46,XX Testicular Disorder of Sex Development (DSD): A Case Report and Systematic Review


Abstract
1. Introduction
Case Report
2. Materials and Methods
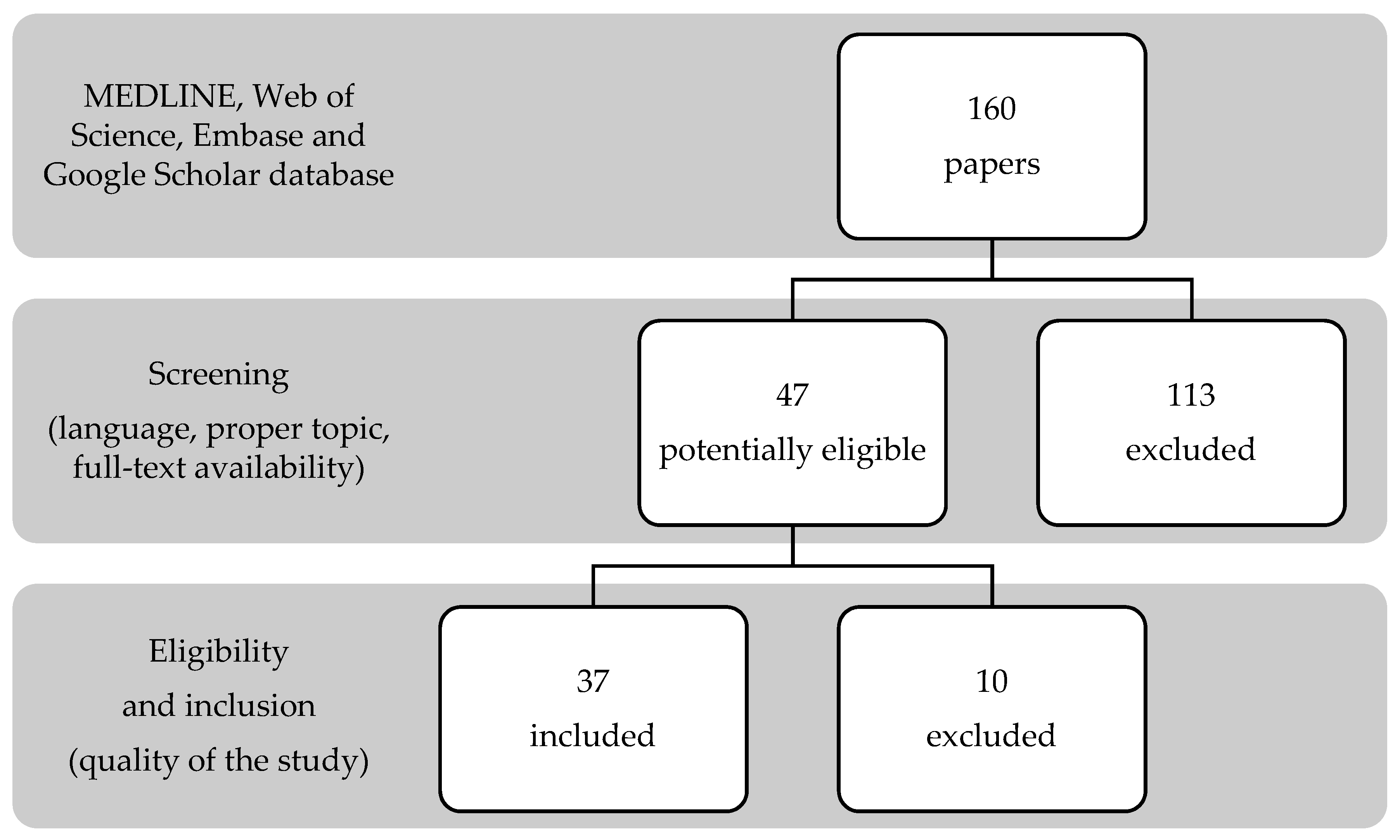
2.1. Search Strategy
2.2. Inclusion and Exclusion Criteria
2.3. Data Extraction
3. Results
3.1. Search Results
3.2. Synthesis of Results
4. Discussion
- DSD with atypical sex chromosome configurations, including Turner syndrome, Klinefelter syndrome, and conditions with 46,XX or 46,XY karyotypes;
- XY DSD characterized by 46,XY karyotype;
- XX DSD comprised of conditions characterized by 46,XX karyotype and androgen excess, such as congenital adrenal hyperplasia, P450 oxidoreductase deficiency, or exogenous causes [42].
- The hypothesis of target gene mutation [57]. It supposes the structural gene that determines human gender may be located in autosome, which is regulated by the inhibition of the X chromosome and the activation of the Y chromosome. 46,XX individuals, due to defects in the inhibition of the X chromosome, which results in spontaneous activation of the downstream gene in the absence of the SRY gene, transform into 46,XX males.
