A Robust Approach for Identification of Cancer Biomarkers and Candidate Drugs


Abstract
:1. Introduction
2. Materials and Methods
2.1. Selection of Top Differential Gene Expressions
- Calculate β-FC and β-SAM values for each gene.
- Rank both |β-FC| and |β-SAM| values separately in descending order, where |β-FC| indicates the absolute value of β-FC.
- Calculate the average of both ranks for each gene.
- Arrange the average rank values in ascending order.
- Select first N ordered genes as top DE genes such that β-SAM of Nth ordered gene produces p-value < 0.1.
2.2. Performance Assessment
3. Results
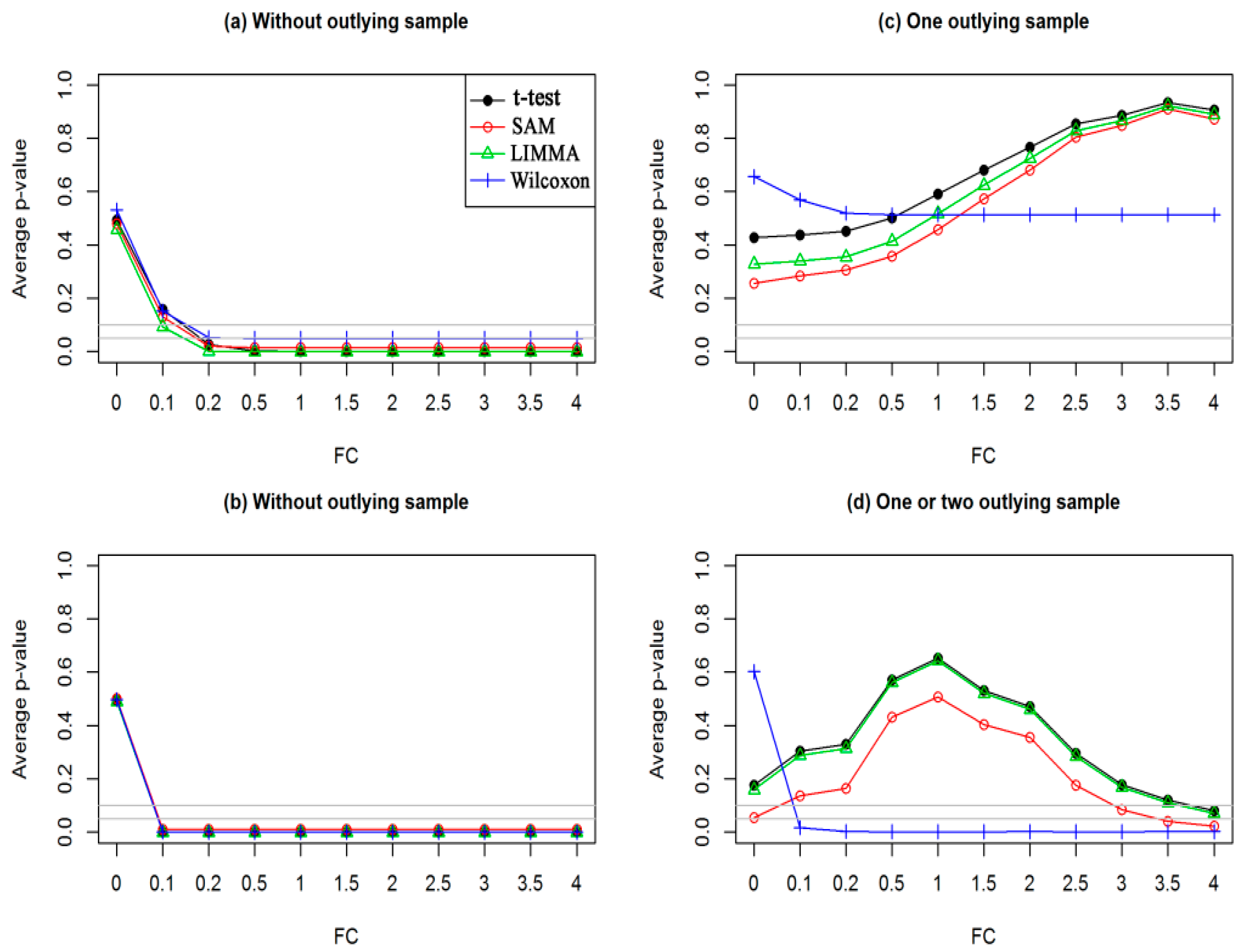
3.1. Performance Evaluation Based on Simulated Gene Expression Profiles
Performance Evaluation Based on Simulated Gene Expression Profiles Using Data Type 1
3.2. Performance Evaluation Based on Real Gene Expression Profiles
3.2.1. Performance Evaluation Based on Platinum Spike Gene Expression Profiles
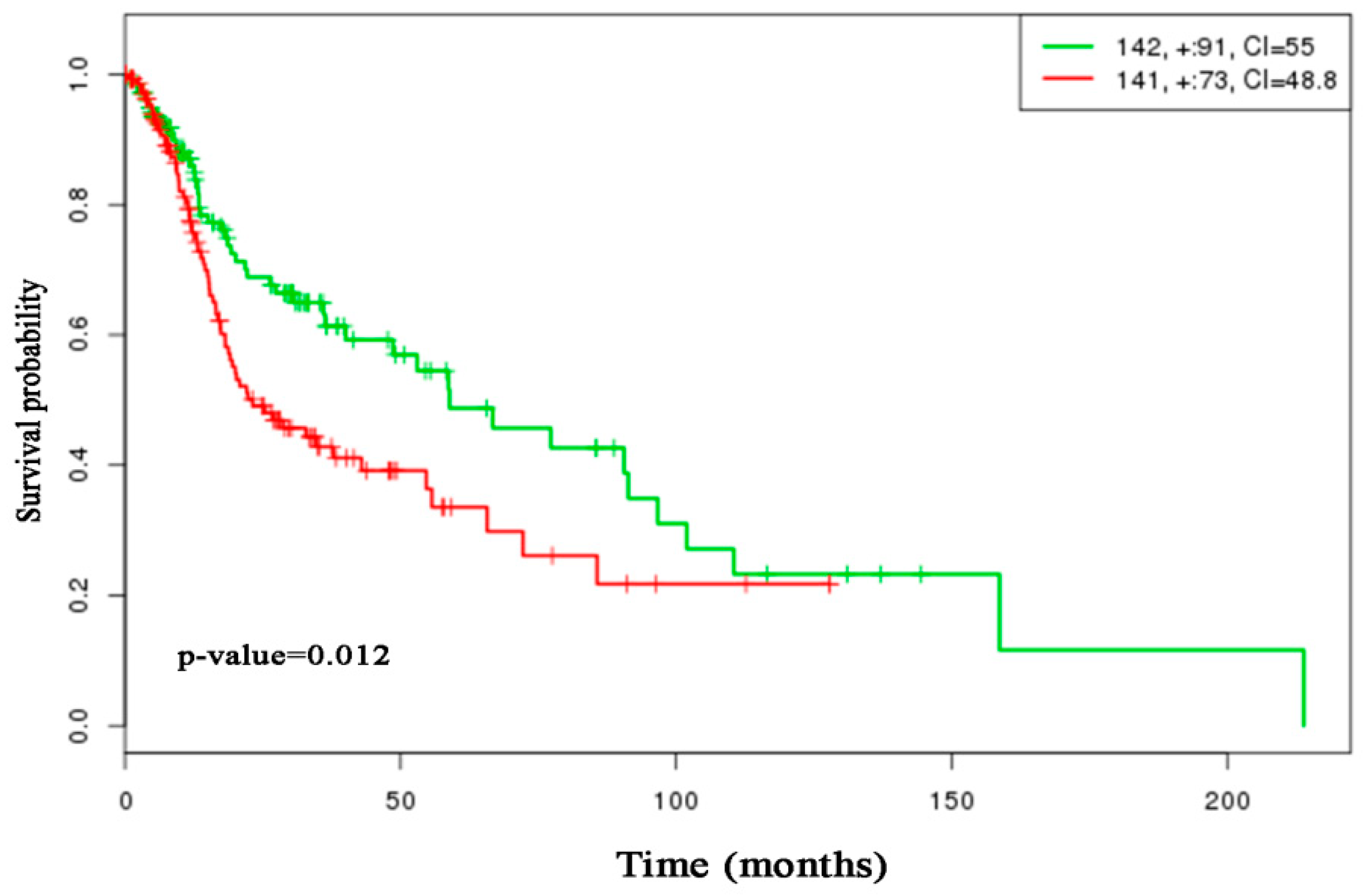
3.2.2. Performance Evaluation Based on Head and Neck Cancer Gene Expression Profiles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- DeRisi, J.; Penland, L.; Bittner, M.L.; Meltzer, P.S.; Ray, M.; Chen, Y.; Su, Y.A.; Trent, J.M. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat. Genet. 1996, 14, 457–460. [Google Scholar] [PubMed]
- Schena, M.; Shalon, D.; Heller, R.; Chai, A.; Brown, P.O.; Davis, R.W. Parallel human genome analysis: Microarray-based expression monitoring of 1000 genes. Proc. Natl. Acad. Sci. USA 1996, 93, 10614–10619. [Google Scholar] [CrossRef] [PubMed]
- Farztdinov, V.; McDyer, F. Distributional fold change test-a statistical approach for detecting differential expression in Microarray experiments. Algorithms Mol. Biol. 2012, 7, 29. [Google Scholar] [CrossRef]
- Kadota, K.; Nakai, Y.; Shimizu, K. A weighted average difference method for detecting differentially expressed genes from microarray data. Algorithms Mol. Biol. 2008, 3, 1748–7188. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 3. [Google Scholar] [CrossRef]
- Smyth, G.K.; Thorne, N.P.; Wettenhall, J. Limma: Linear Models for Microarray Data User’s Guide. Software Manual. Available online: http://www.bioconductor.org (accessed on 23 January 2019).
