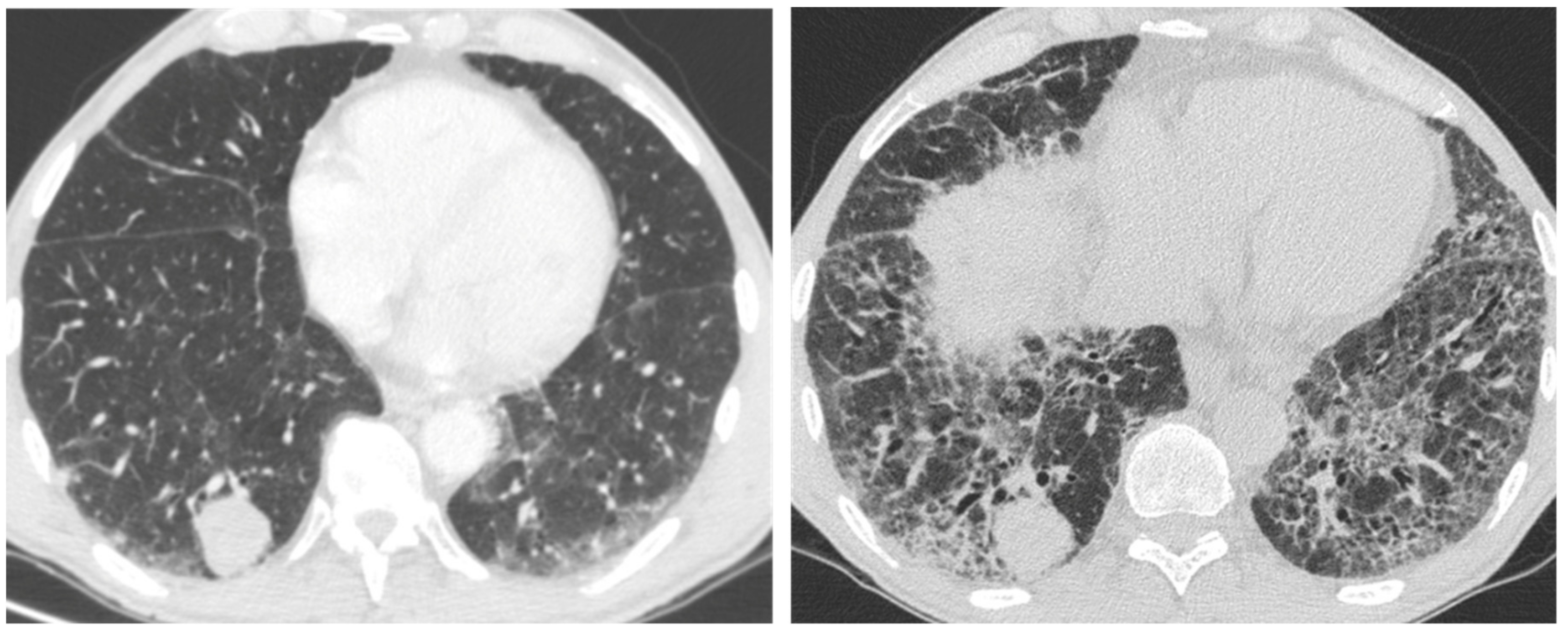
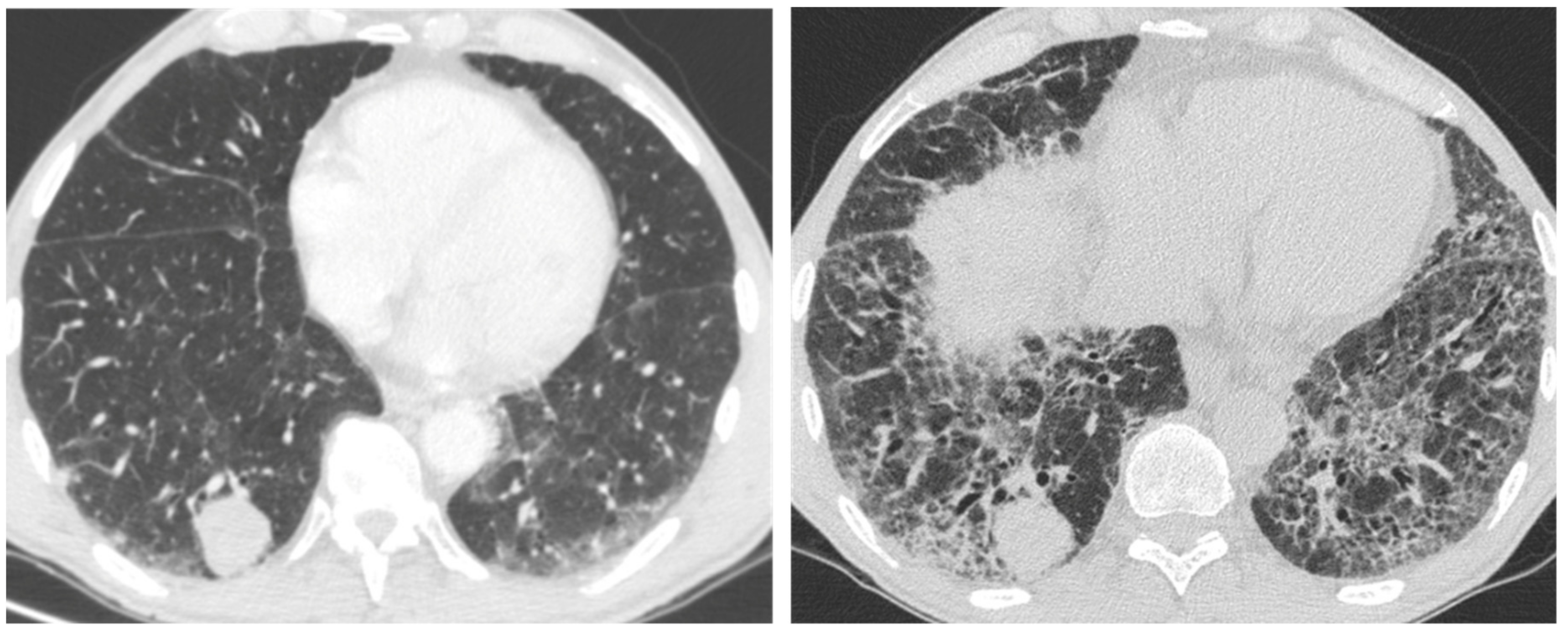
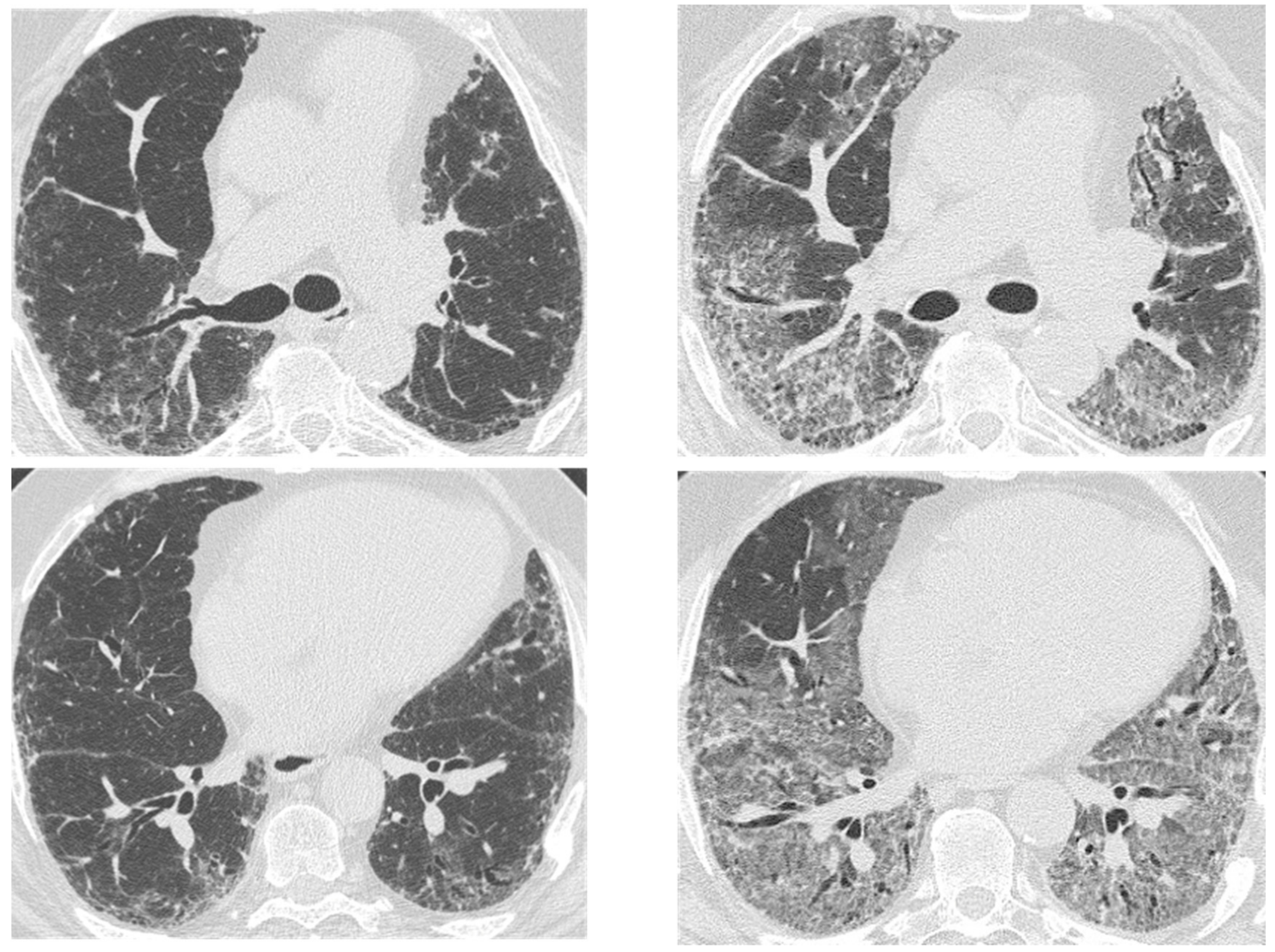
2.1. Infection
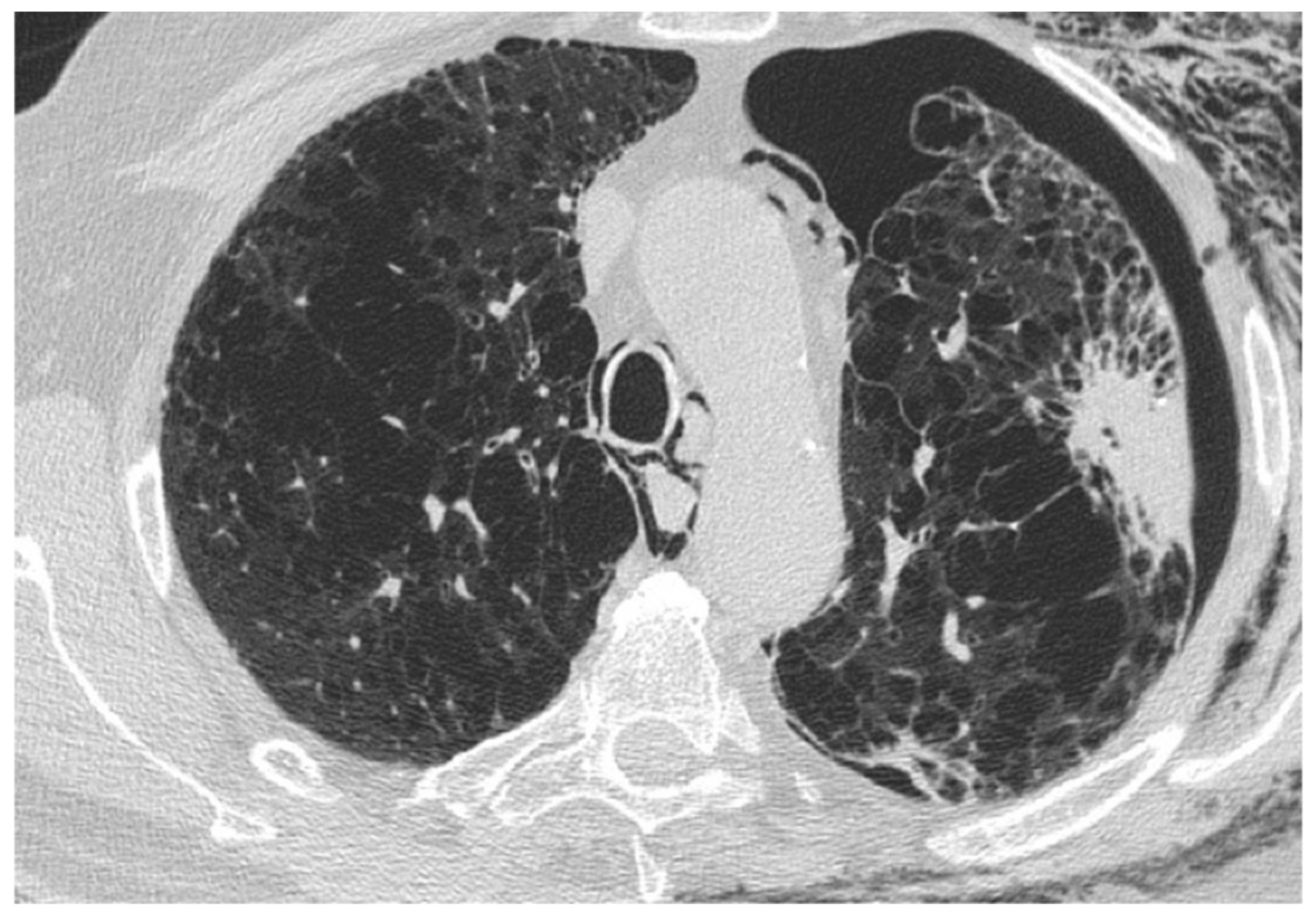
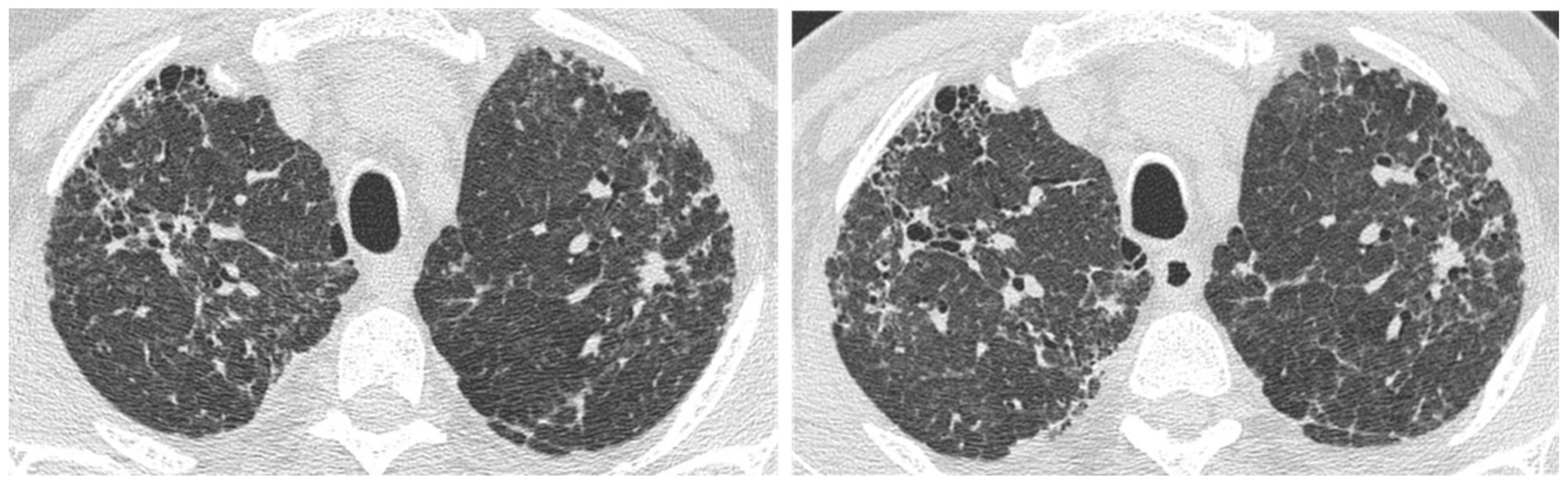
2.2. Lung Cancer
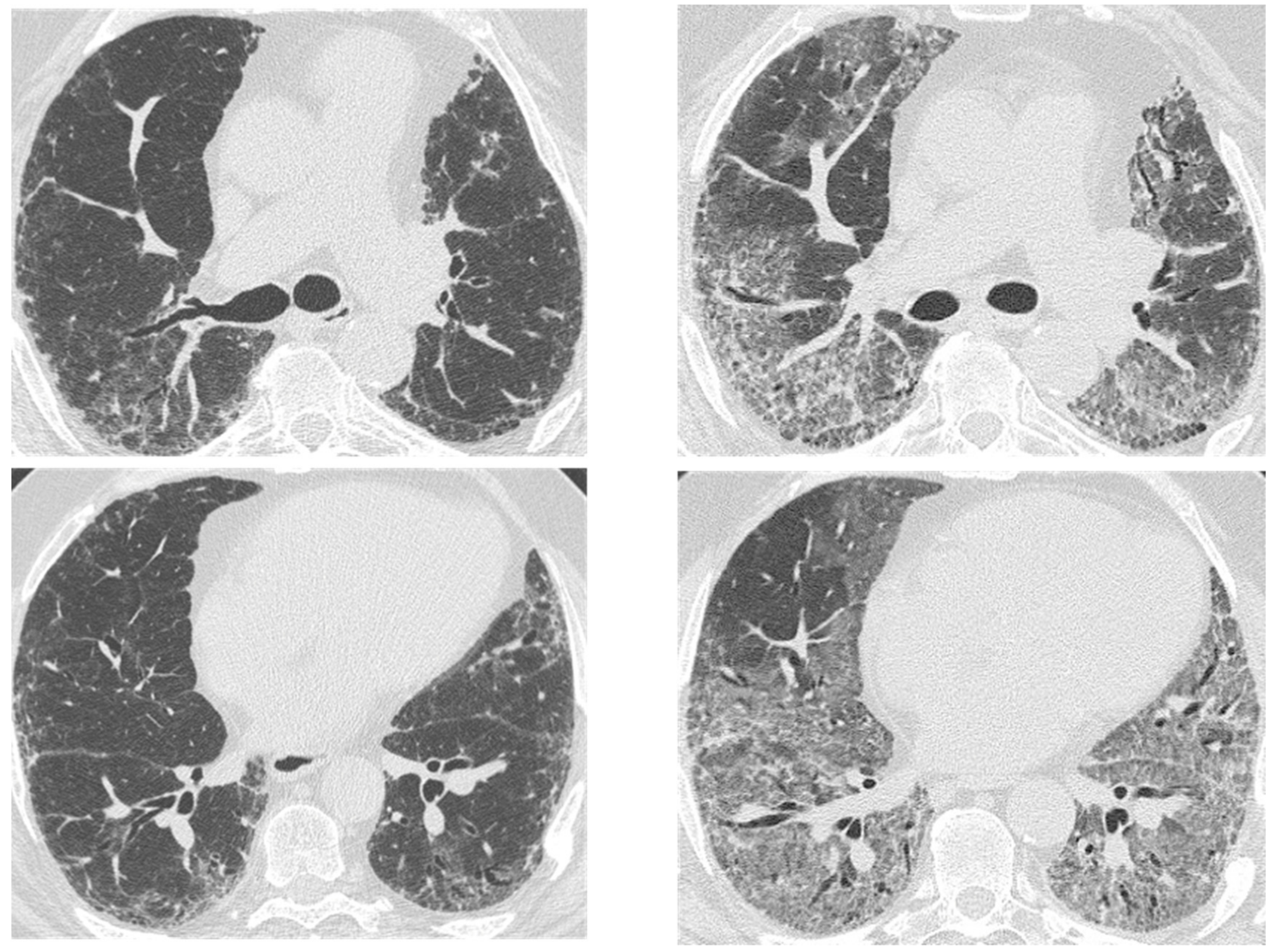
2.3. Acute Exacerbation
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Suzuki, A.; Kondoh, Y. The clinical impact of major comorbidities on idiopathic pulmonary fibrosis. Respir. Investig. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, C.R.; Walsh, S.L.F.; Hansell, D.M. High-resolution CT of complications of idiopathic fibrotic lung disease. Br. J. Radiol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Cano-Jiménez, E.; Hernández González, F.; Peloche, G. Comorbidities and Complications in Idiopathic Pulmonary Fibrosis. Med. Sci. 2018, 71. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of idiopathic pulmonary fibrosis An Official ATS/ERS/JRS/ALAT Clinical practice guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.A.; Sverzellati, N.; Travis, W.D.; Brown, K.K.; Colby, T.V.; Galvin, J.R.; Goldin, J.G.; Hansell, D.M.; Inoue, Y.; Johkoh, T.; et al. Diagnostic criteria for idiopathic pulmonary fibrosis: A Fleischner Society White Paper. Lancet Respir. Med. 2018. [Google Scholar] [CrossRef]
- Kusmirek, J.E.; Martin, M.D.; Kanne, J.P. Imaging of Idiopathic Pulmonary Fibrosis. Radiol. Clin. North Am. 2016. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Ward, A.J.; Lanes, S.; Cortney Hayflinger, D.; Rosenberg, D.M.; Hunsche, E. Burden of illness in idiopathic pulmonary fibrosis. J. Med. Econ. 2012. [Google Scholar] [CrossRef]
- Tsukamoto, K.; Hayakawa, H.; Sato, A.; Chida, K.; Nakamura, H.; Miura, K. Involvement of Epstein-Barr virus latent membrane protein 1 in disease progression in patients with idiopathic pulmonary fibrosis. Thorax 2000. [Google Scholar] [CrossRef]