- The hypothesis of SOX9 gene (SRY box-related gene 9) overexpression. SOX are a large gene family, in which SOX9 is located in 17q24.3–q25.1 and homologous with SRY High-Mobility Group box (HMG)-box as high as 60%. Early studies confirmed that SOX9 was mainly involved in bone formation and the regulation of Sertoli cell differentiation [58], which was also expressed in the precursor cell of supporting cells with the SRY gene expression. In addition, the fact that SOX9 can be found in spinal animals and mammals, compared with the SRY gene which can be found only in mammals, might indicate that SOX9 was a more ancient gene involved in sex differentiation than the SRY gene. Huang et al. [59] reported the case of a SRY-negative 46,XX male patient, and chromosome analysis showed the existence of overlapping of the SOX9 gene. Malki et al. [60] found that prostaglandin D2 (PGD2) could induce the expression of SOX9 in a normal female rat gonad in vitro. Thus, up-regulation of SOX9 expression caused by chromosomal abnormalities or mediated by other bypass activation (e.g., PGD2) may result in the occurrence of SRY-negative 46,XX male patients.
- The hypothesis of Xp-Yp translocation. It supposes that the ends of the XY chromosomes’ abnormal exchange (Xp-Yp translocation) occurs during paternal sperm meiosis and results in X-type sperm containing the SRY gene, which could lead to 46,XX offspring when combined with eggs. PCR can detect the SRY gene, however, only to find it translocated in the X chromosome by FISH. In Brazil, Domenice et al. [61] found that 90% of 46,XX males carried Y chromosome material, including the SRY gene, in most cases. It indicated that the Y chromosome fragment containing the SRY gene translocation may be an important factor for the occurrence of SRY+ 46,XX male patients.
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nordenstrom, B.C.; Rohle, A.R.; Thyen, R.U. Hormone therapy and patient satisfaction with treatment, in a large cohort of diverse disorders of sex development. Clin. Endocrinol. 2018, 88, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Vilain, E. 46, XX Testicular Disorder of Sex Development. Gene Rev. Last Updat. 2009, 26, 38–41. [Google Scholar]
- Wang, T.; Bai, J.; Sakai, S.; Ohno, Y.; Ohno, H.T. Magnetotransport studies of AlGaN/GaN heterostructures grown on sapphire substrates: Effective mass and scattering time. Appl. Phys. Lett. 2000, 76, 2737–2739. [Google Scholar] [CrossRef]
- De la Chapelle, A.; Hortling, H.; Niemi, M. XX sex chromosome in a human male, first case. Acta Med. Scand. 1964, 175 (Suppl. 412), 25–28. [Google Scholar] [CrossRef]
- Guzman, J.M.P.; Navarro, H.P.; Mata, Q.M.; Lopez, P.C.; Ruiz, J.M.; Sanchiz, C.M.; Rodriguez, J.A.V. 46, XX Testicular Disorder of sex development. Case report. Arch. Esp. Urol. 2011, 64, 468–472. [Google Scholar]
- Gunes, S.; Asci, R.; Okten, G.; Atac, F.; Onat, O.E.; Ogur, G.; Aydin, O.; Ozcelik, T. Two males with SRY positive 46 XX testicular disorder of sex development. Syst. Biol. Reprod. Med. 2013, 59, 42–47. [Google Scholar] [CrossRef]
- Valetto, A.; Bertini, V.; Rapalini, E. A 46, XX SRY negative man with complete virilization and infertility as the main anomaly. Fertil. Steril. 2005, 83, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Lee, B.Y.; Oh, A.R.; Park, S.Y.; Lee, H.S.; Seo, J.T. Clinical, Hormonal, and Genetic Evaluation of Idiopathic Nonobstructive Azoospermia and Klinefelter Syndrome Patients. Cytogenet. Genome Res. 2018, 153, 190–197. [Google Scholar] [CrossRef]
- Xiao, B.; Ji, X.; Xing, Y.; Chen, Y.W. A rare case of 46, XX SRY negative male with approximately 74 kb duplication in a region upstream of SOX9. Eur. J. Med. Genet. 2013, 56, 695–698. [Google Scholar] [CrossRef]