- Tibshirani, R.; Chu, G.; Narasimhan, B.; Li, J. Samr: Significance Analysis of Microarrays. Available online: http://cran.r-project.org/web/packages/samr/index.html (accessed on 16 October 2018).
- Tusher, V.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [Green Version]
- Kerr, M.K.; Martin, M.; Churchill, G.A. Analysis of variance for gene expression microarray data. J. Comput. Biol. 2000, 7, 819–837. [Google Scholar] [CrossRef] [PubMed]
- Kerr, M.K.; Churchill, G.A. Experimental design for gene expression microarrays. Biostatistics 2001, 2, 183–201. [Google Scholar] [CrossRef] [PubMed]
- Kruskal, W.H.; Wallis, W.A. Use of Ranks in One-Criterion Variance Analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Wilcoxon, F. Individual Comparisons by Ranking Methods. Biom. Bull. 1945, 1, 80–83. [Google Scholar] [CrossRef]
- Vavoulis, D.V.; Francescatto, M.; Heutink, P.; Gough, J. DGEclust: Differential expression analysis of clustered count data. Genome Biol. 2015, 16, 39. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Krishna, N.S.; Grigor, K.M.; Bartlett, J.M.S. Androgen receptor gene amplification and protein expression in hormone refractory prostate cancer. Br. J. Cancer 2003, 89, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Popławski, A.B.; Jankowski, M.; Erickson, S.W.; Díaz de Ståhl, T.; Partridge, E.C.; Crasto, C.; Guo, J.; Gibson, J.; Menzel, U.; Bruder, C.E.; et al. Frequent genetic differences between matched primary and metastatic breast cancer provide an approach to identification of biomarkers for disease progression. Eur. J. Hum. Genet. 2010, 18, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Haney, S.; Kam, M.; Hrebien, L. Benefits of using paired controls for analysing gene expression of prostate cancer. In Proceedings of the 8th IEEE International Conference on BioInformatics and BioEngineering (BIBE 2008), Athens, Greece, 8–10 October 2008; pp. 1–3. [Google Scholar]
- Tan, Q.; Thomassen, M.; Kruse, T.A. Feature selection for predicting tumours metastases in microarray experiments using paired design. Cancer Inf. 2007, 3, 213–218. [Google Scholar]
- Dudoit, S.; Shaffer, J.P.; Boldrick, J.C. Multiple hypothesis testing in microarray experiments. Stat. Sci. 2003, 18, 71–103. [Google Scholar] [CrossRef]