- Tang, Y.W.; Johnson, J.E.; Browning, P.J.; Cruz-Gervis, R.A.; Davis, A.; Graham, B.S.; Brigham, K.L.; Oates Jr, J.A.; Loyd, J.E.; Stecenko, A.A. Herpesvirus DNA is consistently detected in lungs of patients with idiopathic pulmonary fibrosis. J. Clin. Microbiol. 2003. [Google Scholar] [CrossRef]
- Lawson, W.E.; Crossno, P.F.; Polosukhin, V.V.; Roldan, J.; Cheng, D.S.; Lane, K.B.; Blackwell, T.R.; Xu, C.; Markin, C.; Ware, L.B.; et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: Association with altered surfactant protein processing and herpesvirus infection. Am. J. Physiol. Lung Cell Mol. Physiol. 2008. [Google Scholar] [CrossRef]
- Richter, A.G.; Stockley, R.A.; Harper, L.; Thickett, D.R. Pulmonary infection in Wegener granulomatosis and idiopathic pulmonary fibrosis. Thorax 2009. [Google Scholar] [CrossRef] [PubMed]
- Molyneaux, P.L.; Cox, M.J.; Willis-Owen, S.A.; Mallia, P.; Russell, K.E.; Russell, A.M.; Murphy, E.; Johnston, S.L.; Schwartz, D.A.; Wells, A.U.; et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am. J. Respir. Crit Care Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Han, M.K.; Zhou, Y.; Murray, S.; Tayob, N.; Noth, I.; Lama, V.N.; Moore, B.B.; White, E.S.; Flaherty, K.R.; Huffnagle, G.B.; et al. Lung microbiome and disease progression in idiopathic pulmonary fibrosis: An analysis of the COMET study. Lancet Respir. Med. 2014. [Google Scholar] [CrossRef]
