Tools for the Quality Control of Pharmaceutical Heparin

 ,
,
Abstract
:1. Introduction
2. Heparin Structure
3. Original Screening Techniques for Pharmaceutical-Grade Heparin
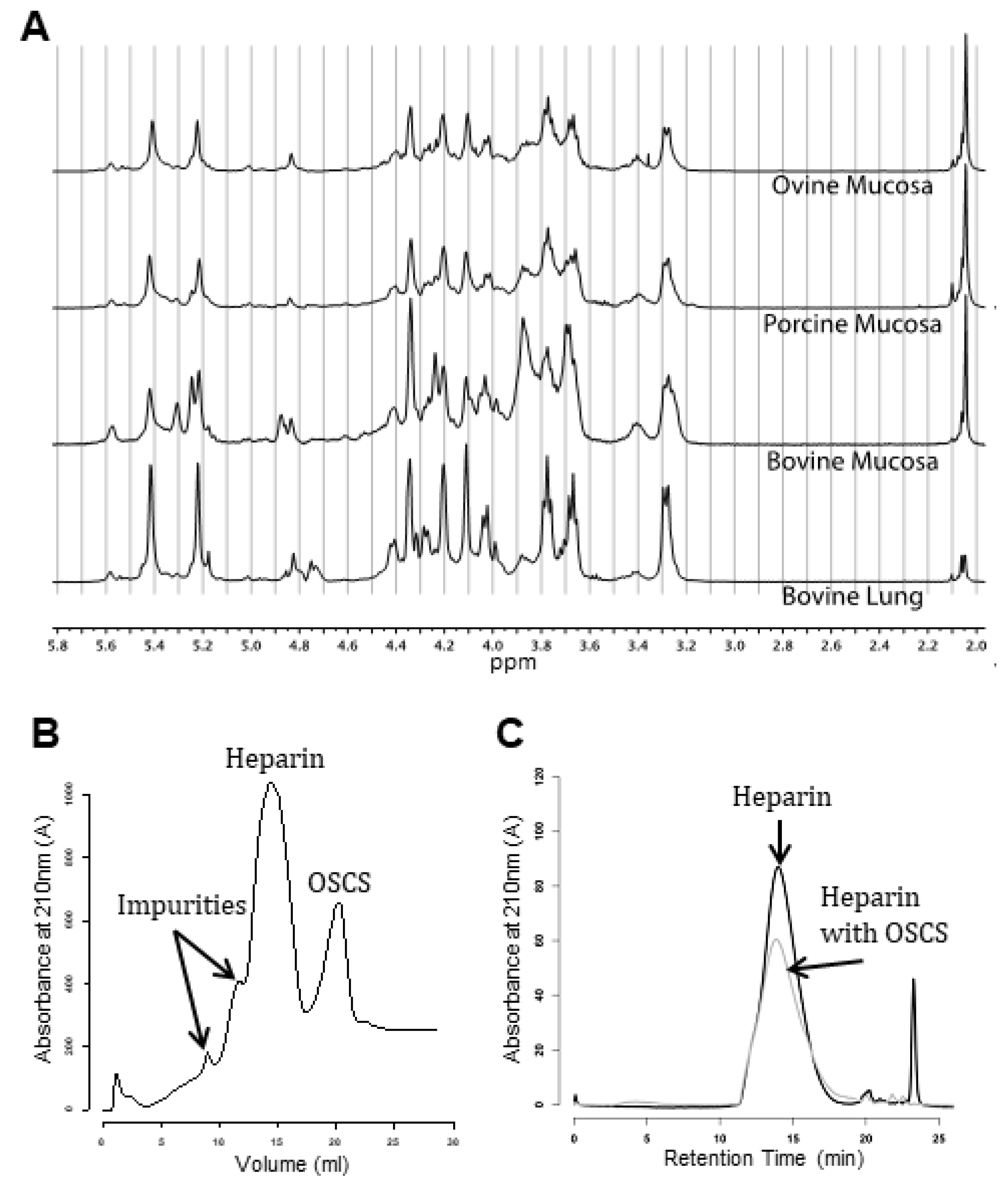
4. Contamination of Pharmaceutical Heparin, 2007–2008
5. Changes to the Heparin Monograph Post-Contamination
6. Alternative Techniques that May Assist in Heparin Quality Assurance
6.1. Species Separation
6.2. Structural Elucidation
6.3. Size Definition
6.4. Other Methods
7. Novel Issues Facing Heparin Quality Control
8. The Future of Pharmaceutical Heparin
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Van Der Meer, J.Y.; Kellenbach, E.; Van Den Bos, L.J. From farm to pharma: An overview of industrial heparin manufacturing methods. Molecules 2017, 22, 1025. [Google Scholar] [CrossRef] [PubMed]
- Casu, B. Structure and Biological Activity of Heparin. Adv. Carbohydr. Chem. Biochem. 1985, 43, 51–134. [Google Scholar] [PubMed]
- Casu, B.; Oreste, P.; Torri, G.; Zopetti, G. The structure of heparin oligosaccharide fragments with high anti-(factor Xa) activity containing the minimal antithrombin III-binding sequence Chemical and 13C nuclear-magnetic-resonance studies. Biochem. J. 1981, 197, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Thunberg, L.; Bäckström, G.; Lindahl, U. Further Characterization of the Antithrombin-Binding Sequence in Heparin. Carbohydr. Res. 1982, 100, 393–410. [Google Scholar] [CrossRef]
- Cheng, J.W.M. Fondaparinux: A New Antithrombotic Agent. Clin. Theraoeutics 2002, 24, 1757–1769. [Google Scholar] [CrossRef]
- Mulloy, B.; Heath, A.; Shriver, Z.; Jameison, F.; Al Hakim, A.; Morris, T.S.; Szajek, A.Y. USP compendial methods for analysis of heparin: Chromatographic determination of molecular weight distributions for heparin sodium. Anal. Bioanal. Chem. 2014, 406, 4815–4823. [Google Scholar] [CrossRef]
- Mulloy, B.; Hogwood, J.; Gray, E.; Lever, R.; Page, C.P. Pharmacology of Heparin and Related Drugs. Pharmacol. Rev. 2015, 68, 76–141. [Google Scholar] [CrossRef]
- Kreuger, J.; Kjellén, L. Heparan Sulfate Biosynthesis: Regulation and Variability. J. Histochem. Cytochem. 2012, 60, 898–907. [Google Scholar] [CrossRef]
- Esko, J.D.; Selleck, S.B. Order Out of Chaos: Assembly of Ligand Binding Sites in Heparan Sulfate 1. Annu. Rev. Biochem. 2002, 71, 435–471. [Google Scholar] [CrossRef]
- Rabenstein, D.L. Heparin and heparan sulfate: Structure and function. Nat. Prod. Rep. 2002, 19, 312–331. [Google Scholar] [CrossRef]
- Stringer, S.E.; Gallagher, J.T. Heparan sulphate. Int. J. Biochem. Cell Biol. 1997, 29, 709–714. [Google Scholar] [CrossRef]
- Casu, B.; Naggi, A.; Torri, G. Re-visiting the structure of heparin. Carbohydr. Res. 2015, 403, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Lyon, M.; Rushton, G.; Askari, J.A.; Humphries, M.J.; Gallagher, J.T. Elucidation of the structural features of heparan sulfate important for interaction with the Hep-2 domain of fibronectin. J. Biol. Chem. 2000, 275, 4599–4606. [Google Scholar] [CrossRef] [PubMed]
- Bertini, S.; Risi, G.; Guerrini, M.; Carrick, K.; Szajek, A.Y.; Mulloy, B. Molecular Weights of Bovine and Porcine Heparin Samples: Comparison of Chromatographic Methods and Results of a Collaborative Survey. Molecules 2017, 22, 1214. [Google Scholar] [CrossRef] [PubMed]
- St Ange, K.; Onishi, A.; Fu, L.; Sun, X.; Lin, L.; Mori, D.; Zhang, F.; Dordick, J.S.; Fareed, J.; Hoppensteadt, D.; et al. Analysis of Heparins Derived From Bovine Tissues and Comparison to Porcine Intestinal Heparins. Clin. Appl. Thromb. 2016, 22, 520–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Yu, Y.; Fareed, J.; Hoppensteadt, D.; Jeske, W.; Kouta, A.; Caijuan, J.; Jin, Y.; Yao, Y.; Xia, K.; et al. Comparison of Low-Molecular-Weight Heparins Prepared From Ovine Heparins with Enoxaparin. Clin. Appl. Thromb. 