Assessment of Drugs Toxicity and Associated Biomarker Genes Using Hierarchical Clustering

Abstract
:1. Introduction
2. Methods and Materials
2.1. Data Processing
2.2. Hierarchical Clustering (HC) Algorithms and Distance Measures
2.2.1. Single Linkage
2.2.2. Complete Linkage
2.2.3. Average Linkage
2.2.4. Centroid
2.2.5. Median
2.2.6. Ward’s Algorithm
2.2.7. Distance Measures for HC
2.3. Selection of the Suitable Combination of Distance and HC Method
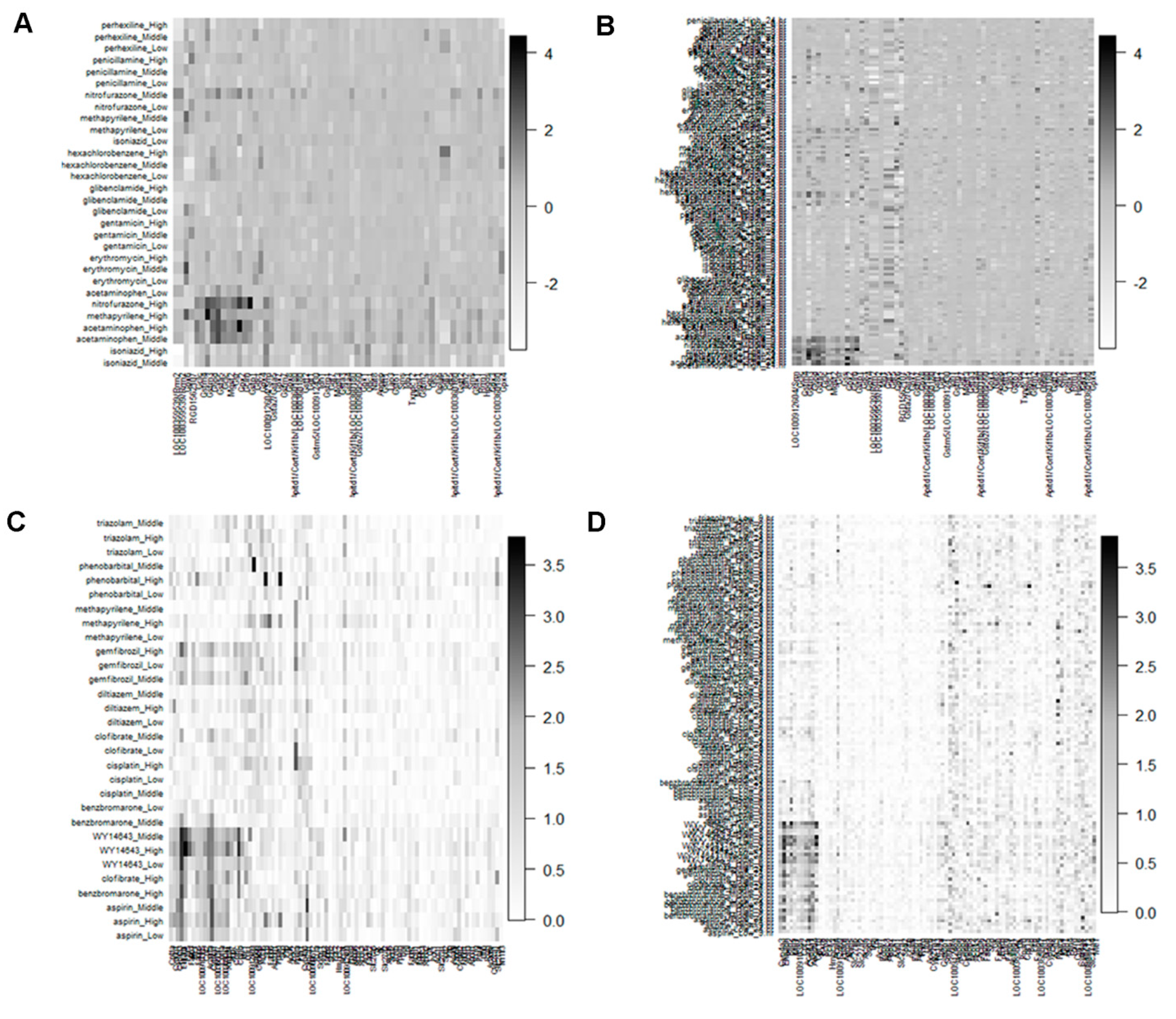
2.4. Co-Clustering between Genes and DDs and Detection of Toxic DDs and Associated Biomarker Genes Using HC
- Step 1:
- Fix the number of clusters in the genes as well as in DDs observing the dendrogram produced by HC according to the researchers’ interest.