- Park, S.W.; Song, J.W.; Shim, T.S.; Park, M.S.; Lee, H.L.; Uh, S.T.; Park, C.S.; Kim, D.S. Mycobacterial pulmonary infections in patients with idiopathic pulmonary fibrosis. J. Korean Med. Sci. 2012. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.J.; Goo, J.M.; Im, J.G. Pulmonary tuberculosis in patients with idiopathic pulmonary fibrosis. Eur. J. Radiol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Shachor, Y.; Schindler, D.; Siegal, A.; Lieberman, D.; Mikulski, Y.; Bruderman, I. Increased incidence of pulmonary tuberculosis in chronic interstitial lung disease. Thorax 1989, 44, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Saraceno, J.L.; Phelps, D.T.; Ferro, T.J.; Futerfas, R.; Schwartz, D.B. Chronic necrotizing pulmonary aspergillosis: Approach to management. Chest 1997. [Google Scholar] [CrossRef]
- Roberts, C.M.; Citron, K.M.; Strickland, B. Intrathoracic aspergilloma: Role of CT in diagnosis and treatment. Radiology 1987. [Google Scholar] [CrossRef]
- Yamazaki, R.; Nishiyama, O.; Sano, H.; Iwanaga, T.; Higashimoto, Y.; Kume, H.; Tohda, Y. Clinical features and outcomes of IPF patients hospitalized for pulmonary infection: A Japanese cohort study. PLoS ONE 2016. [Google Scholar] [CrossRef]
- Ozawa, Y.; Suda, T.; Naito, T.; Enomoto, N.; Hashimoto, D.; Fujisawa, T.; Nakamura, Y.; Inui, N.; Nakamura, H.; Chida, K. Cumulative incidence of and predictive factors for lung cancer in IPF. Respirology 2009. [Google Scholar] [CrossRef]
- Yoon, J.H.; Nouraie, M.; Chen, X.; Zou, R.H.; Sellares, J.; Veraldi, K.L.; Chiarchiaro, J.; Lindell, K.; Wilson, D.O.; Kaminski, N.; et al. Characteristics of lung cancer among patients with idiopathic pulmonary fibrosis and interstitial lung disease—Analysis of institutional and population data. Respir. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- JafariNezhad, A.; YektaKooshali, M.H. Lung cancer in idiopathic pulmonary fibrosis: A systematic review and meta-analysis. PLoS ONE 2018. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.; Venn, A.; Lewis, S.; Britton, J. Lung cancer and cryptogenic fibrosing alveolitis: A population-based cohort study. Am. J. Respir Crit. Care Med. 2000. [Google Scholar] [CrossRef] [PubMed]