- Mollah, M.M.H.; Jamal, R.; Mokhtar, N.M.; Harun, R.; Mollah, M.N.H. A Hybrid One-Way ANOVA Approach for the Robust and Efficient Estimation of Differential Gene Expression with Multiple Patterns. PLoS ONE 2015, 10, e0138810. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Smyth, G.K. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics 2009, 25, 765–771. [Google Scholar] [CrossRef] [Green Version]
- Breitling, R.; Armengaud, P.; Amtmann, A.; Herzyk, P. Rank products: A simple, yet powerful, new method to detect differentially expressed genes in replicatedmicroarray experiments. FEBS Lett. 2004, 573, 83–92. [Google Scholar] [CrossRef]
- Dembele, D.; Kastner, P. Fold change rank ordering statistics: A new method for detecting differentially expressed genes. BMC Bioinform. 2014, 15, 14. [Google Scholar] [CrossRef]
- Xiao, Y.; Hsiao, T.H.; Suresh, U.; Chen, H.H.; Wu, X.; Wolf, S.E.; Chen, Y. A novel significance score for gene selection and ranking. Bioinformatics 2014, 30, 801–807. [Google Scholar] [CrossRef]
- Mollah, M.N.H.; Minami, M.; Eguchi, S. Robust prewhitening for ICA by minimizing β-divergence and its application to FastICA. Neural Process. Lett. 2007, 25, 91–110. [Google Scholar] [CrossRef]
- Mollah, M.N.H.; Sultana, N.; Minami, M.; Eguchi, S. Robust Extraction of Local Structures by the Minimum β-Divergence method. Neural Netw. 2010, 23, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Shahjaman, M.; Kumar, N.; Mollah, M.M.H.; Ahmed, M.S.; Begum, A.A.; Islam, S.M.S.; Mollah, M.N.H. Robust Significance Analysis of Microarrays by Minimum β-Divergence Method. BioMed Res. Int. 2017, 2017, 5310198. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Nishiyama, T.; Shimizu, K.; Kadota, K. TCC: An R package for comparing tag count data with robust normalization strategies. BMC Bioinform. 2013, 14, 219. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Miecznikowski, J.C.; Halfon, M.S. Preferred analysis methods for affymetrix geneChips. II. An expanded, balanced, wholly-defined spike-in dataset. BMC Bioinform. 2010, 11, 1471–2105. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, M.A.; Chen, W.T.; He, Z.M.; Sikora, A.G.; Zhang, P.; Zhang, Z.Y.; Qiu, W.L.; Hsu, D.F.; McMunn-Coffran, C.; Brown, S.M.; et al. Selection and validation of differentially expressed genes in head and neck cancer. Cell. Mol. Life Sci. 2004, 61, 1372–1383. [Google Scholar] [CrossRef] [PubMed]