- Step 2:
- Take absolute of the FCGE values within intersection areas for all pairs of genes and DDs clusters to give them equal weight in average calculations. Since the FCGE value for upregulated and downregulated genes consists of positive and negative expression values, respectively.
- Step 3:
- Compute the average of the absolute FCGE value for intersection areas of all pairs of genes and DDs clusters.
- Step 4:
- Rank the average FCGE values (computed in step 3) and the respective genes and DDs clusters simultaneously.
- Step 5:
- Assign cluster numbers for genes and DDs newly, based on the ranked average FCGE values which we get from step 4. For example, the gene and DD cluster intersection which produces the largest average FCGE value; we assign both of these gene and DD clusters as cluster 1. Simultaneously, the genes and DDs in cluster 1 together with form co-cluster 1. Similarly, we assign both of the gene and DD cluster as cluster 2 which produces the second largest average FCGE value and they form co-cluster 2 accordingly.
2.5. Real TGP Datasets to Investigate Clustering Performance
3. Results
3.1. Selection of Suitable Combination of Distance and HC Methods
3.2. Detection of Biomarker Genes and Their Regulatory DDs from the Co-Clusters
4. Discussion
5. Conclusions
- Detect the biomarker genes and the regulatory (associated) DDs simultaneously.
- The method safe time, since it requires less time for preparing results compared to the other EM based iterative co-clustering methods.
- The results produced by the method conform to the literature and database results.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Waters, M.D.; Fostel, J.M. Toxicogenomics and systems toxicology: Aims and prospects. Nat. Rev. Genet. 2004, 5, 936–948. [Google Scholar] [CrossRef] [PubMed]
- Aardema, M.J.; MacGregor, J.T. Toxicology and genetic toxicology in the new era of “toxicogenomics”: Impact of “-omics” technologies. Mutat. Res. 2002, 499, 13–25. [Google Scholar] [CrossRef]
- Afshari, C.A. Perspective: Microarray Technology, Seeing More Than Spots. Endocrinology 2002, 143, 1983–1989. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, R.; Friend, S.H. Toxicogenomics and drug discovery: Will new technologies help us produce better drugs? Nat. Rev. Drug Discov. 2001, 1, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Zacharewski, T.R.; Fielden, M.R. Challenges and Limitations of Gene Expression Profiling in Mechanistic and Predictive Toxicology. Toxicol. Sci. 2001, 60, 6–10. [Google Scholar] [Green Version]
- Olden, K.; Guthrie, J. Genomics: Implications for toxicology. Mutat. Res. 2001, 473, 3–10. [Google Scholar] [CrossRef]
- Knall, C.M.; Davis, J.W.; Paules, R.S.; Boggs, S.E.; Afshari, C.A.; Burchiel, S.W. Analysis of Genetic and Epigenetic Mechanisms of Toxicity: Potential Roles of Toxicogenomics and Proteomics in Toxicology. Toxicol. Sci. 2001, 59, 193–195. [Google Scholar]