- Aubry, M.C.; Myers, J.L.; Douglas, W.W.; Tazelaar, H.D.; Stephens, T.L.W.; Hartman, T.E.; Deschamps, C.; Pankratz, V.S. Primary pulmonary carcinoma in patients with idiopathic pulmonary fibrosis. Mayo Clin. Proc. 2002. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Nyberg, F.; Morgan, G. The epidemiology of interstitial lung disease and its association with lung cancer. Br. J. Cancer 2004. [Google Scholar] [CrossRef]
- Matsushita, H.; Tanaka, S.; Saiki, Y.; Hara, M.; Nakata, K.; Tanimura, S.; Banba, J. Iung cancer associated with usual interstitial pneumonia. Pathol. Int. 1995. [Google Scholar] [CrossRef]
- Hironaka, M.; Fukayama, M. Pulmonary fibrosis and lung carcinoma: A comparative study of metaplastic epithelia in honeycombed areas of usual interstitial pneumonia with or without lung carcinoma. Pathol. Int. 1999. [Google Scholar] [CrossRef]
- Kreuter, M.; Ehlers-Tenenbaum, S.; Schaaf, M.; Oltmanns, U.; Palmowski, K.; Hoffmann, H.; Schnabel, P.A.; Heußel, C.P.; Puderbach, M.; Herth, F.J.; et al. Treatment and outcome of lung cancer in idiopathic interstitial pneumonias. Sarcoidosis Vasc. Diffuse Lung Dis. 2014, 31, 266–274. [Google Scholar]
- Tomassetti, S.; Gurioli, C.; Ryu, J.H.; Decker, P.A.; Ravaglia, C.; Tantalocco, P.; Buccioli, M.; Piciucchi, S.; Sverzellati, N.; Dubini, A.; et al. The impact of lung cancer on survival of idiopathic pulmonary fibrosis. Chest 2015. [Google Scholar] [CrossRef]
- Kato, E.; Takayanagi, N.; Takaku, Y.; Kagiyama, N.; Kanauchi, T.; Ishiguro, T.; Sugita, Y. Incidence and predictive factors of lung cancer in patients with idiopathic pulmonary fibrosis. ERS Monogr. 2018. [Google Scholar] [CrossRef]
