Differentiation of Tumorigenic C6 Glioma Cells Induced by Enhanced IL-6 Signaling

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. RNA Extraction and Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.3. Western Blot Analysis
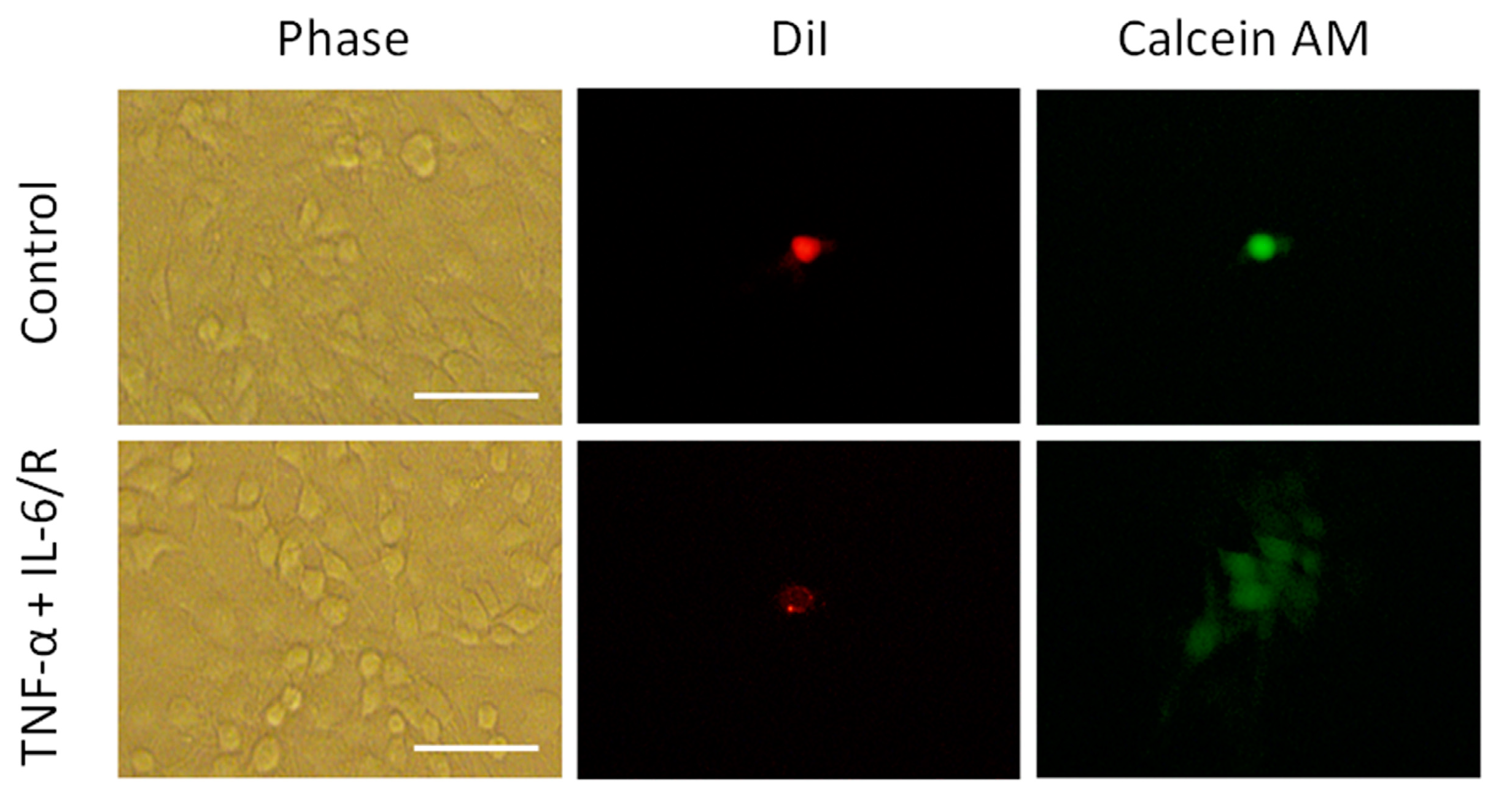
2.4. Dye-Coupling Assay
2.5. Cell Proliferation Assay
2.6. Apoptosis and Necrosis Detection
2.7. Soft Agar Assay
2.8. Sphere Formation Assay
2.9. Statistical Analysis
3. Results
3.1. The Expression of Il-6 Is Efficiently Triggered by TNF-α/IL-6/sIL-6R
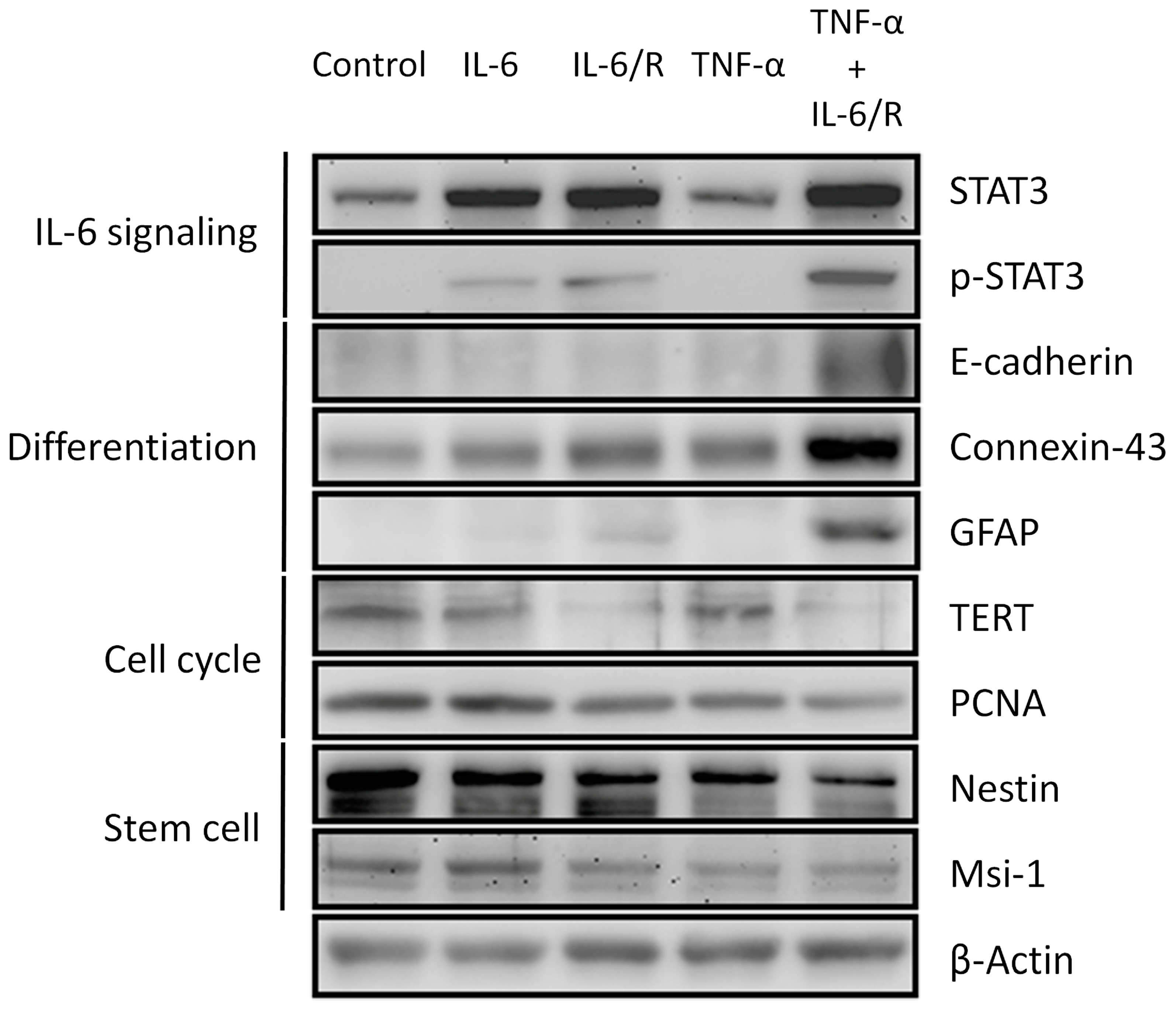
3.2. Differentiation of C6 Glioma Cells Is Induced by TNF-α/IL-6/sIL-6R as Evidenced by Changes in Biomarker Levels
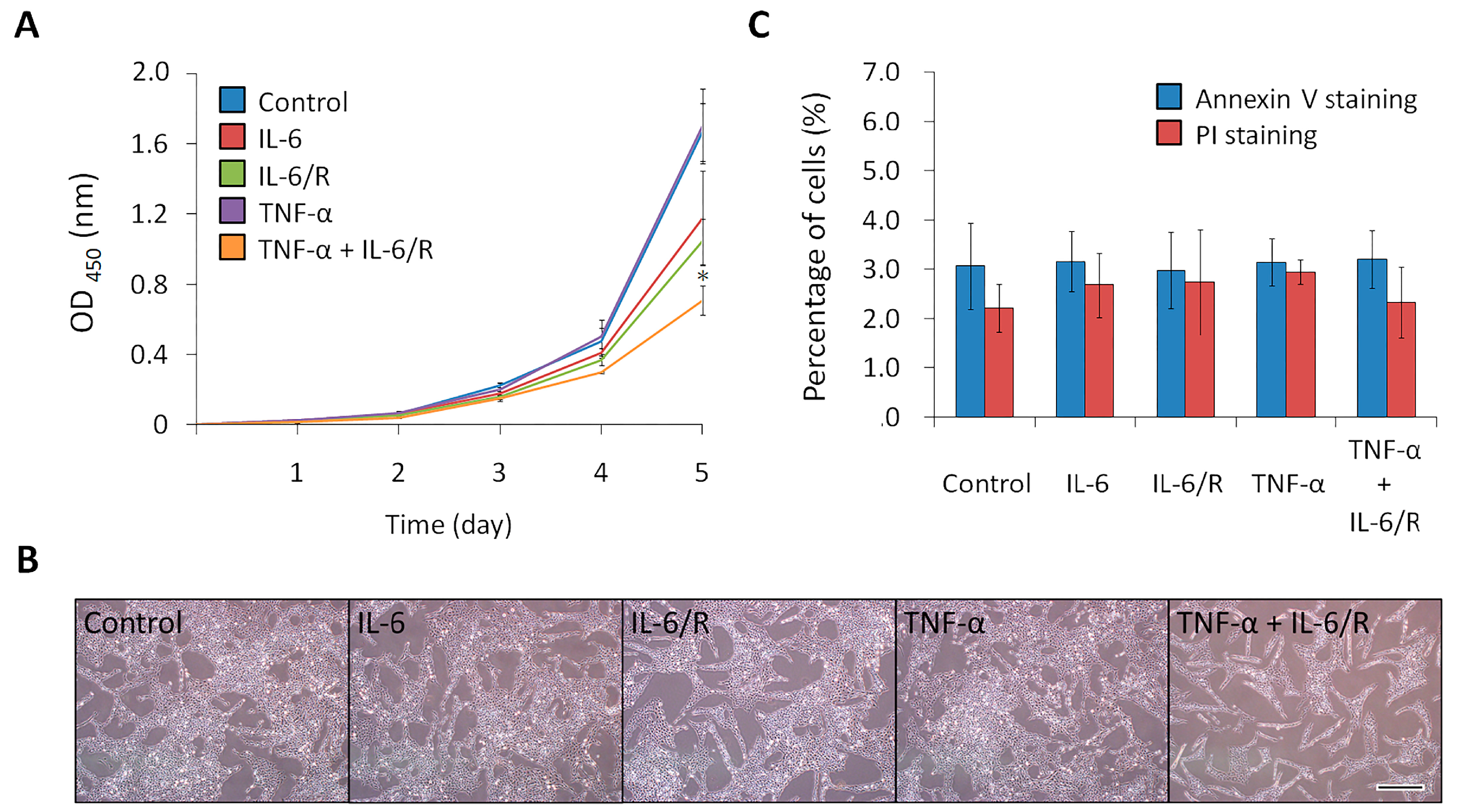
3.3. TNF-α/IL-6/sIL-6R Decreases the Proliferation Rate of C6 Glioma Cells
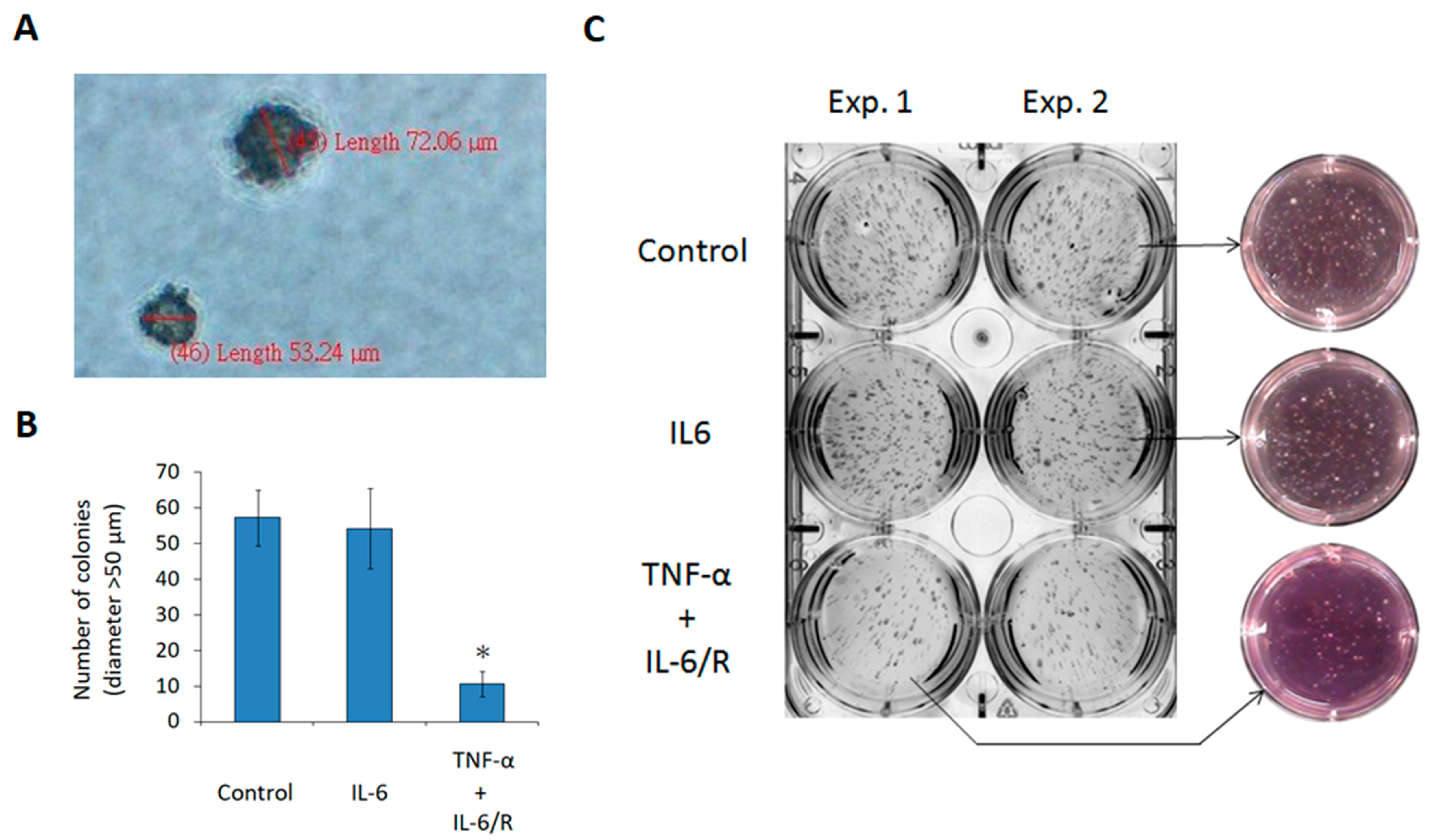
3.4. TNF-α/IL-6/sIL-6R Reduces the Tumorigenicity of C6 Glioma Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, A.I.; Câtoi, C. Chapter 3 Tumor Cell Morphology. In Comparative Oncology; The Publishing House of the Romanian Academy: Bucharest, Romania, 2007. [Google Scholar]
- Weissman, I.L.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nat. Cell Biol. 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Polyak, K.; Hahn, W.C. Roots and stems: Stem cells in cancer. Nat. Med. 2006, 12, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Bomken, S.; Fišer, K.; Heidenreich, O.; Vormoor, J. Understanding the cancer stem cell. Br. J. Cancer 2010, 103, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Ghiaur, G.; Gerber, J.; Jones, R.J. Concise Review: Cancer Stem Cells and Minimal Residual Disease. Stem Cells 2012, 30, 