- Queralt, R.; Madrigal, I.; Vallecillos, M.A.; Morales, C.; Ballesca, J.L.; Oliva, R.; Soler, A.; Sánchez, A.; Margarit, E. Atypical XX male with the SRY gene located at the long arm of chromosome 1 and a 1qter microdeletion. Am. J. Med. Genet. Part A 2008, 146, 1335–1340. [Google Scholar] [CrossRef]
- Baziz, M.; Hamouli-Said, Z.; Ratbi, I.; Habel, M.; Guaoua, S.; Sbiti, A.; Sefiani, A. Cytogenetic investigation in a group of ten infertile men with non-obstructive azoospermia: First Algerian 46, xx syndrome. Iran. J. Public Health 2016, 45, 739–747. [Google Scholar] [PubMed]
- Tomomasa, H. XX-male syndrome bearing the sex-determining region Y. Arch. Androl. 1999, 42, 89–96. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chung Jung, W.; Song, Y.M.; Sheu, W.H. Short stature in a 46 XX Male Adolescent. South Med. J. 1999, 92, 921–924. [Google Scholar]
- Wang, T.; Liu, J.H.; Yang, J.; Chen, J. 46, XX male sex reversal syndrome: A case report and review of the genetic basis. Andrologia 2009, 41, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, T.; Saleem, M.; Ahmed, A. 46 XX male: A case of sex reversal syndrome. J. Pak. Med. Assoc. 1998, 48, 19–20. [Google Scholar] [PubMed]
- Jain, M.; Veeramohan, V.; Chaudhary, I. The Sertoli cell only syndrome and glaucoma in a sex—Determining region Y (SRY) positive XX infertile male. J. Clin. Diagn. Res. 2013, 7, 1457–1459. [Google Scholar] [CrossRef] [PubMed]
- Yencilek, F.; Baykal, C. 46, XX male syndrome: A case report. Clin. Exp. Obs. Gyn. 2005, 32, 263–264. [Google Scholar]
- Pepene, C.E.; Coman, I.; Mihu, D.; Militaru, M. Infertility in a new 46, XX male with positive SRY confirmed by fluorescence in situ hybridization: A case report. Clin. Exp. Obs. Gynecol. 2008, 35, 299–300. [Google Scholar]
- Mustafa, O.A. 46, XX SRY—Negative man with infertility, and co existing with chronic autoimmune thyroiditis. Gynecol. Endocrinol. 2010, 26, 413–415. [Google Scholar] [CrossRef]
- Majzoub, A.; Arafa, M.; Starks, C.; Elbardisi, H.; Al Said, S.; Sabanegh, E. 46 XX karyotype during male fertility evaluation; case series and literature review. Asian J. Androl. 2017, 19, 168. [Google Scholar] [CrossRef]
- Onrat, S.T.; Söylemez, Z. 46, XX der (15), t (Y;15) (q12; p11) karyotype in an azoospermic male. Indian J. Hum. Genet. 2012, 18, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Hado, H.S.; Helmy, S.W.; Klemm, K.; Miller, P. XX male: A rare cause of short stature, infertility, gynaecomastia and carcinoma of the breast. Int. J. Clin. Pract. 2003, 57, 844–845. [Google Scholar] [PubMed]
- Rigola, M.A.; Carrera, M.; Ribas, I.; Egozcue, J.; Miro, R.; Fuster, C. A comparative genomic hybridization study in a 46, XX male. Fertil. Steril. 2002, 78, 186–188. [Google Scholar] [CrossRef]
- Dauwerse, J.G.; Hansson, K.B.; Brouwers, A.A.; Peters, D.J. An XX male with the sex determining region Y gene inserted in the long arm of chromosome 16. Fertil. Steril. 2006, 86, 463. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.A. A case report of an incidental finding of a 46, XX, SRY negative male with masculine phenotype during standard fertility workup with review of the literature and proposed immediate and long term management guidance. Fertil. Steril. 2013, 99, 1273–1276. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Chen, G.; Huang, J.; Bai, Q.; Zhao, N.; Shao, M.; Jiao, L.; Wei, Y.; Chang, L.; Li, D.; et al. Clinical, cytogenetic, and molecular analysis with 46, XX male sex reversal syndrome: Case reports. J. Assist. Reprod. Genet. 2013, 30, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, A.A. 46, XX man with SRY gene translocation: Cytogenetic characteristics, clinical features and management. Am. J. Med. Sci. 2008, 335, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Minor, A.; Mohammed, F.; Farouk, A.; Hatakeyama, C.; Johnson, K.; Chow, V.; Ma, S. Genetic characterization of two 46, XX males without gonadal ambiguities. J. Assist. Reprod. Genet. 2008, 25, 547–552. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rajender, S.; Rajani, V.; Gupta, N.J.; Chakravarty, B.; Singh, L.; Thangaraj, K. SRY negative 46, XX male with normal genitals, complete masculinization and infertility. Mol. Hum. Reprod. 2006, 12, 341–346. [Google Scholar] [CrossRef][Green Version]