- Lee, H.J.; Im, J.-G.; Ahn, J.M.; Yeon, K.M. Lung Cancer in Patients with Idiopathic Pulmonary Fibrosis: CT Findings. J. Comput. Assist. Tomogr. 1996, 20, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Sakai, S.; Ono, M.; Nishio, T.; Kawarada, Y.; Nagashima, A.; Toyoshima, S. Lung Cancer Associated with Diffuse Pulmonary Fibrosis: CT-Pathologic Correlation. J. Thorac. Imagin. 2003. [Google Scholar] [CrossRef]
- Jang, H.J.; Lee, K.S.; Kwon, O.J.; Rhee, C.H.; Shim, Y.M.; Han, J. Bronchioloalveolar carcinoma: Focal area of ground-glass attenuation at thin-section CT as an early sign. Radiology 1996. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; Kim, M.Y.; Kim, J.E.; Kim, S.S.; Park, T.S.; Kim, D.S.; Choi, C.M. Evolving early lung cancers detected during follow-up of idiopathic interstitial pneumonia: Serial CT features. Am. J. Roentgenol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, R.B.; Smith, C.; Le Jeune, I.; Gribbin, J.; Fogarty, A.W. The association between idiopathic pulmonary fibrosis and vascular disease: A population-based study. Am. J. Respir Crit. Care Med. 2008. [Google Scholar] [CrossRef] [PubMed]
- Dalleywater, W.; Powell, H.A.; Fogarty, A.W.; Hubbard, R.B.; Navaratnam, V. Venous thromboembolism in people with idiopathic pulmonary fibrosis: A population-based study. Eur. Respir. J. 2014. [Google Scholar] [CrossRef] [PubMed]
- Panos, R.J.; Mortenson, R.L.; Niccoli, S.A.; King, T.E. Clinical deterioration in patients with idiopathic pulmonary fibrosis: Causes and assessment. Am. J. Med. 1990. [Google Scholar] [CrossRef]
- Frazier, A.A.; Galvin, J.R.; Franks, T.J.; Rosado-de-Christenson, M.L. Pulmonary Vasculature: Hypertension and Infarction. Radiographics 2000, 20, 491–524. [Google Scholar] [CrossRef]