2019, 25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Yang, B.; Ly, M.; Solakyildirim, K.; Xiao, Z.; Wang, Z.; Beaudet, J.M.; Torelli, A.Y.; Dordick, J.S.; Linhardt, R.J. Structural characterization of heparins from different commercial sources. Anal. Bioanal. Chem. 2011, 401, 2793–2803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeske, W.; Kouta, A.; Farooqui, A.; Siddiqui, F.; Rangnekar, V.; Niverthi, M.; Laddu, R.; Hoppensteadt, D.; Iqbal, O.; Walenga, J.; et al. Bovine Mucosal Heparins Are Comparable to Porcine Mucosal Heparin at USP Potency Adjusted Levels. Front. Med. 2019, 5, 360. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Xu, X.; Liu, X.; Sheng, A.; Jin, L.; Linhardt, R.J.; Chi, L. Comparison of Low-Molecular-Weight Heparins Prepared From Bovine Lung Heparin and Porcine Intestine Heparin. J. Pharm. Sci. 2016, 105, 1843–1850. [Google Scholar] [CrossRef]
- Bianchini, P.; Liverani, L.; Mascellani, G.; Parma, B. Heterogeneity of unfractionated heparins studied in connection with species, source, and production processes. Semin. Thromb. Hemost. 1997, 23, 3–10. [Google Scholar] [CrossRef]
- Mulloy, B.; Gray, E.; Barrowcliffe, T.W. Characterization of unfractionated heparin: Comparison of materials from the last 50 years. Thromb. Haemost. 2000, 84, 1052–1056. [Google Scholar] [PubMed]
- Fasciano, J.M.; Danielson, N.D. Ion chromatography for the separation of heparin and structurally related glycoaminoglycans: A review. J. Sep. Sci. 2016, 39, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Li, G.; Yang, B.; Onishi, A.; Li, L.; Sun, P.; Zhang, F.; Linhardt, R.J. Structural Characterization of Pharmaceutical Heparins Prepared from Different Animal Tissues. J. Pharm. Sci. 2013, 102, 1447–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watt, D.K.; Yorke, S.C.; Slim, G.C. Comparison of ovine, bovine and porcine mucosal heparins and low molecular weight heparins by disaccharide analyses and 13C NMR. Carbohydr. Polym. 1997, 33, 5–11. [Google Scholar] [CrossRef]
- Tovar, A.M.F.; Capillé, N.V.M.; Santos, G.R.C.; Vairo, B.C.; Oliveira, S.N.M.C.G.; Fonseca, R.J.C.; Mourão, P.A.S. Heparin from bovine intestinal mucosa: Glycans with multiple sulfation patterns and anticoagulant effects. Thromb. Haemost. 2012, 107, 903–915. [Google Scholar] [PubMed]
- Turnbull, J.E.; Gallagher, J.T. Distribution of iduronate 2-sulphate residues in heparan sulphate. Evidence for an ordered polymeric structure. Biochem. J. 2015, 273, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Bourin, M.C.; Lindahl, U. Glycosaminoglycans and the regulation of blood coagulation. Biochem. J. 2015, 289, 313–330. [Google Scholar] [CrossRef] [PubMed]
- Stevic, I.; Parmar, N.; Paredes, N.; Berry, L.R.; Chan, A.K.C. Binding of Heparin to Metals. Cell Biochem. Biophys. 2011, 59, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Mulloy, B.; Forster, M.J.; Jones, C.; Drake, A.F.; Johnson, E.A.; Davies, D.B. The effect of variation of substitution on the solution conformation of heparin: A spectroscopic and molecular modelling study. Carbohydr. Res. 1994, 255, 1–26. [Google Scholar] [CrossRef]
- Grant, D.; Long, W.F.; Moffat, C.F.; Williamson, F.B. Infrared spectroscopy of chemically modified heparins. Biochem. J. 1989, 261, 1035–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Boeckel, C.A.A.; van Aelst, S.F.; Wagenaars, G.N.; Mellema, J.R. Conformational analysis of synthetic heparin-like oligosaccharides containing a-L-idopyranosyluronic acid. Recl. Trav. Chim. Pays-Bas 1987, 106, 19–29. [Google Scholar] [CrossRef]
- Stancanelli, E.; Elli, S.; Hsieh, P.H.; Liu, J.; Guerrini, M. Recognition and Conformational Properties of an Alternative Antithrombin Binding Sequence Obtained by Chemoenzymatic Synthesis. ChemBioChem 2018, 19, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Hricovíni, M.; Guerrini, M.; Bisio, A.; Torri, G.; Naggi, A.; Casu, B. Active conformations of glycosaminoglycans. NMR determination of the conformation of heparin sequences complexed with antithrombin and fibroblast growth factors in solution. Semin. Thromb. Hemost. 2002, 28, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Rudd, T.; Yates, E.; Hricovini, M. Spectroscopic and Theoretical Approaches for the Determination of Heparin Saccharide Structure and the Study of Protein-Glycosaminoglycan Complexes in Solution. Curr. Med. Chem. 2009, 16, 4750–4766. [Google Scholar] [CrossRef]