- Tan, T.T. Primary infertility in a phenotypic male with 46XX chromosomal constitution. Postgrad. Med. J. 1983, 69, 315–317. [Google Scholar] [CrossRef][Green Version]
- Zakharia, G. Sex reversal syndrome (XX male). Urology 1990, 36, 322–324. [Google Scholar] [CrossRef]
- Chiang, H.S.; Wu, Y.N.; Wu, C.C. Cytogenic and molecular analyses of 46, XX male syndrome with clinical comparison to other groups with testicular azoospermia of genetic origin. J. Formos. Med. Assoc. 2013, 112, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.Y.; Li, N.; Li, W.W.; Li, T.F.; Zhang, C.; Cui, Y.X.; Xia, X.Y.; Zhai, J.S. Clinical, molecular and cytogenetic analysis of 46, XX testicular disorder of sex development with SRY positive. BMC Urol. 2014, 14, 70. [Google Scholar] [CrossRef]
- Chernykh, V.B.; Kurilo, L.F.; Shilova, N.V.; Zolotukhina, T.V.; Ryzhkova, O.P.; Bliznetz, E.A.; Polyakov, A.V. X chromosomal mosaicism in a 46, XX male. Sex Dev. 2009, 3, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Walzak, M.P.; Sanger, W.G. A possible etiology of the infertile 46XX male subject. J. Urol. 1983, 130, 154–156. [Google Scholar] [CrossRef]
- Castineyra, G.; Copelli, S. 46, XX male: Clinical, hormonal/genetic findings. Arch. Androl. 2002, 48, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Fuse, H.; Satomi, S.; Kazama, T.; Katayama, T.; Nagabuchi, S.; Tamura, T.; Nakahori, Y.; Nakagome, Y. DNA hybridization study using Y specific probes in an XX male. Andrologia 1991, 23, 237–239. [Google Scholar] [CrossRef]
- Pais, V.M. Infertility in an XX male. J. Urol. 1977, 118, 690–691. [Google Scholar] [CrossRef]
- Wegner, R.D. Clinical, cytological, and biochemical investigations in a case of an XX male. Andrologia 1983, 15, 253–258. [Google Scholar] [CrossRef]
- Micic, S.; Micic, M. An example of a 46, XX infertile man and his permanent tooth sizes. Int. J. Fertil. 1983, 28, 165–168. [Google Scholar]
- Matthews, C.D.; Ford, J. The XX male. Clinical and theoretical aspects. Reprod. Fertil. 1983, 2, 207–215. [Google Scholar]
- Lee, P.A.; Nordenström, A.; Houk, C.P.; Ahmed, S.F.; Auchus, R.; Baratz, A.; Baratz Dalke, K.; Liao, L.M.; Lin-Su, K.; Looijenga, L.H., 3rd; et al. Global Disorders of Sex Development Update since 2006: Perceptions, Approach and Care. Horm. Res. Paediatr. 2016, 85, 158–180. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Huang, T.H.; Jiang, X.W. 46, XX male sex reversal syndrome. Asian J. Androl. 2004, 6, 165–167. [Google Scholar] [PubMed]
- Kolon, T.T. Bases moleculares de los trastornos intersexuales. AUA Updat. Ser. 2001, 34, 39–50. [Google Scholar]
- Zenteno-Ruiz, J.C.; Kofman-Alfaro, S.; Méndez, J.P. 46, XX sex reversal. Arch. Med. Res. 2001, 32, 559–566. [Google Scholar] [CrossRef]
- Lopez, M.; Torres, L.; Mendez, J.P.; Cervantes, A.; Alfaro, G.; Pérez-Palacios, G.; Erickson, R.P.; Kofman-Alfaro, S. SRY alone can induce normal male sexual differentiation. Am. Med. Genet. 1995, 55, 356–358. [Google Scholar] [CrossRef]
- Bouayed Abdelmoula, N.; Portnoi, M.F.; Keskes, L.; Recan, D.; Bahloul, A.; Boudawara, T.; Saad, A.; Rebai, T. Skewed X-chromosome inactivation pattern in SRY positive XX maleness: A case report and review of literature. Ann. Genet. 2003, 46, 11–18. [Google Scholar] [CrossRef]
- Kusz, K.; Kotecki, M.; Wodja, A.; Szarras-Czapnik, M.; Latos-Bielenska, A.; Warenik-Szymankiewicz, A.; Ruszczynska-Wolskab, A.; Jaruzelskaa, J. Incomplete masculinisation of XX subjects carrying the SRY gene on an inactive X chromosome. J. Med. Genet. 1999, 36, 452–456. [Google Scholar]