- Collard, H.R.; Ryerson, C.J.; Corte, T.J.; Jenkins, G.; Kondoh, Y.; Lederer, D.J.; Lee, J.S.; Maher, T.M.; Wells, A.U.; Antoniou, K.M.; et al. Acute exacerbation of idiopathic pulmonary fibrosis an international working group report. Am. J. Respir. Crit. Care Med. 2016. [Google Scholar] [CrossRef]
- Dalpiaz, G. Fibrosi polmonare idiopatica. Radiol Med. 2010, 115, S130–S138. [Google Scholar]
- Natsuizaka, M.; Chiba, H.; Kuronuma, K.; Otsuka, M.; Kudo, K.; Mori, M.; Bando, M.; Sugiyama, Y.; Takahashi, H. Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am. J. Respir. Crit. Care Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kishaba, T.; Tamaki, H.; Shimaoka, Y.; Fukuyama, H.; Yamashiro, S. Staging of Acute Exacerbation in Patients with Idiopathic Pulmonary Fibrosis. Lung 2014, 192, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Song, J.W.; Hong, S.B.; Lim, C.M.; Koh, Y.; Kim, D.S. Acute exacerbation of idiopathic pulmonary fibrosis: Incidence, risk factors and outcome. Eur. Respir. J. 2011. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Jindal, S.K. Acute exacerbation of idiopathic pulmonary fibrosis: A systematic review. Eur. J. Intern. Med. 2008. [Google Scholar] [CrossRef] [PubMed]
- Huie, T.J.; Olson, A.L.; Cosgrove, G.P.; Janssen, W.J.; Lara, A.R.; Lynch, D.A.; Groshong, S.D.; Moss, M.; Schwarz, M.I.; Brown, K.K.; et al. A detailed evaluation of acute respiratory decline in patients with fibrotic lung disease: Aetiology and outcomes. Respirology 2010. [Google Scholar] [CrossRef] [PubMed]