- Mobli, M.; Nilsson, M.; Almond, A. The structural plasticity of heparan sulfate NA-domains and hence their role in mediating multivalent interactions is confirmed by high-accuracy 15N-NMR relaxation studies. Glycoconj. J. 2008, 25, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Ferro, D.R.; Provasoli, A.; Ragazzi, M.; Casu, B.; Torri, G.; Bossennec, V.; Perly, B.; Sinaÿ, P.; Petitou, M.; Choay, J. Conformer populations of l-iduronic acid residues in glycosaminoglycan sequences. Carbohydr. Res. 1990, 195, 157–167. [Google Scholar] [CrossRef]
- Hricovíni, M. Solution Structure of Heparin Pentasaccharide: NMR and DFT Analysis. J. Phys. Chem. B 2015, 119, 12397–12409. [Google Scholar] [CrossRef] [PubMed]
- Rudd, T.R.; Guimond, S.E.; Skidmore, M.A.; Duchesne, L.; Guerrini, M.; Torri, G.; Cosentino, C.; Brown, A.; Clarke, D.T.; Turnbull, J.E.; et al. Influence of substitution pattern and cation binding on conformation and activity in heparin derivatives. Glycobiology 2007, 17, 983–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabenstein, D.L.; Robert, J.M.; Peng, J. Multinuclear magnetic resonance studies of the interaction of inorganic cations with heparin. Carbohydr. Res. 1995, 278, 239–256. [Google Scholar] [CrossRef]
- Turnbull, J.E.; Hopwood, J.J.; Gallagher, J.T. A strategy for rapid sequencing of heparan sulfate and heparin saccharides. Proc. Natl. Acad. Sci. USA 2002, 96, 2698–2703. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Z.; Linhardt, R.J. Lessons learned from the contamination of heparin. Nat. Prod. Rep. 2009, 26, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrini, M.; Bisio, A.; Torri, G. Combined Quantitative 1H and 13C Nuclear Magnetic Resonance Spectroscopy for Characterization of Heparin Preparationsy. Semin. Thromb. Hemost. 2001, 27, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Volpi, N.; Maccari, F.; Suwan, J.; Linhardt, R.J. Electrophoresis for the analysis of heparin purity and quality. Electrophoresis 2012, 33, 1531–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishimoto, T.K.; Viswanathan, K.; Ganguly, T.; Elankumaran, S.; Smith, S.; Pelzer, K.; Lansing, J.C.; Sriranganathan, N.; Zhao, G.; Galcheva-Gargova, Z.; et al. Contaminated Heparin Associated with Adverse Clinical Events and Activation of the Contact System. N. Engl. J. Med. 2008, 358, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramacciotti, E.; Clark, M.; Sadeghi, N.; Hoppensteadt, D.; Thethi, I.; Gomes, M.; Fareed, J. Contaminants in heparin: Review of the literature, molecular profiling, and clinical implications. Clin. Appl. Thromb. 2011, 17, 126–135. [Google Scholar] [CrossRef]
- Guerrini, M.; Beccati, D.; Shriver, Z.; Naggi, A.; Viswanathan, K.; Bisio, A.; Capila, I.; Lansing, J.C.; Guglieri, S.; Fraser, B.; et al. Oversulfated chondroitin sulfate is a contaminant in heparin associated with adverse clinical events. Nat. Biotechnol. 2008, 26, 669–675. [Google Scholar] [CrossRef]
- Beyer, T.; Diehl, B.; Randel, G.; Humpfer, E.; Schäfer, H.; Spraul, M.; Schollmayer, C.; Holzgrabe, U. Quality assessment of unfractionated heparin using 1H nuclear magnetic resonance spectroscopy. J. Pharm. Biomed. Anal. 2008, 48, 13–19. [Google Scholar] [CrossRef]
- Fernandes, A. Heparin Sodium Monograph. Pharmacopeial Forum 2009, 34, 1–4. [Google Scholar]
- Volpi, N.; Maccari, F.; Linhardt, R.J. Quantitative capillary electrophoresis determination of oversulfated chondroitin sulfate as a contaminant in heparin preparations. Anal. Biochem. 2009, 388, 140–145. [Google Scholar] [CrossRef] [Green Version]
- Alban, S.; Lühn, S.; Schiemann, S.; Beyer, T.; Norwig, J.; Schilling, C.; Rädler, O.; Wolf, B.; Matz, M.; Baumann, K.; et al. Comparison of established and novel purity tests for the quality control of heparin by means of a set of 177 heparin samples. Anal. Bioanal. Chem. 2011, 399, 605–620. [Google Scholar] [CrossRef]