89–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaccio, F.; Paino, F.; Regad, T.; Papaccio, G.; Desiderio, V.; Tirino, V. Concise Review: Cancer Cells, Cancer Stem Cells, and Mesenchymal Stem Cells: Influence in Cancer Development. Stem Cells Transl. Med. 2017, 6, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Donnenberg, V.S.; Donnenberg, A.D. Multiple Drug Resistance in Cancer Revisited: The Cancer Stem Cell Hypothesis. J. Clin. Pharmacol. 2005, 45, 872–877. [Google Scholar] [CrossRef]
- Rich, J.N. Cancer Stem Cells in Radiation Resistance: Figure. Cancer Res. 2007, 67, 8980–8984. [Google Scholar] [CrossRef] [Green Version]
- Frank, N.Y.; Schatton, T.; Frank, M.H. The therapeutic promise of the cancer stem cell concept. J. Clin. Investig. 2010, 120, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Piccirillo, S.G.M.; Reynolds, B.A.; Zanetti, N.; Lamorte, G.; Binda, E.; Broggi, G.; Brem, H.; Olivi, A.; DiMeco, F.; Vescovi, A.L. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nat. Cell Biol. 2006, 444, 761–765. [Google Scholar] [CrossRef]
- Lotem, J.; Sachs, L.M. Epigenetics and the plasticity of differentiation in normal and cancer stem cells. Oncogene 2006, 25, 7663–7672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J.; et al. let-7 Regulates Self Renewal and Tumorigenicity of Breast Cancer Cells. Cell 2007, 131, 1109–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of Selective Inhibitors of Cancer Stem Cells by High-Throughput Screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takanaga, H.; Yoshitake, T.; Hara, S.; Yamasaki, C.; Kunimoto, M. cAMP-induced Astrocytic Differentiation of C6 Glioma Cells Is Mediated by Autocrine Interleukin-6. J. Biol. Chem. 2004, 279, 15441–15447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taga, T.; Fukuda, S. Role of IL-6 in the Neural Stem Cell Differentiation. Clin. Rev. Allergy Immunol. 2005, 28, 249–256. [Google Scholar] [CrossRef]