- Simon-blancal, V.; Freynet, O. Acute Exacerbation of Idiopathic Pulmonary Fibrosis: Outcome and Prognostic Factors. Respiration 2012, 83, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Mallick, S. Outcome of patients with idiopathic pulmonary fibrosis (IPF) ventilated in intensive care unit. Respir. Med. 2008. [Google Scholar] [CrossRef]
- Luppi, F.; Cerri, S.; Taddei, S.; Ferrara, G.; Cottin, V. Acute exacerbation of idiopathic pulmonary fibrosis: a clinical review. Intern. Emerg. Med. 2015. [Google Scholar] [CrossRef]
- Collard, H.R.; Yow, E.; Richeldi, L.; Anstrom, K.J.; Glazer, C. Suspected acute exacerbation of idiopathic pulmonary fibrosis as an outcome measure in clinical trials. Respir. Res. 2013, 14, 1. [Google Scholar] [CrossRef]
- Olson, A.L.; Swigris, J.J.; Raghu, G.; Brown, K.K. Seasonal variation: Mortality from pulmonary fibrosis is greatest in the winter. Chest 2009, 136, 16–22. [Google Scholar] [CrossRef]
- Ryerson, C.J.; Cottin, V.; Brown, K.K.; Collard, H.R. Acute exacerbation of idiopathic pulmonary fibrosis: Shifting the paradigm. Eur. Respir. J. 2015, 46, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.I.S.; Müller, N.L.; Fujimoto, K.; Kato, S.; Ichikado, K.; Taniguchi, H.; Kondoh, Y.; Johkoh, T.; Churg, A. Acute Exacerbation of Chronic Interstitial Pneumonia: High-resolution Computed Tomography and Pathologic Findings. J. Thorac. Imagin. 2007, 22, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Sokai, A.; Tanizawa, K.; Handa, T.; Kubo, T.; Hashimoto, S.; Ikezoe, K.; Nakatsuka, Y.; Aihara, K.; Taguchi, Y.; Muro, S.; et al. Asymmetry in acute exacerbation of idiopathic pulmonary fibrosis. ERS Monogr. 2017, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acute/Chronic | Complication | HRCT Features | |
|---|---|---|---|
| Acute | Infections | Mycobacterium species | Peripheral mass-like lesion/subpleural nodules/segmental or lobar coalescent consolidation with or without necrotising cavitation. |