- Li, W. Volcano plots in analyzing differential expressions with mRNA microarrays. J. Bioinform. Comput. Biol. 2012, 10, 1231003. [Google Scholar] [CrossRef]
- Bergemann, T.L.; Wilson, J. Proportion statistics to detect differentially expressed genes: A comparison with log-ratio statistics. BMC Bioinform. 2011, 12, 228. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Kirov, S.; Snoddy, J. WebGestalt: An integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005, 33, W741–W748. [Google Scholar] [CrossRef] [PubMed]
- Oughtred, R.; Stark, C.; Breitkreutz, B.J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019, 47, D529–D541. [Google Scholar] [CrossRef]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martínez-Ledesma, E.; Martínez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Peña, J.G.; Treviño, V.; Trevino, V. SurvExpress: An Online Biomarker Validation Tool and Database for Cancer Gene Expression Data Using Survival Analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Average Performance Results for Small Sample Case (n1 = n2 = 3) | ||||||||
| Methods | TPR | FPR | TNR | FNR | MER | FDR | AUC | pAUC |
| t-test | 0.461 (0.132) | 0.017 (0.027) | 0.983 (0.973) | 0.539 (0.868) | 0.033 (0.052) | 0.538 (0.864) | 0.459 (0.131) | 0.090 (0.026) |
| Wilcoxon | 0.889 (0.141) | 0.003 (0.026) | 0.997 (0.974) | 0.111 (0.859) | 0.007 (0.052) | 0.112 (0.854) | 0.889 (0.141) | 0.178 (0.028) |
| SAM | 0.802 (0.145) | 0.006 (0.026) | 0.994 (0.974) | 0.198 (0.855) | 0.012 (0.051) | 0.202 (0.850) | 0.802 (0.145) | 0.160 (0.029) |
| Limma | 0.924 (0.145) | 0.002 (0.026) | 0.998 (0.974) | 0.076 (0.855) | 0.005 (0.051) | 0.081 (0.850) | 0.924 (0.145) | 0.185 (0.029) |
| WAD | 0.785 (0.002) | 0.007 (0.031) | 0.993 (0.969) | 0.215 (0.998) | 0.013 (0.060) | 0.214 (0.998) | 0.785 (0.002) | 0.157 (0.000) |
| RP | 0.926 (0.010) | 0.002 (0.031) | 0.998 (0.969) | 0.074 (0.990) | 0.005 (0.060) | 0.080 (0.989) | 0.926 (0.010) | 0.185 (0.002) |
| FCROS | 0.909 (0.146) | 0.003 (0.026) | 0.997 (0.974) | 0.091 (0.854) | 0.006 (0.051) | 0.096 (0.849) | 0.909 (0.146) | 0.182 (0.029) |
| Proposed | 0.924 (0.855) | 0.002 (0.010) | 0.998 (0.990) | 0.076 (0.145) | 0.005 (0.019) | 0.081 (0.153) | 0.924 (0.850) | 0.185 (0.166) |