- Uehara, T.; Hirode, M.; Ono, A.; Kiyosawa, N.; Omura, K.; Shimizu, T.; Mizukawa, Y.; Miyagishima, T.; Nagao, T.; Urushidani, T. A toxicogenomics approach for early assessment of potential non-genotoxic hepatocarcinogenicity of chemicals in rats. Toxicology 2008, 250, 15–26. [Google Scholar] [CrossRef]
- Igarashi, Y.; Nakatsu, N.; Yamashita, T.; Ono, A.; Ohno, Y.; Urushidani, T.; Yamada, H. Open TG-GATEs: A large-scale toxicogenomics database. Nucleic Acids Res. 2015, 43, D921–D927. [Google Scholar] [CrossRef]
- Yildirimman, R.; Brolén, G.; Vilardell, M.; Eriksson, G.; Synnergren, J.; Gmuender, H.; Kamburov, A.; Ingelman-Sundberg, M.; Castell, J.; Lahoz, A.; et al. Human Embryonic Stem Cell Derived Hepatocyte-Like Cells as a Tool for In Vitro Hazard Assessment of Chemical Carcinogenicity. Toxicol. Sci. 2011, 124, 278–290. [Google Scholar] [CrossRef] [Green Version]
- Hofree, M.; Shen, J.P.; Carter, H.; Gross, A.; Ideker, T. Network-based stratification of tumor mutations. Nat. Methods 2013, 10, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Hardt, C.; Beber, M.; Rasche, A.; Kamburov, A.; Hebels, D.; Kleinjans, J.; Herwig, R. ToxDB: Pathway-level interpretation of drug-treatment data. Database 2016, 2016, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kim, S. Identifying dynamic pathway interactions based on clinical information. Comput. Boil. Chem. 2017, 68, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.N.; Rana, M.M.; Begum, A.A.; Rahman, M.R.R.; Mollah, M.N.H. Robust Co-clustering to Discover Toxicogenomic Biomarkers and Their Regulatory Doses of Chemical Compounds Using Logistic Probabilistic Hidden Variable Model. Front. Genet. 2018, 9, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyström-Persson, J.; Igarashi, Y.; Ito, M.; Morita, M.; Nakatsu, N.; Yamada, H.; Mizuguchi, K. Toxygates: Interactive toxicity analysis on a hybrid microarray and linked data platform. Bioinformatics 2013, 29, 3080–3086. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.N.; Akond, Z.; Alam, M.J.; Begum, A.A.; Rahman, M.; Mollah, M.N.H. Toxic Dose prediction of Chemical Compounds to Biomarkers using an ANOVA based Gene Expression Analysis. Bioinformation 2018, 14, 369–377. [Google Scholar] [CrossRef]
- Otava, M.; Shkedy, Z.; Kasim, A. Prediction of gene expression in human using rat in vivo gene expression in Japanese Toxicogenomics Project. Syst. Biomed. 2014, 2, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Okuno, Y.; Tsujimoto, G.; Mamitsuka, H. A probabilistic model for mining implicit ’chemical compound-gene’ relations from literature. Bioinformatics 2005, 21 (Suppl. 2), 245–251. [Google Scholar] [CrossRef]