- Takanaga, H.; Yoshitake, T.; Yatabe, E.; Hara, S.; Kunimoto, M. beta-Naphthoflavone disturbs astrocytic differentiation of C6 glioma cells by inhibiting autocrine interleukin-6. J. Neurochem. 2004, 90, 750–757. [Google Scholar] [CrossRef]
- Shu, M.; Zhou, Y.; Zhu, W.; Wu, S.; Zheng, X.; Yan, G. Activation of a pro-survival pathway IL-6/JAK2/STAT3 contributes to glial fibrillary acidic protein induction during the cholera toxin-induced differentiation of C6 malignant glioma cells. Mol. Oncol. 2011, 5, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T.; Yasukawa, K.; Harada, H.; Taga, T.; Watanabe, S.; Matsuda, T.; Kashiwamura, S.-I.; Nakajima, K.; Koyama, K.; Iwamatsu, A.; et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nat. Cell Biol. 1986, 324, 73–76. [Google Scholar] [CrossRef]
- Gruol, D.L.; Nelson, T.E. Physiological and pathological roles of interleukin-6 in the central nervous system. Mol. Neurobiol. 1997, 15, 307–339. [Google Scholar] [CrossRef]
- Kishimoto, T. IL-6: From its discovery to clinical applications. Int. Immunol. 2010, 22, 347–352. [Google Scholar] [CrossRef] [Green Version]
- Hama, T.; Miyamoto, M.; Tsukui, H.; Nishio, C.; Hatanaka, H. Interleukin-6 as a neurotrophic factor for promoting the survival of cultured basal forebrain cholinergic neurons from postnatal rats. Neurosci. Lett. 1989, 104, 340–344. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta Bioenerg. 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Shen, G.; Yang, X.; Liu, W. Most C6 Cells Are Cancer Stem Cells: Evidence from Clonal and Population Analyses. Cancer Res. 2007, 67, 3691–3697. [Google Scholar] [CrossRef] [Green Version]