- Trehy, M.L.; Reepmeyer, J.C.; Kolinski, R.E.; Westenberger, B.J.; Buhse, L.F. Analysis of heparin sodium by SAX/HPLC for contaminants and impurities. J. Pharm. Biomed. Anal. 2009, 49, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Szajek, A.Y.; Chess, E.; Johansen, K.; Gratzl, G.; Gray, E.; Keire, D.; Linhardt, R.J.; Liu, J.; Morris, T.; Mulloy, B.; et al. The US regulatory and pharmacopeia response to the global heparin contamination crisis. Nat. Biotechnol. 2016, 34, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Volpi, N.; Maccari, F. Electrophoretic approaches to the analysis of complex polysaccharides. J. Chromatogr. B 2006, 834, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Edens, R.E.; Al-Hakim, A.; Weiler, J.M.; Rethwisch, D.G.; Fareed, J.; Linhardt, R.J. Gradient polyacrylamide gel electrophoresis for determination of molecular weights of heparin preparations and low-molecular-weight heparin derivatives. J. Pharm. Sci. 1992, 81, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, B.; Suwan, J.; Zhang, F.; Wang, Z.; Liu, H.; Mulloy, B.; Linhardt, R.J. Analysis of Pharmaceutical Heparins and Potential Contaminants Using 1H-NMR and PAGE. J. Pharm. Sci. 2009, 98, 4017–4026. [Google Scholar] [CrossRef]
- Guerrini, M.; Zhang, Z.; Shriver, Z.; Naggi, A.; Masuko, S.; Langer, R.; Casu, B.; Linhardt, R.J.; Torri, G.; Sasisekharan, R. Orthogonal analytical approaches to detect potential contaminants in heparin. Proc. Natl. Acad. Sci. USA 2009, 106, 16956–16961. [Google Scholar] [CrossRef] [Green Version]
- Keire, D.A.; Mans, D.J.; Ye, H.; Kolinski, R.E.; Buhse, L.F. Assay of possible economically motivated additives or native impurities levels in heparin by 1H NMR, SAX-HPLC, and anticoagulation time approaches. J. Pharm. Biomed. Anal. 2010, 52, 656–664. [Google Scholar] [CrossRef]
- Spencer, J.A.; Kauffman, J.F.; Reepmeyer, J.C.; Gryniewicz, C.M.; Yi, W.; Duckhee, T.; Buhse, L.F.; Westenberger, B.J. Screening of Heparin API by Near Infrared Reflectance and Raman Spectroscopy. J. Pharm. Sci. 2009, 98, 3540–3547. [Google Scholar] [CrossRef]
- Walenga, J.M.; Prechel, M.; Jeske, W.P.; Bakhos, M. Unfractionated heparin compared with low-molecular-weight heparin as related to heparin-induced thrombocytopenia. Curr. Opin. Pulm. Med. 2005, 11, 385–391. [Google Scholar] [CrossRef]
- Kakoi, N.; Kinoshita, M.; Kawasaki, N.; Yamaguchi, T.; Hayakawa, T.; Kakehi, K. Capillary Electrophoresis Analysis of Contaminants in Heparin Sodium for the Japanese Pharmacopoeia Purity Test. Yakugaku Zasshi 2009, 129, 1255–1264. [Google Scholar] [CrossRef] [Green Version]
- Wielgos, T.; Havel, K.; Ivanova, N.; Weinberger, R. Determination of impurities in heparin by capillary electrophoresis using high molarity phosphate buffers. J. Pharm. Biomed. Anal. 2009, 49, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Somsen, G.W.; Tak, Y.H.; Toraño, J.S.; Jongen, P.M.J.M.; de Jong, G.J. Determination of oversulfated chondroitin sulfate and dermatan sulfate impurities in heparin by capillary electrophoresis. J. Chromatogr. A 2009, 1216, 4107–4112. [Google Scholar] [CrossRef] [PubMed]
- Beni, S.; Limtiaco, J.F.K.; Larive, C.K. Analysis and characterization of heparin impurities. Anal. Bioanal. Chem. 2011, 399, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Volpi, N.; Buzzega, D. Agarose-gel electrophoresis for the quality assurance and purity of heparin formulations. J. Pharm. Biomed. Anal. 2012, 67–68, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Hashii, N.; Kawasaki, N.; Itoh, S.; Qin, Y.; Fujita, N.; Hattori, T.; Miyata, K.; Bando, A.; Sekimoto, Y.; Hama, T.; et al. Heparin identification test and purity test for OSCS in heparin sodium and heparin calcium by weak anion-exchange high-performance liquid chromatography. Biologicals 2010, 38, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Solakyildirim, K.; Chang, Y.; Linhardt, R.J. Hyphenated techniques for the analysis of heparin and heparan sulfate. Anal. Bioanal. Chem. 2011, 399, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wickramasekara, S.; Nemes, P. One-Hour Screening of Adulterated Heparin by Simplified Peroxide Digestion and Fast RPIP-LC-MS2. Anal. Chem. 2015, 87, 8424–8432. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Calero, V.; Moyano, E.; Puignou, L.; Galceran, M.T. Pressure-assisted capillary electrophoresis-electrospray ion trap mass spectrometry for the analysis of heparin depolymerised disaccharides. J. Chromatogr. A 2001, 914, 277–291. [Google Scholar] [CrossRef]