| p-value | 0.000 (0.000) | 0.000 (0.040) | 0.000 (0.000) | 0.000 (0.000) | 0.000 (0.000) | (0.000) (0.000) | 0.000 (0.000) | 0.000 (0.000) |
| Average Performance Results for Small Sample Case (n1 = n2 = 15) | ||||||||
| Methods | TPR | FPR | TNR | FNR | MER | FDR | AUC | pAUC |
| t-test | 0.927 (0.144) | 0.003 (0.026) | 0.997 (0.974) | 0.073 (0.856) | 0.005 (0.051) | 0.089 (0.852) | 0.927 (0.144) | 0.185 (0.028) |
| Wilcoxon | 0.927 (0.893) | 0.003 (0.004) | 0.997 (0.996) | 0.073 (0.107) | 0.005 (0.007) | 0.089 (0.122) | 0.927 (0.893) | 0.185 (0.178) |
| SAM | 0.927 (0.144) | 0.003 (0.026) | 0.997 (0.974) | 0.073 (0.856) | 0.005 (0.051) | 0.089 (0.521) | 0.927 (0.144) | 0.185 (0.029) |
| Limma | 0.927 (0.145) | 0.003 (0.026) | 0.997 (0.974) | 0.073 (0.855) | 0.005 (0.051) | 0.089 (0.851) | 0.927 (0.145) | 0.185 (0.029) |
| WAD | 0.909 (0.012) | 0.006 (0.031) | 0.993 (0.969) | 0.090 (0.988) | 0.011 (0.059) | 0.102 (0.990) | 0.909 (0.011) | 0.181 (0.002) |
| RP | 0.927 (0.926) | 0.003 (0.003) | 0.997 (0.997) | 0.073 (0.074) | 0.005 (0.005) | 0.089 (0.090) | 0.927 (0.926) | 0.185 (0.185) |
| FCROS | 0.927 (0.927) | 0.003 (0.003) | 0.997 (0.997) | 0.073 (0.073) | 0.005 (0.005) | 0.089 (0.089) | 0.927 (0.927) | 0.185 (0.185) |
| Proposed | 0.927 (0.927) | 0.003 (0.003) | 0.997 (0.997) | 0.073 (0.073) | 0.005 (0.005) | 0.089 (0.089) | 0.927 (0.927) | 0.185 (0.185) |
| p-value | 0.352 (0.000) | 0.989 (0.000) | 0.999 (0.000) | 0.820 (0.000) | 0.983 (0.000) | 0.970 (0.000) | 0.352 (0.000) | 0.999 (0.000) |
| Performance Results for Sample Size n1 = n2 = 9 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Methods | TPR | TNR | FPR | FNR | FDR | MER | AUC | pAUC |
| t-test | 0.818 (0.217) | 0.979 (0.909) | 0.021 (0.091) | 0.182 (0.783) | 0.182 (0.783) | 0.038 (0.163) | 0.816 (0.216) | 0.161 (0.043) |
| Wilcoxon | 0.810 (0.540) | 0.978 (0.947) | 0.022 (0.053) | 0.190 (0.460) | 0.190 (0.460) | 0.040 (0.096) | 0.805 (0.534) | 0.158 (0.102) |
| SAM | 0.833 (0.215) | 0.981 (0.909) | 0.019 (0.091) | 0.167 (0.785) | 0.167 (0.785) | 0.035 (0.163) | 0.832 (0.214) | 0.165 (0.042) |
| LIMMA | 0.830 (0.217) | 0.980 (0.909) | 0.020 (0.091) | 0.170 (0.783) | 0.170 (0.783) | 0.035 (0.163) | 0.828 (0.216) | 0.164 (0.043) |