- Fang, K.-M.; Yang, C.-S.; Lin, T.-C.; Chan, T.-C.; Tzeng, S.-F. Induced interleukin-33 expression enhances the tumorigenic activity of rat glioma cells. Neuro-Oncology 2014, 16, 552–566. [Google Scholar] [CrossRef] [Green Version]
- Van Wagoner, N.J.; Oh, J.-W.; Repovic, P.; Benveniste, E.N. Interleukin-6 (IL-6) Production by Astrocytes: Autocrine Regulation by IL-6 and the Soluble IL-6 Receptor. J. Neurosci. 1999, 19, 5236–5244. [Google Scholar] [CrossRef] [Green Version]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Rathinam, M.K.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The Soft Agar Colony Formation Assay. J. Vis. Exp. 2014, 92, e51998. [Google Scholar] [CrossRef] [Green Version]
- Niehof, M.; Streetz, K.; Rakemann, T.; Bischoff, S.C.; Manns, M.P.; Horn, F.; Trautwein, C. Interleukin-6-induced Tethering of STAT3 to the LAP/C/EBPβ Promoter Suggests a New Mechanism of Transcriptional Regulation by STAT. J. Biol. Chem. 2000, 276, 9016–9027. [Google Scholar] [CrossRef] [Green Version]
- Vleminckx, K.; Vakaet, L.; Mareel, M.; Fiers, W.; Van Roy, F. Genetic manipulation of E-cadherin expression by epithelial tumor cells reveals an invasion suppressor role. Cell 1991, 66, 107–119. [Google Scholar] [CrossRef]
- Yu, S.; Xiao, H.-L.; Jiang, X.-F.; Wang, Q.-L.; Li, Y.; Yang, X.-J.; Ping, Y.-F.; Duan, J.J.; Jiang, J.-Y.; Ye, X.-Z.; et al. Connexin 43 Reverses Malignant Phenotypes of Glioma Stem Cells by Modulating E-Cadherin. Stem Cells 2012, 30, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.P.; Hossain, M.Z.; Sehgal, A.; Boynton, A.L. Reduced connexin43 expression in high-grade human brain glioma cells. J. Surg. Oncol. 1999, 70, 21–24. [Google Scholar] [CrossRef]