- Skidmore, M.A.; Guimond, S.E.; Dumax-Vorzet, A.F.; Yates, E.A.; Turnbull, J.E. Disaccharide compositional analysis of heparan sulfate and heparin polysaccharides using UV or high-sensitivity fluorescence (BODIPY) detection. Nat. Protoc. 2010, 5, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Holman, J.; Skidmore, M.A.; Rudd, T.R.; Yates, E.A. The latent ampholytic nature of glycosaminoglycan (GAG) oligosaccharides facilitates their separation by isoelectric focusing. Anal. Methods 2010, 2, 1550–1554. [Google Scholar] [CrossRef]
- Volpi, N.; Maccari, F.; Linhardt, R.J. Capillary electrophoresis of complex natural polysaccharides. Electrophoresis 2008, 29, 3095–3106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamfir, A.; Peter-Katalinić, J. Capillary electrophoresis-mass spectrometry for glycoscreening in biomedical research. Electrophoresis 2004, 25, 1949–1963. [Google Scholar] [CrossRef]
- Nemes, P.; Hoover, W.J.; Keire, D.A. High-throughput differentiation of heparin from other glycosaminoglycans by pyrolysis mass spectrometry. Anal. Chem. 2013, 85, 7405–7412. [Google Scholar] [CrossRef]
- Harris, G.A.; Galhena, A.S.; Fern, F.M. Ambient Sampling/Ionization Mass Spectrometry: Applications and Current Trends. Anal. Chem. 2011, 83, 4508–4538. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chi, L. Recent advances in mass spectrometry analysis of low molecular weight heparins. Chin. Chem. Lett. 2018, 29, 11–18. [Google Scholar] [CrossRef]
- Li, G.; Cai, C.; Li, L.; Fu, L.; Chang, Y.; Zhang, F.; Toida, T.; Xue, C.; Linhardt, R.J. Method to detect contaminants in heparin using radical depolymerization and liquid chromatography-mass spectrometry. Anal. Chem. 2014, 86, 326–330. [Google Scholar] [CrossRef]
- Brustkern, A.M.; Buhse, L.F.; Nasr, M.; Al-Hakim, A.; Keire, D.A. Characterization of currently marketed heparin products: Reversed-phase ion-pairing liquid chromatography mass spectrometry of heparin digests. Anal. Chem. 2010, 82, 9865–9870. [Google Scholar] [CrossRef] [PubMed]
- Lamari, F.N.; Kuhn, R.; Karamanos, N.K. Derivatization of carbohydrates for chromatographic, electrophoretic and mass spectrometric structure analysis. J. Chromatogr. B 2003, 793, 15–36. [Google Scholar] [CrossRef]
- Yates, E.A.; Santini, F.; Guerrini, M.; Naggi, A.; Torri, G.; Casu, B. 1H and 13C NMR spectral assignments of the major sequences of twelve systematically modified heparin derivatives. Carbohydr. Res. 1996, 294, 15–27. [Google Scholar] [CrossRef]
- Langeslay, D.J.; Beecher, C.N.; Naggi, A.; Guerrini, M.; Torri, G.; Larive, C.K. Characterizing the microstructure of heparin and heparan sulfate using N-sulfoglucosamine 1H and 15N NMR chemical shift analysis. Anal. Chem. 2013, 85, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Langeslay, D.J.; Beni, S.; Larive, C.K. A closer look at the nitrogen next door: 1H-15N NMR methods for glycosaminoglycan structural characterization. J. Magn. Reson. 2012, 216, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Neely, B.W. Infrared Spectra of Carbohydrates. Adv. Carbohyd. Chem. 1957, 12, 13–33. [Google Scholar]
- Grant, D.; Long, W.F.; Williamson, F.B. Infrared spectroscopy of heparin-cation complexes. Biochem. J. 1987, 244, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Rudd, T.R.; Gaudesi, D.; Skidmore, M.A.; Ferro, M.; Guerrini, M.; Mulloy, B.; Torri, G.; Yates, E.A. Construction and use of a library of bona fide heparins employing 1H NMR and multivariate analysis. Analyst 2011, 136, 1380–1389. [Google Scholar] [CrossRef]
- Devlin, A.; Mycroft-West, C.; Guerrini, M.; Yates, E.; Skidmore, M. Analysis of solid-state heparin samples by ATR-FTIR spectroscopy. bioRxiv 2019, 538074. [Google Scholar] [CrossRef]