- Conte, F.A.; Grumbach, M.M. Disorders of Sex Determination and Differentiation. Greenspan’s Basic & Clinical Endocrinology, 9th ed.; David, G., Dolores, S., Eds.; McGraw-Hill: New York, NY, USA, 2011. [Google Scholar]
- Ferguson-Smith, N.; Cooke, M.A.; Affara, A.; Boyd, E.; Tolmie, J.L. Genotype- phenotype correlations in XX males and their bearing on current theories of sex determination. Hum. Genet. 1990, 84, 198–202. [Google Scholar] [CrossRef]
- She, Z.Y.; Yang, W.X. Sry and SoxE genes: How they participate in mammalian sex determination and gonadal development? In Seminars in Cell & Developmental Biology; Academic Press: New York NY, USA, 2017; Volume 63, pp. 13–22. [Google Scholar]
- Cox, M.C.; Maffei, L.; Buffolino, S.; Del Poeta, G.; Venditti, A.; Cantonetti, M.; Aronica, G.; Aquilina, P.; Masi, M.; Amadori, S. A Comparative Analysis of FISH, RT-PCR, and Cytogenetics for the Diagnosis of bcr-abl Positive Leukemias. Am. J. Clin. Pathol. 1998, 109, 24–31. [Google Scholar] [CrossRef]
- Petit, C.; de la Chapelle, A.; Levilliers, J.; Castillo, S.; Noë, B.; Weissenbach, J. An abnormal XY interchange accounts for most but not all the cases of human XX maleness. Cell 1997, 49, 595–602. [Google Scholar] [CrossRef]
- Mizuno, K.; Kojima, Y.; Kamisawa, H.; Moritoki, Y.; Nishio, H.; Kohri, K.; Hayashi, Y. Gene expression profile during testicular development in patients with SRY-negative 46, XX testicular disorder of sex development. Urology 2013, 82, 1453. [Google Scholar] [CrossRef] [PubMed]
- Ergun-Longmire, N.M.; Vinci, G.; Alonso, L.; Matthew, S.; Tansil, S.; Lin-Su, K.; McElreavey, K. Clinical, hormonal and cytogenetic evaluation of 46, XX males and review of the literature. J. Pediatr. Endocrinol. Metab. 2005, 18, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Hazama, M.; Kondo, K. Male infertility with chromosomal abnormalities. II. XX-male syndrome. Hinyokika Kiyo Acta Urologica Japonica 1987, 33, 193–203. [Google Scholar] [PubMed]
- Liu, L.; Feng, L.N. The phenotype and genetics of 46, XX male syndrome. Endocrinol. Foreign Med. Sci. 2005, 25, 283–285. [Google Scholar]
- Foster, J.W.; Dominguez-Steglich, M.A.; Guioli, S.; Kowk, G.; Weller, P.A.; Stevanovic, M.; Weissenbach, J.; Mansour, S.; Young, I.D.; Goodfellow, P.N.; et al. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRY- related gene. Nature 1994, 372, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Wang, S.; Ning, Y.; Lamb, A.N. Autosomal XX sex reversal caused by duplication of SOX9. Am. J. Med. Genet. 1999, 87, 349–353. [Google Scholar] [CrossRef]
- Malki, S.; Nef, S.; Notarnicola, C.; Thevenet, L.; Gasca, S.; Méjean, C.; Berta, P.; Poulat, F. Prostaglandin D2 induces nuclear import of the sex-determining factor SOX9 via its cAMP-PKA phosphorylation. EMBO J. 2005, 24, 1798–1809. [Google Scholar] [CrossRef]
- Domenice, S.; Correa, R.V.; Costa, E.M.; Nishi, M.Y.; Vilain, E.; Arn-Hold, I.J. Mutations in the SRY, DAX1, SF1 and WNT4 genes in Brazilian sex-reversed patients. Braz. J. Med. Biol. Res. 2004, 37, 145–150. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Authors | Age Years | Weight kg | Height cm | HD | GM | Testes Volume | Penis Size | Pubic Hair | ED | Libido |
|---|---|---|---|---|---|---|---|---|---|---|
| Guzman et al. [5] | 20 | 76 | 169 | Normal | No | Small | Normal | Normal | No | Normal |
| Gunes et al. [6] | 30 | 70 | 155 | Poor | Yes | Small | Normal | Normal | No | Normal |
| Gunes et al. [6] | 16 | 65 | 152 | Normal | No | Small | Normal | Normal | No | Normal |
| Valetto et al. [7] | 35 | 48 | 152 | Normal | No | NA | Normal | NA | No | Normal |
| Kim et al. [8] | 29 | 62 | 165 | Normal | No | NA | NA | NA | Yes | NA |
| Xiao et al. [9] | 27 | NA | 170 | NA | NA | NA | Small | NA | No | NA |
| Queralt et al. [10] | 31 | 58 | 170 | Normal | No | NA | NA | NA | NA | NA |
| Baziz et al. [11] | 44 | NA | NA | Normal | Yes | Small | Normal | NA | No | Normal |
| Tomomasa et al. [12] | 25 | 55 | 177 | Normal | No | Small | NA | Normal | NA | Normal |