- Soroceanu, L.; Manning, T.J., Jr.; Sontheimer, H. Reduced expression of connexin-43 and functional gap junction coupling in human gliomas. Glia 2001, 33, 107–117. [Google Scholar] [CrossRef]
- Eng, L.F. Glial fibrillary acidic protein (GFAP): The major protein of glial intermediate filaments in differentiated astrocytes. J. Neuroimmunol. 1985, 8, 203–214. [Google Scholar] [CrossRef]
- Xu, D.; Wang, Q.; Gruber, A.; Björkholm, M.; Chen, Z.; Zaid, A.; Selivanova, G.; Peterson, C.; Wiman, K.G.; Pisa, P. Downregulation of telomerase reverse transcriptase mRNA expression by wild type p53 in human tumor cells. Oncogene 2000, 19, 5123–5133. [Google Scholar] [CrossRef] [Green Version]
- Kurki, P.; Vanderlaan, M.; Dolbeare, F.; Gray, J.; Tan, E.M. Expression of proliferating cell nuclear antigen (PCNA)/cyclin during the cell cycle. Exp. Cell Res. 1986, 166, 209–219. [Google Scholar] [CrossRef]
- Johansson, C.B.; Momma, S.; Clarke, D.L.; Risling, M.; Lendahl, U.; Frisén, J. Identification of a Neural Stem Cell in the Adult Mammalian Central Nervous System. Cell 1999, 96, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Sakakibara, S.-I.; Imai, T.; Hamaguchi, K.; Okabe, M.; Aruga, J.; Nakajima, K.; Yasutomi, D.; Nagata, T.; Kurihara, Y.; Uesugi, S.; et al. Mouse-Musashi-1, a Neural RNA-Binding Protein Highly Enriched in the Mammalian CNS Stem Cell. Dev. Biol. 1996, 176, 230–242. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nat. Cell Biol. 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Tanabe, K.; Matsushima-Nishiwaki, R.; Yamaguchi, S.; Iida, H.; Dohi, S.; Kozawa, O. Mechanisms of tumor necrosis factor-α-induced interleukin-6 synthesis in glioma cells. J. Neuroinflamm. 