- Barlow, G.H. The determination of molecular weight distributions on heparin samples. Semin. Thromb. Hemost. 1985, 11, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Malsch, R.; Harenberg, J. High-performance size exclusion chromatography and polyacrylamide gel electrophoresis for characterization of unfractionated and low molecular mass glycosaminoglycans. Semin. Thromb. Hemost. 1994, 20, 135–143. [Google Scholar] [CrossRef]
- Bigler, P.; Brenneisen, R. Improved impurity fingerprinting of heparin by high resolution 1H NMR spectroscopy. J. Pharm. Biomed. Anal. 2009, 49, 1060–1064. [Google Scholar] [CrossRef]
- Sitkowski, J.; Bednarek, E.; Bocian, W.; Kozerski, L. Assessment of Oversulfated Chondroitin Sulfate in Low Molecular Weight and Unfractioned Heparins Diffusion Ordered Nuclear Magnetic Resonance Spectroscopy Method. J. Med. Chem. 2008, 51, 7663–7665. [Google Scholar] [CrossRef]
- Spelta, F.; Liverani, L.; Peluso, A.; Marinozzi, M.; Urso, E.; Guerrini, M.; Naggi, A. SAX-HPLC and HSQC NMR Spectroscopy: Orthogonal Methods for Characterizing Heparin Batches Composition. Front. Med. 2019, 6, 78. [Google Scholar] [CrossRef]
- Stanley, F.E.; Stalcup, A.M. The use of circular dichroism as a simple heparin-screening strategy. Anal. Bioanal. Chem. 2011, 399, 701–706. [Google Scholar] [CrossRef]
- Stivala, S.S.; Herbst, M.; Kratky, O.; Pilz, I. Physico-chemical studies of fractionated bovine heparin. Arch. Biochem. Biophys. 2004, 127, 795–802. [Google Scholar] [CrossRef]
- Ly, M.; Wang, Z.; Laremore, T.N.; Zhang, F.; Zhong, W.; Pu, D.; Zagorevski, D.V.; Dordick, J.S.; Linhardt, R.J. Analysis of E. coli K5 capsular polysaccharide heparosan. Anal. Bioanal. Chem. 2011, 399, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Sommers, C.D.; Ye, H.; Kolinski, R.E.; Nasr, M.; Buhse, L.F.; Al-Hakim, A.; Keire, D.A. Characterization of currently marketed heparin products: Analysis of molecular weight and heparinase-I digest patterns. Anal. Bioanal. Chem. 2011, 401, 2445–2454. [Google Scholar] [CrossRef]
- Rodriguez, H.J.; Vanderwielen, A.J. Molecular weight determination of commercial heparin sodium USP and its sterile solutions. J. Pharm. Sci. 1979, 68, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, A.; Jeske, W.; Hoppensteadt, D.; Lormeau, J.C.; Wolf, H. Molecular profiling and weight determination of heparins and depolymerized heparins. J. Pharm. Sci. 1995, 84, 724–727. [Google Scholar] [CrossRef]
- Guo, X.; Condra, M.; Kimura, K.; Berth, G.; Dautzenberg, H.; Dubin, P.L. Determination of molecular weight of heparin by size exclusion chromatography with universal calibration. Anal. Biochem. 2003, 312, 33–39. [Google Scholar] [CrossRef]
- Knobloch, J.E.; Shaklee, P.N. Absolute molecular weight distribution of low-molecular-weight heparins by size-exclusion chromatography with multiangle laser light scattering detection. Anal. Biochem. 1997, 245, 231–241. [Google Scholar] [CrossRef]
- Beirne, J.; Truchan, H.; Rao, L. Development and qualification of a size exclusion chromatography coupled with multiangle light scattering method for molecular weight determination of unfractionated heparin. Anal. Bioanal. Chem. 2011, 399, 717–725. [Google Scholar] [CrossRef]
- Barlow, G.H.; Sanderson, N.D.; McNeill, P.D. Macromolecular properties and biological activity of heparin. Arch. Biochem. Biophys. 1961, 94, 518–525. [Google Scholar] [CrossRef]
- Desai, U.R.; Linhardt, R.J. Molecular Weight of Heparin Using. J. Pharm. Sci. 1995, 84, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Juhasz, P.; Biemann, K. Mass spectrometric molecular-weight determination of highly acidic compounds of biological significance via their complexes with basic polypeptides. Proc. Natl. Acad. Sci. USA 1994, 91, 4333–4337. [Google Scholar] [CrossRef] [PubMed]
- Chi, L.; Amster, J.; Linhardt, R. Mass Spectrometry for the Analysis of Highly Charged Sulfated Carbohydrates. Curr. Anal. Chem. 2005, 1, 223–240. [Google Scholar] [CrossRef]