- Chung, M.-H.; Wang, Y.; Tang, H.; Zou, W.; Basinger, J.; Xu, X.; Tong, W. Asymmetric author-topic model for knowledge discovering of big data in toxicogenomics. Front. Pharmacol. 2015, 6, 1–7. [Google Scholar] [CrossRef]
- Dempster, A.P.; Laird, N.M.; Rubin, D.B. Maximum Likelihood from Incomplete Data via the EM Algorithm. J. R. Stat. Soc. Ser. B 1977, 39, 1–22. [Google Scholar] [CrossRef]
- Afshari, C.A.; Hamadeh, H.K.; Bushel, P.R. The evolution of bioinformatics in toxicology: Advancing toxicogenomics. Toxicol. Sci. 2011, 120, S225–S237. [Google Scholar] [CrossRef] [PubMed]
- Uehara, T.; Ono, A.; Maruyama, T.; Kato, I.; Yamada, H.; Ohno, Y.; Urushidani, T. The Japanese toxicogenomics project: Application of toxicogenomics. Mol. Nutr. Food Res. 2010, 54, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Kiyosawa, N.; Shiwaku, K.; Hirode, M.; Omura, K.; Uehara, T.; Shimizu, T.; Mizukawa, Y.; Miyagishima, T.; Ono, A.; Nagao, T.; et al. Utilization of a one-dimensional score for surveying chemical-induced changes in expression levels of multiple biomarker gene sets using a large-scale toxicogenomics database. J. Toxicol. Sci. 2006, 31, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Distance Measure | Mathematical Form |
|---|---|
| Euclidean | |
| Minkowski | |
| Manhattan | |
| Canbera | |
| Maximum |
| Sl | Combination of Distance and HC Clustering Methods | Drug Clustering ER for GMP Data | Drug Clustering ER for PPAR-SP Data | DDs Clustering ER for GMP Data | DDs Clustering ER for PPAR-SP Data |
|---|---|---|---|---|---|
| 1 | euclidean:ward | 10 | 0 | 6.666666667 | 20 |
| 2 | euclidean:single | 10 | 40 | 16.66666667 | 36.66666667 |
| 3 | euclidean:complete | 10 | 30 | 26.66666667 | 20 |
| 4 | euclidean:average | 10 | 40 | 26.66666667 | 20 |
| 5 | euclidean:mcquitty | 40 | 40 | 26.66666667 | 13.33333333 |
| 6 | euclidean:median | 40 | 40 | 3.333333333 | 26.66666667 |
| 7 | euclidean:centroid | 40 | 40 | 16.66666667 | 30 |
| 8 | maximum:ward | 10 | 0 | 16.66666667 | 10 |
| 9 | maximum:single | 10 | 40 | 16.66666667 | 36.66666667 |
| 10 | maximum:complete | 20 | 0 | 16.66666667 | 26.66666667 |
| 11 | maximum:average | 10 | 40 | 26.66666667 | 36.66666667 |
| 12 | maximum:mcquitty | 10 | 40 | 26.66666667 | 36.66666667 |
| 13 | maximum:median | 40 | 40 | 26.66666667 | 36.66666667 |
| 14 | maximum:centroid | 40 | 40 | 16.66666667 | 30 |
| 15 | manhattan:ward | 10 | 0 | 6.666666667 | 20 |
| 16 | manhattan:single | 40 | 40 | 16.66666667 | 36.66666667 |
| 17 | manhattan:complete | 10 | 30 | 3.333333333 | 20 |
| 18 | manhattan:average | 10 | 40 | 26.66666667 | 20 |
| 19 | manhattan:mcquitty | 10 | 40 | 3.333333333 | 20 |
| 20 | manhattan:median | 40 | 40 | 26.66666667 | 36.66666667 |
| 21 | manhattan:centroid | 40 | 40 | 16.66666667 | 30 |
| 22 | canberra:ward | 50 | 10 | 30 | 20 |
| 23 | canberra:single | 50 | 10 | 23.33333333 | 36.66666667 |
| 24 | canberra:complete | 50 | 10 | 30 | 20 |