| Chung Jung et al. [13] | 17 | NA | 154 | Normal | No | Small | Normal | Normal | No | Normal |
| Wang et al. [14] | 20 | NA | NA | Poor | No | Small | Normal | Inverted | No | Normal |
| Ahsan T et al. [15] | 24 | NA | NA | Poor | Yes | Small | Normal | Normal | No | Normal |
| Jain et al. [16] | 38 | 63 | 162 | Normal | Yes | NA | Normal | NA | Yes | NA |
| Yencilek et al. [17] | 26 | 72 | 165 | Normal | No | NA | Small | NA | No | NA |
| Pepene et al. [18] | 28 | 65 | 167 | Normal | Yes | Small | Normal | NA | No | Normal |
| Mustafa et al. [19] | 30 | 75 | 170 | Normal | Yes | NA | Normal | NA | NA | Normal |
| Majzoub et al. [20] | 40 | 84 | 175 | Normal | No | NA | Normal | NA | No | Normal |
| Majzoub et al. [20] | 31 | NA | NA | Normal | No | NA | Normal | NA | No | Normal |
| Majzoub et al. [20] | 35 | NA | NA | Poor | Yes | NA | Normal | NA | Yes | Normal |
| Majzoub et al. [20] | 39 | 74 | 160 | Normal | No | Small | Normal | NA | No | Normal |
| Majzoub et al. [20] | 29 | 77 | 181 | Normal | No | NA | Normal | NA | No | Normal |
| Majzoub et al. [20] | 32 | 86 | 170 | Normal | Yes | NA | Normal | NA | No | Normal |
| Onrat et al. [21] | 23 | NA | NA | Normal | No | Small | Normal | NA | No | Normal |
| Hado et al. [22] | 76 | NA | 157 | Normal | Yes | NA | NA | NA | No | Normal |
| Rigola et al. [23] | 33 | NA | NA | Normal | No | NA | Normal | NA | NA | NA |
| Dauwerse et al. [24] | 61 | NA | 171 | NA | No | Small | Normal | NA | NA | NA |
| Ryan et al. [25] | 40 | NA | NA | Poor | No | NA | 3.6 | NA | No | NA |
| Gao et al. [26] | NA | NA | 163 | NA | NA | NA | NA | NA | NA | NA |
| Gao et al. [26] | NA | NA | 163 | NA | NA | NA | NA | NA | NA | NA |
| Gao et al. [26] | NA | NA | 162 | NA | NA | NA | NA | NA | NA | NA |
| Gao et al. [26] | NA | NA | 161 | NA | NA | NA | NA | NA | NA | NA |
| Gao et al. [26] | NA | NA | 158 | NA | NA | NA | NA | NA | NA | NA |
| Gao et al. [26] | NA | NA | 162 | NA | NA | NA | NA | NA | NA | NA |
| Gao et al. [26] | NA | NA | 162 | NA | NA | NA | NA | NA | NA | NA |
| Gao et al. [26] | NA | NA | 161 | NA | NA | NA | NA | NA | NA | NA |
| Gao et al. [26] | NA | NA | 160 | NA | NA | NA | NA | NA | NA | NA |
| Gao et al. [26] | NA | NA | 160 | NA | NA | NA | NA | NA | NA | NA |
| Gao et al. [26] | NA | NA | 161 | NA | NA | NA | NA | NA | NA | NA |
| Rizvi et al. [27] | 33 | 85.8 | 177 | NA | NA | NA | Normal | NA | No | NA |
| Minor et al. [28] | 24 | NA | NA | NA | Yes | NA | NA | NA | NA | NA |
| Rajender et al. [29] | 34 | 64 | 156 | Normal | No | NA | Normal | NA | NA | NA |
| Tan et al. [30] | NA | NA | 176 | Normal | Yes | NA | Small | NA | No | NA |
| Zakharia et al. [31] | 65 | 65 | 165 | NA | Yes | NA | Normal | NA | No | NA |
| Chiang et al. [32] | 33 | NA | NA | NA | NA | NA | NA | NA | No | NA |
| Chiang et al. [32] | 34 | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Chiang et al. [32] | 52 | NA | NA | NA | NA | NA | Small | NA | NA | NA |
| Wu et al. [33] | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Wu et al. [33] | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Wu et al. [33] | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Wu et al. [33] | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Wu et al. [33] | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| Chernykh et al. [34] | 37 | 74 | 160 | Poor | Yes | NA | NA | NA | NA | NA |
| Butler et al. [35] | 31 | 72 | 169 | Normal | Yes | NA | Normal | NA | No | NA |
| Castineyra et al. [36] | 28 | NA | 180 | Normal | Yes | NA | NA | NA | NA | NA |
| Castineyra et al. [36] | 35 | NA | 170 | Normal | NA | Small | NA | NA | NA | NA |
| Castineyra et al. [36] | 28 | NA | 160 | Poor | NA | NA | NA | NA | NA | NA |
| Castineyra et al. [36] | 39 | NA | 174 | Poor | NA | NA | NA | NA | NA | NA |
| Castineyra et al. [36] | 24 | NA | 172 | Normal | NA | NA | NA | NA | NA | NA |