- Bertini, S.; Bisio, A.; Torri, G.; Bensi, D.; Terbojevich, M. Molecular weight determination of heparin and dermatan sulfate by size exclusion chromatography with a triple detector array. Biomacromolecules 2005, 6, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Viskov, C.; Bouley, E.; Hubert, P.; Martinez, C.; Herman, F.; Jeske, W.; Hoppensteadt, D.; Walenga, J.M.; Fareed, J. Isolation and characterization of contaminants in recalled unfractionated heparin and low-molecular-weight heparin. Clin. Appl. Thromb. 2009, 15, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Tami, C.; Puig, M.; Reepmeyer, J.C.; Ye, H.; Avignon, D.A.; Buhse, L.; Verthelyi, D. Inhibition of Taq polymerase as a method for screening heparin for oversulfated contaminants. Biomaterials 2008, 29, 4808–4814. [Google Scholar] [CrossRef] [PubMed]
- Sommers, C.D.; Mans, D.J.; Mecker, L.C.; Keire, D.A. Sensitive detection of oversulfated chondroitin sulfate in heparin sodium or crude heparin with a colorimetric microplate based assay. Anal. Chem. 2011, 83, 3422–3430. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Shi, L.; Wei, H. A “turn on” fluorescent probe for heparin and its oversulfated chondroitin sulfate contaminant. Chem. Sci. 2015, 6, 6361–6366. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Zhou, M.; Wei, H. A supercharged fluorescent protein based FRET sensing platform for detection of heparin contamination. Anal. Methods 2017, 9, 5593–5597. [Google Scholar] [CrossRef]
- Hu, Y.; Guo, W.; Ding, Y.; Cheng, H.; Wei, H. Modulating luminescence of Tb3+ with biomolecules for sensing heparin and its contaminant OSCS. Biosens. Bioelectron. 2016, 86, 858–863. [Google Scholar] [CrossRef]
- Kalita, M.; Balivada, S.; Swarup, V.P.; Mencio, C.; Raman, K.; Desai, U.R.; Troyer, D.; Kuberan, B. A nanosensor for ultrasensitive detection of oversulfated chondroitin sulfate contaminant in heparin. J. Am. Chem. Soc. 2014, 136, 554–557. [Google Scholar] [CrossRef] [PubMed]
- Lühn, S.; Schiemann, S.; Alban, S. Simple fluorescence assay for quantification of OSCS in heparin. Anal. Bioanal. Chem. 2011, 399, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Alban, S.; Lühn, S.; Schiemann, S. Combination of a two-step fluorescence assay and a two-step anti-Factor Xa assay for detection of heparin falsifications and protein in heparins. Anal. Bioanal. Chem. 2011, 399, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.; Tseng, W.L. Molecular beacon-based fluorescent assay for specific detection of oversulfated chondroitin sulfate contaminants in heparin without enzyme treatment. Anal. Chem. 2015, 87, 5031–5035. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Buchanan, S.; Meyerhoff, M.E. Detection of high-charge density polyanion contaminants in biomedical heparin preparations using potentiometric polyanion sensors. Anal. Chem. 2008, 80, 9845–9847. [Google Scholar] [CrossRef] [PubMed]
- Rudd, T.R.; MacChi, E.; Gardini, C.; Muzi, L.; Guerrini, M.; Yates, E.A.; Torri, G. How to find a needle (or anything else) in a haystack: Two-dimensional correlation spectroscopy-filtering with iterative random sampling applied to pharmaceutical heparin. Anal. Chem. 2012, 84, 6841–6847. [Google Scholar] [CrossRef] [PubMed]
- Zang, Q.; Keire, D.A.; Wood, R.D.; Buhse, L.F.; Moore, C.M.V.; Nasr, M.; Al-Hakim, A.; Trehy, M.L.; Welsh, W.J. Combining 1H NMR spectroscopy and chemometrics to identify heparin samples that may possess dermatan sulfate (DS) impurities or oversulfated chondroitin sulfate (OSCS) contaminants. J. Pharm. Biomed. Anal. 2011, 54, 1020–1029. [Google Scholar] [CrossRef]
- Lima, M.A.; Rudd, T.R.; de Farias, E.H.C.; Ebner, L.F.; Gesteira, T.F.; de Souza, L.M.; Mendes, A.; Córdula, C.R.; Martins, J.R.M.; Hoppensteadt, D.; et al. A new approach for heparin standardization: Combination of scanning UV spectroscopy, nuclear magnetic resonance and principal component analysis. PLoS ONE 2011, 6, e15970. [Google Scholar] [CrossRef]
- Zang, Q.; Keire, D.A.; Buhse, L.F.; Wood, R.D.; Mital, D.P.; Haque, S.; Srinivasan, S.; Moore, C.M.V.; Nasr, M.; Al-Hakim, A.; et al. Identification of heparin samples that contain impurities or contaminants by chemometric pattern recognition analysis of proton NMR spectral data. Anal. Bioanal. Chem. 2011, 401, 939–955. [Google Scholar] [CrossRef]