2010, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.B.; Chaffer, C.L.; Weinberg, R.A. Cancer stem cells: Mirage or reality? Nat. Med. 2009, 15, 1010–1012. [Google Scholar] [CrossRef]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in Cancer: Cancer Stem Cells versus Clonal Evolution. Cell 2009, 138, 822–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aboody, K.S.; Brown, A.B.; Rainov, N.G.; Bower, K.A.; Liu, S.; Yang, W.; Small, J.E.; Herrlinger, U.; Ourednik, V.; Black, P.M.; et al. Neural stem cells display extensive tropism for pathology in adult brain: Evidence from intracranial gliomas. Proc. Natl. Acad. Sci. USA 2000, 97, 12846–12851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Kim, C.G.; Shim, J.K.; Kim, J.H.; Lee, H.; Lee, J.E.; Kim, M.H.; Haam, K.; Jung, I.; Park, S.H.; et al. Effect of combined anti-PD-1 and temozolomide therapy in glioblastoma. Oncoimmunology 2018, 8, e1525243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cammarata, F.P.; Torrisi, F.; Forte, G.I.; Minafra, L.; Bravatà, V.; Pisciotta, P.; Savoca, G.; Calvaruso, M.; Petringa, G.; Cirrone, G.A.P.; et al. Proton Therapy and Src Family Kinase Inhibitor Combined Treatments on U87 Human Glioblastoma Multiforme Cell Line. Int. J. Mol. Sci. 2019, 20, 4745. [Google Scholar] [CrossRef] [Green Version]
- Islam, O.; Gong, X.; Rose-John, S.; Heese, K. Interleukin-6 and Neural Stem Cells: More Than Gliogenesis. Mol. Biol. Cell 2009, 20, 188–199. [Google Scholar] [CrossRef]
- Taga, T.; Nakashima, K. Mechanisms Underlying Cytokine-Mediated Cell-Fate Regulation in the Nervous System. Mol. Neurobiol. 2002, 25, 233–244. [Google Scholar] [CrossRef]