| Fuse et al. [37] | 30 | 90 | 172 | Normal | No | NA | NA | NA | NA | NA |
| Pais et al. [38] | 29 | 82 | 170 | Normal | Yes | NA | Small | NA | No | NA |
| Wegner et al. [39] | 35 | 81 | 167 | Normal | No | NA | Normal | NA | NA | NA |
| Micic et al. [40] | 25 | 63 | 171 | Poor | No | NA | NA | NA | No | NA |
| Matthews et al. [41] | 27 | 68 | 166 | Poor | No | NA | NA | NA | NA | |
| Our case | 36 | 74 | 165 | NA | Yes | Small | Normal | NA | No | Normal |
| Authors | FSH mIU/mL | LH mIU/mL | TT ng/mL | E2 pg/mL | PRL ng/mL |
|---|---|---|---|---|---|
| Guzman et al. [5] | 27.9 | 16.5 | 2.3 | 24.8 | 24.5 |
| Gunes et al. [6] | 37.88 | 18.96 | 0.51 | 17.57 | 17.54 |
| Gunes et al. [6] | 41.05 | 14.55 | 2.16 | 32 | 24.11 |
| Valetto et al. [7] | 23.9 | 17.7 | 3.06 | NA | 7.13 |
| Kim et al. [8] | 76 | 41 | 1.79 | NA | NA |
| Xiao et al. [9] | 47 | 18.7 | 1.80 | NA | 14.6 |
| Queralt et al. [10] | 62.2 | 25.8 | 3.23 | 17 | NA |
| Baziz et al. [11] | 51 | 11.71 | NA | NA | NA |
| Tomomasa et al. [12] | 19.7 | 10.3 | 4.28 | NA | NA |
| Chung Jung et al. [13] | 25.1 | 11.4 | 4.3 | 30.1 | 16.8 |
| Wang et al. [14] | 77.5 | 40.75 | 4.64 | NA | 15.59 |
| Ahsan T et al. [15] | 35 | 21 | 1.8 | NA | NA |
| Jain et al. [16] | 76.6 | 36.3 | 1.20 | NA | NA |
| Yencilek et al. [17] | 45.6 | 48.9 | 2.70 | NA | 9.4 |
| Pepene et al. [18] | 43.9 | 25.3 | 3.33 | NA | NA |
| Mustafa et al. [19] | NA | 40.7 | 2.11 | 16.6 | 8.5 |
| Majzoub et al. [20] | 38 | 12 | 3.35 | 29 | 13.6 |
| Majzoub et al. [20] | 14 | 6 | 1.29 | 25 | 3.2 |
| Majzoub et al. [20] | 10 | 23 | 0.74 | 5.7 | NA |
| Majzoub et al. [20] | 28 | 15 | 0.74 | NA | NA |
| Majzoub et al. [20] | 13.4 | 12 | 2.46 | 19 | NA |
| Majzoub et al. [20] | 29.7 | 16.9 | 0.95 | NA | 12 |
| Onrat et al. [21] | 9.95 | 17.3 | 0.20 | NA | NA |
| Hado et al. [22] | 27.8 | 21 | 2.59 | NA | NA |
| Rigola et al. [23] | NA | NA | NA | NA | NA |
| Dauwerse et al. [24] | 13 | 10 | 3.25 | 31.3 | NA |
| Ryan et al. [25] | 1 | NA | NA | 10 | NA |
| Gao et al. [26] | 93.6 | 19.4 | 3.08 | 33 | 17.9 |
| Gao et al. [26] | 24.7 | 14.4 | 2.77 | 43 | 18.5 |
| Gao et al. [26] | NA | NA | 1.29 | NA | NA |
| Gao et al. [26] | 81.6 | 27.7 | 1.37 | 19.8 | 22.9 |
| Gao et al. [26] | 13.1 | 3.61 | 2.44 | 34 | 9.67 |
| Gao et al. [26] | 54.7 | 19.4 | 1.72 | 27 | 10.08 |
| Gao et al. [26] | 37.1 | 16.5 | 3.19 | 28 | 9.88 |
| Gao et al. [26] | 43 | 33.9 | 2.16 | 22 | 7.28 |
| Gao et al. [26] | 72 | 34.6 | 3.36 | 19.8 | 10 |
| Gao et al. [26] | 49 | 26.8 | 1.80 | 19.8 | 15.8 |
| Gao et al. [26] | 87.7 | 31.4 | 5.21 | 30.5 | 49.6 |
| Rizvi et al. [27] | 46 | 23 | 2.07 | NA | NA |
| Minor et al. [28] | 55.4 | 28.4 | 0.119 | NA | NA |
| Rajender et al. [29] | 25.8 | 15.8 | 5.8 | NA | NA |
| Tan et al. [30] | 21 | 34 | 2.63 | 25 | NA |
| Zakharia et al. [31] | 72 | 61 | 2.40 | NA | 16.3 |
| Chiang et al. [32] | 46.5 | 17.6 | 2.03 | NA | 27.05 |
| Chiang et al. [32] | 54.3 | 19.6 | 2.17 | NA | 8.15 |
| Chiang et al. [32] | 64.3 | 20.2 | 1.44 | NA | 16.08 |
| Wu et al. [33] | 35.5 | 13.8 | 1.95 | 30.5 | 4.6 |
| Wu et al. [33] | 29.2 | 12.9 | 1.55 | 19.1 | 3.6 |
| Wu et al. [33] | 45.9 | 25.1 | 2.56 | 26.7 | 7.8 |
| Wu et al. [33] | 33.7 | 22.3 | 2.41 | 29.1 | 10.9 |
| Wu et al. [33] | 31.4 | 19.6 | 2.01 | 22.1 | 7.8 |
| Chernykh et al. [34] | 26.9 | 13.5 | 2.90 | NA | NA |
| Butler et al. [35] | 51 | NA | 4.77 | NA | NA |
| Castineyra et al. [36] | 50 | 16 | 3.00 | 28 | 14 |
| Castineyra et al. [36] | 3.5 | 6.2 | 7.00 | 38 | 3.4 |
| Castineyra et al. [36] | 21 | 5.2 | 1.40 | 19 | 8.1 |
| Castineyra et al. [36] | 6.7 | 4.2 | 5.60 | 30 | 6.2 |
| Castineyra et al. [36] | 45 | 40 | 3.00 | 20 | 5.4 |
| Fuse et al. [37] | 47 | 60 | 1.60 | NA | NA |
| Pais et al. [38] | 53 | 45 | 2.67 | NA | NA |
| Wegner et al. [39] | 23.7 | 37.1 | 6.30 | NA | 3.8 |
| Micic et al. [40] | 31 | 18 | 3.19 | 47 | 6.8 |