- Rudd, T.R.; Mauri, L.; Marinozzi, M.; Stancanelli, E.; Yates, E.; Naggi, A.; Guerrini, M. Multivariate analysis applied to complex biological medicines. Faraday Discuss. 2019, 218, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Monakhova, Y.B.; Fareed, J.; Yao, Y.; Diehl, B.W.K. Improving reliability of chemometric models for authentication of species origin of heparin by switching from 1D to 2D NMR experiments. J. Pharm. Biomed. Anal. 2018, 153, 168–174. [Google Scholar] [CrossRef]
- Mauri, L.; Marinozzi, M.; Mazzini, G.; Kolinski, R.E.; Karfunkle, M.; Keire, D.A.; Guerrini, M. Combining NMR Spectroscopy and Chemometrics to Monitor Structural Features of Crude Hep-arin. Molecules 2017, 22, 1146. [Google Scholar] [CrossRef] [PubMed]
- Keire, D.A.; Trehy, M.L.; Reepmeyer, J.C.; Kolinski, R.E.; Ye, W.; Dunn, J.; Westenberger, B.J.; Buhse, L.F. Analysis of crude heparin by 1H NMR, capillary electrophoresis, and strong-anion-exchange-HPLC for contamination by over sulfated chondroitin sulfate. J. Pharm. Biomed. Anal. 2010, 51, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Rudd, T.R.; Skidmore, M.A.; Guimond, S.E.; Guerrini, M.; Cosentino, C.; Edge, R.; Brown, A.; Clarke, D.T.; Torri, G.; Turnbull, J.E.; et al. Site-specific interactions of copper (II) ions with heparin revealed with complementary (SRCD, NMR, FTIR and EPR) spectroscopic techniques. Carbohydr. Res. 2008, 343, 2184–2193. [Google Scholar] [CrossRef]
- Schwçrer, R.; Zubkova, O.V.; Turnbull, J.E.; Tyler, P.C. Synthesis of a Targeted Library of Heparan Sulfate Hexa- to Dodecasaccharides as Inhibitors of β-Secretase: Potential Therapeutics for Alzheimer s Disease. Chem. Eur. J. 2013, 19, 6817–6823. [Google Scholar]
- Wang, Z.; Hsieh, P.; Xu, Y.; Thieker, D.; En, E.J.; Xie, S.; Cooley, B.; Woods, R.J.; Chi, L.; Liu, J.; et al. Synthesis of 3-O-sulfated oligosaccharides to understand the relationship between structures and functions of heparan sulfate. J. Am. Chem. Soc. 2018, 139, 5249–5256. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Xu, Y.; Liu, J.; Ho, M. Epitope mapping by a Wnt-blocking antibody: Evidence of the Wnt binding domain in heparan sulfate. Sci. Rep. 2016, 6, 26245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Mccallum, S.A.; Xie, J.; Nieto, L.; Corzana, F.; Chen, M.; Liu, J.; Linhardt, R.J. Solution Structures of Chemoenzymatically Synthesized Heparin and Its Precursors. J. Am. Chem. Soc. 2009, 130, 12998–13007. [Google Scholar] [CrossRef]
- Keire, D.A.; Ye, H.; Trehy, M.L.; Ye, W.; Kolinski, R.E.; Westenberger, B.J.; Buhse, L.F.; Nasr, M.; Al-Hakim, A. Characterization of currently marketed heparin products: Key tests for quality assurance. Anal. Bioanal. Chem. 2011, 399, 581–591. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Ido2S-GlcNS,6S | Specific Activity IU/mg | Mw/Da | |||
|---|---|---|---|---|---|
| Anti-Xa | Anti-IIa | APTT | |||
| Porcine Mucosal Heparin | 51.5–85 (68.3) | 145–220 (194) | 172–230 (197) | 145-277 (196) | 12,000–27,090 (19,002) |
| Bovine Lung Heparin | 70–87 (79.8) | 105–156 (133) | 130.6–180 (153) | 89–167 (139) | 12,000–15,240 (14,230) |
| Bovine Mucosal Heparin | 47.4–64.2 (54.5) | 113.6–159 (134) | 92.2–160.7 (126) | 88.1–181 (136) | 14,900–16,417 (15,439) |
| Ovine Mucosal Heparin | 60–89.4 (75.2) | 196–205 (201) | 191–201 (195) | 165–165 (165) | 12,200–20,023 (14,773) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devlin, A.; Mycroft-West, C.; Procter, P.; Cooper, L.; Guimond, S.; Lima, M.; Yates, E.; Skidmore, M. Tools for the Quality Control of Pharmaceutical Heparin. Medicina 2019, 55, 636. https://doi.org/10.3390/medicina55100636
Devlin A, Mycroft-West C, Procter P, Cooper L, Guimond S, Lima M, Yates E, Skidmore M. Tools for the Quality Control of Pharmaceutical Heparin. Medicina. 2019; 55(10):636. https://doi.org/10.3390/medicina55100636
Chicago/Turabian StyleDevlin, Anthony, Courtney Mycroft-West, Patricia Procter, Lynsay Cooper, Scott Guimond, Marcelo Lima, Edwin Yates, and Mark Skidmore. 2019. "Tools for the Quality Control of Pharmaceutical Heparin" Medicina 55, no. 10: 636. https://doi.org/10.3390/medicina55100636