- Barkho, B.Z.; Song, H.; Aimone, J.B.; Smrt, R.D.; Kuwabara, T.; Nakashima, K.; Gage, F.H.; Zhao, X. Identification of Astrocyte-expressed Factors That Modulate Neural Stem/Progenitor Cell Differentiation. Stem Cells Dev. 2006, 15, 407–421. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, D.; Norman, A.A.; Woodard, C.L.; Yang, G.; Gauthier-Fisher, A.; Fujitani, M.; Vessey, J.P.; Cancino, G.I.; Sachewsky, N.; Woltjen, K.; et al. Transient Maternal IL-6 Mediates Long-Lasting Changes in Neural Stem Cell Pools by Deregulating an Endogenous Self-Renewal Pathway. Cell Stem Cell 2013, 13, 564–576. [Google Scholar] [CrossRef] [Green Version]
- Hossain, A.; Gumin, J.; Gao, F.; Figueroa, J.; Shinojima, N.; Takezaki, T.; Priebe, W.; Villarreal, D.; Kang, S.G.; Joyce, C.; et al. Mesenchymal stem cells isolated from human gliomas increase proliferation and maintain stemness of glioma stem cells through the IL-6/gp130/STAT3 pathway. Stem Cells 2015, 33, 2400–2415. [Google Scholar] [CrossRef] [Green Version]
- Plaks, V.; Kong, N.; Werb, Z. The Cancer Stem Cell Niche: How Essential Is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Benveniste, E.N.; Sparacio, S.M.; Norris, J.G.; Grennett, H.E.; Fuller, G.M. Induction and regulation of interleukin-6 gene expression in rat astrocytes. J. Neuroimmunol. 1990, 30, 201–212. [Google Scholar] [CrossRef]
- Benveniste, E.N.; Kwon, J.; Chung, W.J.; Sampson, J.; Pandya, K.; Tang, L.P. Differential modulation of astrocyte cytokine gene expression by TGF-beta. J. Immunol. 1994, 153, 5210–5221. [Google Scholar]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Walling, J.; Kotliarov, Y.; Center, A.; Steed, M.E.; Ahn, S.J.; Rosenblum, M.; Mikkelsen, T.; Zenklusen, J.C.; Fine, H.A. Genomic Changes and Gene Expression Profiles Reveal That Established Glioma Cell Lines Are Poorly Representative of Primary Human Gliomas. Mol. Cancer Res. 2008, 6, 21–30. [Google Scholar] [CrossRef] [Green Version]