| WAD | 0.831 (0.297) | 0.980 (0.918) | 0.020 (0.082) | 0.169 (0.703) | 0.169 (0.703) | 0.035 (0.146) | 0.830 (0.284) | 0.165 (0.046) |
| RP | 0.824 (0.743) | 0.980 (0.970) | 0.020 (0.030) | 0.176 (0.257) | 0.176 (0.257) | 0.037 (0.053) | 0.822 (0.741) | 0.163 (0.146) |
| FCROS | 0.834 (0.799) | 0.981 (0.977) | 0.019 (0.023) | 0.166 (0.201) | 0.166 (0.201) | 0.035 (0.042) | 0.833 (0.798) | 0.166 (0.158) |
| Proposed | 0.837 (0.832) | 0.981 (0.981) | 0.019 (0.019) | 0.163 (0.168) | 0.163 (0.168) | 0.032 (0.035) | 0.837 (0.831) | 0.170 (0.165) |
| p-value | 0.807 (0.000) | 0.999 (0.000) | 0.999) (0.000) | 0.766 (0.000) | 0.766 (0.000) | 0.991 (0.000) | 0.610 (0.000) | 0.998 (0.000) |
| Average Performance Results for Small Sample Case n1 = n2 = 3 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Methods | TPR | TNR | FPR | FNR | FDR | MER | AUC | pAUC |
| t-test | 0.6939 (0.2253) | 0.9645 (0.9102) | 0.0355 (0.0898) | 0.3061 (0.7747) | 0.3061 (0.7747) | 0.0636 (0.1610) | 0.6888 (0.2234) | 0.1337 (0.0432) |
| Wilcoxon | 0.3405 (0.2238) | 0.9235 (0.9100) | 0.0765 (0.0900) | 0.6595 (0.7762) | 0.6595 (0.7762) | 0.1371 (0.1613) | 0.3278 (0.2178) | 0.0553 (0.0388) |
| SAM | 0.7701 (0.1456) | 0.9733 (0.9009) | 0.0267 (0.0991) | 0.2299 (0.8544) | 0.2299 (0.8544) | 0.0478 (0.1776) | 0.7683 (0.1445) | 0.1522 (0.0281) |
| LIMMA | 0.7675 (0.2068) | 0.9730 (0.9080) | 0.0270 (0.0920) | 0.2325 (0.7932) | 0.2325 (0.7932) | 0.0483 (0.1649) | 0.7659 (0.2060) | 0.1519 (0.0405) |
| WAD | 0.7639 (0.2850) | 0.9726 (0.9171) | 0.0274 (0.0829) | 0.2361 (0.7150) | 0.2361 (0.7150) | 0.0491 (0.1486) | 0.7627 (0.2715) | 0.1516 (0.0435) |
| RP | 0.7711 (0.1898) | 0.9735 (0.9060) | 0.0265 (0.0940) | 0.2289 (0.8102) | 0.2289 (0.8102) | 0.0476 (0.1684) | 0.7696 (0.1796) | 0.1527 (0.0277) |
| FCROS | 0.7685 (0.3853) | 0.9732 (0.9287) | 0.0268 (0.0713) | 0.2315 (0.6147) | 0.2315 (0.6147) | 0.0481 (0.1278) | 0.7671 (0.3757) | 0.1523 (0.0675) |
| Proposed | 0.7716 (0.7582) | 0.9735 (0.9720) | 0.0265 (0.0280) | 0.2284 (0.2418) | 0.2284 (0.2418) | 0.0475 (0.0502) | 0.7702 (0.7565) | 0.1529 (0.1499) |
| p-value | 0.000 (0.000) | 0.000 (0.000) | 0.000 (0.000) | 0.000 (0.000) | 0.000 (0.000) | 0.000 (0.000) | 0.000 (0.000) | 0.000 (0.000) |
| KEGG ID | Pathway Name | Adjusted p-Value |
|---|---|---|
| hsa00591 | Linoleic acid metabolism | 0.004 |