| Pneumocystis jirovecii | Ground glass opacity/no change from the baseline in a clinic context of infection. | ||
| Aspergillus | Fungal fronds in a pre-existing cavity in early stages. Subsequent coalescence of the cavity (air crescent sign). | ||
| Acute exacerbation IPF | New bilateral ground glass opacities and/or consolidation on a background of reticular or honeycombing pattern. | ||
| Right heart failure | Profuse septal thickening, ground glass opacities, pleural effusion on a background of reticular or honeycombing pattern. | ||
| Chronic | Lung cancer | Ill-defined rounded lesion, mimicking air space consolidation/nodular lesion developing within peripheral and basal honeycombing areas. Ground glass opacity in fibrosis area (mucinous bronchioloalveolar carcinoma). | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baratella, E.; Fiorese, I.; Marrocchio, C.; Salton, F.; Cova, M.A. Imaging Review of the Lung Parenchymal Complications in Patients with IPF. Medicina 2019, 55, 613. https://doi.org/10.3390/medicina55100613
Baratella E, Fiorese I, Marrocchio C, Salton F, Cova MA. Imaging Review of the Lung Parenchymal Complications in Patients with IPF. Medicina. 2019; 55(10):613. https://doi.org/10.3390/medicina55100613
Chicago/Turabian StyleBaratella, Elisa, Ilaria Fiorese, Cristina Marrocchio, Francesco Salton, and Maria Assunta Cova. 2019. "Imaging Review of the Lung Parenchymal Complications in Patients with IPF" Medicina 55, no. 10: 613. https://doi.org/10.3390/medicina55100613
APA StyleBaratella, E., Fiorese, I., Marrocchio, C., Salton, F., & Cova, M. A. (2019). Imaging Review of the Lung Parenchymal Complications in Patients with IPF. Medicina, 55(10), 613. https://doi.org/10.3390/medicina55100613