| 25 | canberra:average | 50 | 10 | 30 | 23.33333333 |
| 26 | canberra:mcquitty | 50 | 40 | 30 | 23.33333333 |
| 27 | canberra:median | 50 | 40 | 40 | 36.66666667 |
| 28 | canberra:centroid | 50 | 40 | 33.33333333 | 36.66666667 |
| 29 | minkowski:ward | 10 | 0 | 6.666666667 | 20 |
| 30 | minkowski:single | 10 | 40 | 16.66666667 | 36.66666667 |
| 31 | minkowski:complete | 10 | 30 | 26.66666667 | 20 |
| 32 | minkowski:average | 10 | 40 | 26.66666667 | 20 |
| 33 | minkowski:mcquitty | 40 | 40 | 26.66666667 | 13.33333333 |
| 34 | minkowski:median | 40 | 40 | 3.333333333 | 26.66666667 |
| 35 | minkowski:centroid | 40 | 40 | 16.66666667 | 30 |
| Euclidean: ward, Dataset: glutathione metabolism pathway at 24 h time point | |
| Gene and compound co-cluster | Co-cluster mean |
| Gene-Cluster-3(1): Compound-Cluster-3(1) | 2.5550390 |
| Gene-Cluster-2(2): Compound-Cluster-2(2) | 1.6619841 |
| Gene-Cluster-3(1): Compound-Cluster-2(2) | 0.8249199 |
| Gene-Cluster-3(1): Compound-Cluster-1(3) | 0.8129127 |
| Gene-Cluster-2(2): Compound-Cluster-3(1) | 0.5994644 |
| Gene-Cluster-1(3): Compound-Cluster-3(1) | 0.5991663 |
| Gene-Cluster-1(3): Compound-Cluster-2(2) | 0.4653372 |
| Gene-Cluster-2(2): Compound-Cluster-1(3) | 0.3402437 |
| Gene-Cluster-1(3): Compound-Cluster-1(3) | 0.2481545 |
| Euclidean: ward, Dataset: glutathione metabolism pathway at 3 h, 6 h 9 h, and 24 h time points | |
| Gene and compound co-cluster | Co-cluster mean |
| Gene-Cluster-3(1): Compound-Cluster-2(1) | 1.2954907 |
| Gene-Cluster-1(2): Compound-Cluster-1(2) | 0.6118177 |
| Gene-Cluster-2(3): Compound-Cluster-1(2) | 0.5850958 |
| Gene-Cluster-3(1): Compound-Cluster-1(2) | 0.5157947 |
| Gene-Cluster-3(1): Compound-Cluster-3(3) | 0.3513179 |
| Gene-Cluster-1(2): Compound-Cluster-2(1) | 0.3360666 |
| Gene-Cluster-2(3): Compound-Cluster-2(1) | 0.3285539 |
| Gene-Cluster-1(1): Compound-Cluster-3(3) | 0.2478899 |
| Gene-Cluster-2(3): Compound-Cluster-3(3) | 0.2424664 |
| Euclidean: ward, Dataset: PPAR signaling pathway at 24 h time point | |
| Gene and compound co-cluster | Co-cluster mean |
| Gene-Cluster-1(1): Compound-Cluster-1(1) | 1.5972416 |
| Gene-Cluster-3(2): Compound-Cluster-2(2) | 0.6596625 |
| Gene-Cluster-3(2): Compound-Cluster-1(1) | 0.6522308 |
| Gene-Cluster-1(1): Compound-Cluster-2(2) | 0.4973316 |
| Gene-Cluster-2(3): Compound-Cluster-1(1) | 0.3994878 |
| Gene-Cluster-2(3): Compound-Cluster-2(2) | 0.2378871 |
| Euclidean: ward, Dataset: PPAR signaling pathway at 3 h, 6 h 9 h, and 24 h time points | |
| Gene and compound co-cluster | Co-cluster mean |
| Gene-Cluster-3(1): Compound-Cluster-2(1) | 1.5863836 |
| Gene-Cluster-1(2): Compound-Cluster-2(1) | 0.5842037 |
| Gene-Cluster-1(2): Compound-Cluster-1(2) | 0.4385611 |
| Gene-Cluster-3(1): Compound-Cluster-1(2) | 0.4025768 |
| Gene-Cluster-2(3): Compound-Cluster-2(1) | 0.2569643 |