| hsa00983 | Drug metabolism-other enzymes | 0.006 |
| hsa00140 | Steroid hormone biosynthesis | 0.008 |
| hsa00830 | Retinol metabolism | 0.009 |
| hsa00982 | Drug metabolism-cytochrome P450 | 0.009 |
| hsa04976 | Bile secretion | 0.009 |
| hsa00980 | Metabolism of xenobiotics by cytochrome | 0.010 |
| hsa05204 | Chemical carcinogenesis | 0.010 |
| hsa01100 | Metabolic pathways | 0.010 |
| ID | Disease Name | Adjusted p-Value |
|---|---|---|
| umls:C0040479 | Torsades de Pointes | 0.0005 |
| umls:C0019196 | Hepatitis C | 0.0016 |
| umls:C0029463 | Osteosarcoma | 0.0041 |
| umls:C1458155 | Mammary Neoplasms | 0.040 |
| umls:C0033578 | Prostatic Neoplasms | 0.051 |
| ID | Name of the Drug | Adjusted p-Value |
|---|---|---|
| PA163522472 | darunavir | 7.11 × 10−4 |
| PA165111677 | Cremophor EL | 7.11 × 10−4 |
| PA165958385 | nilvadipine | 7.11 × 10−4 |
| PA448333 | alprazolam | 7.11 × 10−4 |
| PA449591 | felodipine | 7.11 × 10−4 |
| PA451753 | triazolam | 7.11 × 10−4 |
| PA164712364 | Androgen, progestogen and estrogen in combination | 8.53 × 10−4 |
| PA164713223 | Quinine and derivatives | 8.53 × 10−4 |
| PA164776964 | desloratadine | 8.53 × 10−4 |
| PA165983955 | acetaminophen glucuronide | 8.53 × 10−4 |
| DB01211 | Clarithromycin | 2.22 × 10−3 |
| DB00199 | Erythromycin | 3.11 × 10−3 |
| DB01267 | Paliperidone | 7.55 × 10−3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahjaman, M.; Rahman, M.R.; Islam, S.M.S.; Mollah, M.N.H. A Robust Approach for Identification of Cancer Biomarkers and Candidate Drugs. Medicina 2019, 55, 269. https://doi.org/10.3390/medicina55060269
Shahjaman M, Rahman MR, Islam SMS, Mollah MNH. A Robust Approach for Identification of Cancer Biomarkers and Candidate Drugs. Medicina. 2019; 55(6):269. https://doi.org/10.3390/medicina55060269
Chicago/Turabian StyleShahjaman, Md., Md. Rezanur Rahman, S. M. Shahinul Islam, and Md. Nurul Haque Mollah. 2019. "A Robust Approach for Identification of Cancer Biomarkers and Candidate Drugs" Medicina 55, no. 6: 269. https://doi.org/10.3390/medicina55060269
APA StyleShahjaman, M., Rahman, M. R., Islam, S. M. S., & Mollah, M. N. H. (2019). A Robust Approach for Identification of Cancer Biomarkers and Candidate Drugs. Medicina, 55(6), 269. https://doi.org/10.3390/medicina55060269