| Matthews et al. [41] | 46 | 19 | 2.82 | 33 | 9.87 |
| Our case | 24.7 | 9.4 | 2.7 | 14 | 12.2 |
| Authors | Presence of SRY | Location of SRY |
|---|---|---|
| Guzman et al. [5] | + | NA |
| Gunes et al. [6] | + | NA |
| Gunes et al. [6] | + | NA |
| Valetto et al. [7] | NA | NA |
| Kim et al. [8] | NA | NA |
| Xiao et al. [9] | − | NA |
| Queralt et al. [10] | + | NA |
| Baziz et al. [11] | + | NA |
| Tomomasa et al. [12] | + | NA |
| Chung Jung et al. [13] | + | NA |
| Wang et al. [14] | + | NA |
| Ahsan T et al. [15] | NA | NA |
| Jain et al. [16] | + | NA |
| Yencilek et al. [17] | NA | NA |
| Pepene et al. [18] | + | NA |
| Mustafa et al. [19] | − | NA |
| Majzoub et al. [20] | + | NA |
| Majzoub et al. [20] | + | NA |
| Majzoub et al. [20] | + | NA |
| Majzoub et al. [20] | − | NA |
| Majzoub et al. [20] | + | NA |
| Majzoub et al. [20] | + | NA |
| Onrat et al. [21] | + | NA |
| Hado et al. [22] | + | NA |
| Rigola et al. [23] | + | Xp |
| Dauwerse et al. [24] | + | 16q |
| Ryan et al. [25] | − | NA |
| Gao et al. [26] | + | Xp |
| Gao et al. [26] | + | Xp |
| Gao et al. [26] | + | Xp |
| Gao et al. [26] | + | Xp |
| Gao et al. [26] | + | Xp |
| Gao et al. [26] | + | Xp |
| Gao et al. [26] | + | Xp |
| Gao et al. [26] | + | Xp |
| Gao et al. [26] | + | Xp |
| Gao et al. [26] | + | Xp |
| Gao et al. [26] | + | Xp |
| Rizvi et al. [27] | + | Xp |
| Minor et al. [28] | + | Xp |
| Rajender et al. [29] | − | NA |
| Tan et al. [30] | NA | NA |
| Zakharia et al. [31] | NA | NA |
| Chiang et al. [32] | + | Xp |
| Chiang et al. [32] | + | Xp |
| Chiang et al. [32] | − | NA |
| Wu et al. [33] | + | Xp |
| Wu et al. [33] | + | Xp |
| Wu et al. [33] | + | Xp |
| Wu et al. [33] | + | Xp |
| Wu et al. [33] | + | Xp |
| Chernykh et al. [34] | + | Xp |
| Butler et al. [35] | + | Xp |
| Castineyra et al. [36] | + | Xp |
| Castineyra et al. [36] | + | Xp |
| Castineyra et al. [36] | + | Xp |
| Castineyra et al. [36] | + | Xp |
| Castineyra et al. [36] | + | Xp |
| Fuse et al. [37] | + | Xp |
| Pais et al. [38] | + | Xp |
| Wegner et al. [39] | + | Xp |
| Micic et al. [40] | NA | NA |
| Matthews et al. [41] | + | Xp |
| Our case | + | Xp |
| Patients | HD% Normal | GM% No | Penis size% Normal | ED% No | FSH mIU/mL Mean (SD) | LH mIU/mL Mean (SD) | TT ng/mL Mean (SD) | PRL ng/mL Mean (SD) |
|---|---|---|---|---|---|---|---|---|
| SRY+ | 73.3 | 55.2 | 90.9 | 90.0 | 38.63 (20.85) | 29.40 (19.09) | 2.64 (1.49) | 12.93 (9.32) |
| SRY− | 66.7 | 57.1 | 60.0 | 85.7 | 51.74 (28.14) | 21.63 (9.56) | 2.49 (1.79) | 14.27 (4.07) |
| p = 0.422 | p = 0.596 | p = 0.144 | p = 0.823 | p = 0.195 | p = 0.326 | p = 0.819 | p = 0.781 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terribile, M.; Stizzo, M.; Manfredi, C.; Quattrone, C.; Bottone, F.; Giordano, D.R.; Bellastella, G.; Arcaniolo, D.; De Sio, M. 46,XX Testicular Disorder of Sex Development (DSD): A Case Report and Systematic Review. Medicina 2019, 55, 371. https://doi.org/10.3390/medicina55070371
Terribile M, Stizzo M, Manfredi C, Quattrone C, Bottone F, Giordano DR, Bellastella G, Arcaniolo D, De Sio M. 46,XX Testicular Disorder of Sex Development (DSD): A Case Report and Systematic Review. Medicina. 2019; 55(7):371. https://doi.org/10.3390/medicina55070371
Chicago/Turabian StyleTerribile, Marco, Marco Stizzo, Celeste Manfredi, Carmelo Quattrone, Francesco Bottone, Dario Ranieri Giordano, Giuseppe Bellastella, Davide Arcaniolo, and Marco De Sio. 2019. "46,XX Testicular Disorder of Sex Development (DSD): A Case Report and Systematic Review" Medicina 55, no. 7: 371. https://doi.org/10.3390/medicina55070371
APA StyleTerribile, M., Stizzo, M., Manfredi, C., Quattrone, C., Bottone, F., Giordano, D. R., Bellastella, G., Arcaniolo, D., & De Sio, M. (2019). 46,XX Testicular Disorder of Sex Development (DSD): A Case Report and Systematic Review. Medicina, 55(7), 371. https://doi.org/10.3390/medicina55070371