| Gene-Cluster-2(3): Compound-Cluster-1(2) | 0.1757952 |
| Biomarker Genes | Regulatory Doses of Drugs |
|---|---|
| Euclidean: ward, Dataset: glutathione metabolism pathway at 24 h time point | |
| Gene-cluster-3: LOC100359539/Rrm2, LOC100359539/Rrm2, Gpx6, RGD1562107 | DCCs-cluster-3: isoniazid_Middle, isoniazid_High |
| Gene-cluster-2: Gclc, Gstm4, Gstm3, G6pd, Gsta5, Gclc, Mgst2, Gsr, Gpx2, Gclm, Gstp1 | DCCs-cluster-2: acetaminophen_Middle, acetaminophen_High, methapyrilene_High, nitrofurazone_High |
| Euclidean:ward, Dataset: glutathione metabolism pathway at 3 h, 6 h 9 h, and 24 h time points | |
| Gene-cluster-2: LOC100912604/Srm, Gclc, Gstm4, Gstm3, G6pd, Gsta5, Gclc, Odc1, Mgst2, Gsr, Gss, Gpx2, Gclm, Gstp1 | DCCs-cluster-3: acetaminophen_High_24.hr, acetaminophen_Middle_24.hr, methapyrilene_High_6.hr, methapyrilene_High_24.hr, methapyrilene_High_9.hr, nitrofurazone_High_24.hr, nitrofurazone_High_6.hr, nitrofurazone_Middle_6.hr, nitrofurazone_High_9.hr, nitrofurazone_Middle_9.hr, |
| Biomarker Genes | Regulatory Doses of Drugs |
|---|---|
| Euclidean: ward, Dataset: PPAR signaling pathway at 24 h time point | |
| Gene-cluster-1: Cpt1a, Cyp8b1, Cyp4a3, Ehhadh, Plin5, Fabp3, Me1, Fabp5, LOC100910385, Cpt2, Acaa1a, Cyp4a1, LOC100365047, Cpt1a, LOC100365047, Angptl4, Aqp7, Cpt1c, Cpt1b, Me1 | DCCs-cluster-1: aspirin_Low, aspirin_High, aspirin_Middle, benzbromarone_High, clofibrate_High, WY14643_Low, WY14643_High, WY14643_Middle |
| Euclidean: ward, Dataset: PPAR signaling pathway at 3 h, 6 h 9 h, and 24 h time points | |
| Gene-cluster-3: Cyp4a3, Ehhadh, Plin2, Plin5, Me1, LOC100910385, Cpt2, Acaa1a, Cyp4a1, Angptl4, Cpt1b | DCCs-cluster-2: aspirin_Low_9.hr, aspirin_Low_24.hr, aspirin_High_9.hr, aspirin_High_24.hr, aspirin_Middle_24.hr, benzbromarone_Middle_6.hr, benzbromarone_High_9.hr, benzbromarone_High_3.hr, benzbromarone_Middle_9.hr, enzbromarone_High_24.hr, benzbromarone_High_6.hr, benzbromarone_Middle_3.hr, clofibrate_Middle_6.hr, clofibrate_High_24.hr, clofibrate_Middle_9.hr, clofibrate_High_6.hr, clofibrate_High_9.hr, gemfibrozil_High_24.hr, gemfibrozil_Middle_24.hr, gemfibrozil_High_9.hr, WY.14643_High_6.hr, WY.14643_Middle_6.hr, WY.14643_Middle_24.hr, WY.14643_Low_3.hr, WY.14643_Low_24.hr, WY.14643_Middle_9.hr, WY.14643_Low_6.hr, WY.14643_High_9.hr, WY.14643_Middle_3.hr, WY.14643_High_3.hr, WY.14643_Low_9.hr, WY.14643_High_24.hr |
| Term | Count | % | p-Value | FDR | Genes |
|---|---|---|---|---|---|
| rno00480: Glutathione metabolism | 2 | 66.66 | 7.48E−3 | 2.04E−38 | RGD1562107, Gpx6 |
| Term | Count | % | p-Value | Genes |
|---|---|---|---|---|
| rno00480: Glutathione metabolism | 10 | 100 | 3.85E−20 | Mgst2, Gpx2, G6pd, Gclm, Gsr, Gsta5, Gclc, Gclc, Gstp1, Gstm3, Gstm4 |
| rno00980: Metabolism of xenobiotics by cytochrome P450 | 5 | 50.0 | 7.43E−7 | Mgst2, Gsta5, Gstp1, Gstm3, Gstm4 |
| rno00982: Drug metabolism—cytochrome P450 | 5 | 50.0 | 7.87E−7 | Mgst2, Gsta5, Gstp1, Gstm3, Gstm4 |
| rno05204: Chemical carcinogenesis | 5 | 50.0 | 2.14E−6 | Mgst2, Gsta5, Gstp1, Gstm3, Gstm4 |
| rno04918: Thyroid hormone synthesis | 2 | 20.0 | 0.076 | Gpx2, Gsr |
| Term | Count | % | p-Value | Genes |
|---|---|---|---|---|
| rno03320: PPAR signaling pathway | 13 | 76.47 | 4.88E−24 | Cpt1b, Aqp7, Cpt1c, Cpt1a, Cyp4a3, Cpt1a, Cpt2, Cyp8b1, Fabp3, Ehhadh, Acaa1a, Cyp4a1, Angptl4, Fabp5 |
| rno00071: Fatty acid degradation | 8 | 47.06 | 3.16E−13 | Cpt1b, Cpt2, Ehhadh, Acaa1a, Cpt1c, Cpt1a, Cyp4a3, Cpt1a, Cyp4a1 |
| rno01212: Fatty acid metabolism | 6 | 35.29 | 1.67E−8 | Cpt1b, Cpt2, Ehhadh, Acaa1a, Cpt1c, Cpt1a, Cpt1a |
| rno04920: Adipocytokine signaling pathway | 3 | 17.65 | 0.0067 | Cpt1b, Cpt1c, Cpt1a, Cpt1a |
| rno04922: Glucagon signaling pathway | 3 | 17.65 | 0.0117 | Cpt1b, Cpt1c, Cpt1a, Cpt1a |
| rno04152: AMPK signaling pathway | 3 | 17.65 | 0.0187 | Cpt1b, Cpt1c, Cpt1a, Cpt1a |
| rno01100: Metabolic pathways | 6 | 35.29 | 0.0500 | Me1, Me1, Cyp8b1, Ehhadh, Acaa1a, Cyp4a3, Cyp4a1 |
| rno00280: Valine, leucine and isoleucine degradation | 2 | 11.76 | 0.0885 | Ehhadh, Acaa1a |
| Term | Count | % | p-Value | Genes |
|---|---|---|---|---|
| rno03320: PPAR signaling pathway | 7 | 63.63 | 5.49E−12 | Cpt1b, Cpt2, Ehhadh, Acaa1a, Cyp4a3, Angptl4, Cyp4a1 |
| rno00071: Fatty acid degradation | 6 | 54.54 | 1.37E−10 | Cpt1b, Cpt2, Ehhadh, Acaa1a, Cyp4a3, Cyp4a1 |
| rno01212: Fatty acid metabolism | 4 | 36.36 | 1.09E−5 | Cpt1b, Cpt2, Ehhadh, Acaa1a |
| rno01100: Metabolic pathways | 5 | 45.45 | 0.0172 | Me1, Ehhadh, Acaa1a, Cyp4a3, Cyp4a1 |
| rno00280: Valine, leucine and isoleucine degradation | 2 | 18.18 | 0.0486 | Ehhadh, Acaa1a |
| rno00590: Arachidonic acid metabolism | 2 | 18.18 | 0.0709 | Cyp4a3, Cyp4a1 |
| rno00830: Retinol metabolism | 2 | 18.18 | 0.0726 | Cyp4a3, Cyp4a1 |
| rno04146: Peroxisome | 2 | 18.18 | 0.0743 | Ehhadh, Acaa1a |
| rno04750: Inflammatory mediator regulation of TRP channels | 2 | 18.18 | 0.0994 | Cyp4a3, Cyp4a1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, M.N.; Malek, M.B.; Begum, A.A.; Rahman, M.; Mollah, M.N.H. Assessment of Drugs Toxicity and Associated Biomarker Genes Using Hierarchical Clustering. Medicina 2019, 55, 451. https://doi.org/10.3390/medicina55080451
Hasan MN, Malek MB, Begum AA, Rahman M, Mollah MNH. Assessment of Drugs Toxicity and Associated Biomarker Genes Using Hierarchical Clustering. Medicina. 2019; 55(8):451. https://doi.org/10.3390/medicina55080451
Chicago/Turabian StyleHasan, Mohammad Nazmol, Masuma Binte Malek, Anjuman Ara Begum, Moizur Rahman, and Md. Nurul Haque Mollah. 2019. "Assessment of Drugs Toxicity and Associated Biomarker Genes Using Hierarchical Clustering" Medicina 55, no. 8: 451. https://doi.org/10.3390/medicina55080451