- Tirino, V.; Desiderio, V.; d’Aquino, R.; De Francesco, F.; Pirozzi, G.; Graziano, A.; Galderisi, U.; Cavaliere, C.; De Rosa, A.; Papaccio, G.; et al. Detection and characterization of CD133+ cancer stem cells in human solid tumours. PLoS ONE 2008, 3, e3469. [Google Scholar] [CrossRef]
- Beier, D.; Hau, P.; Proescholdt, M.A.; Lohmeier, A.; Wischhusen, J.; Oefner, P.J.; Aigner, L.; Brawanski, A.; Bogdahn, U.; Beier, C.P. CD133+ and CD133- Glioblastoma-Derived Cancer Stem Cells Show Differential Growth Characteristics and Molecular Profiles. Cancer Res. 2007, 67, 4010–4015. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Kelly, P.N.; Dakic, A.; Adams, J.M.; Nutt, S.L.; Strasser, A. Tumor Growth Need Not Be Driven by Rare Cancer Stem Cells. Science 2007, 317, 337. [Google Scholar] [CrossRef] [Green Version]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nat. Cell Biol. 2008, 456, 593–598. [Google Scholar] [CrossRef] [Green Version]
- Kreso, A.; Dick, J.E. Evolution of the Cancer Stem Cell Model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Forward Primer, 5′ to 3′ | Reverse Primer, 5′ to 3′ | Tm (°C) |
|---|---|---|---|
| Il-6 | GTCTCGAGATGAAGTTTCTCTCCGCA | GTGGATCCCTAGTGCCGAGTAGA | 50 |
| Gapdh | TGCACCACCAACTGCTTA | GGACAGGGATGATGTTC | 50 |
| Step | Temperature (°C) | Time | Number of Cycles | |
|---|---|---|---|---|
| 1 | Initial denaturation | 94 | 5 min | 1 |
| Denaturation | 94 | 30 s | ||
| 2 | Annealing | 50 | 30 s | 30 |
| Extension | 72 | 45 s | ||
| 3 | Final extension | 72 | 10 min | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, I.-R.; Pan, R.-L.; Yang, C.-S. Differentiation of Tumorigenic C6 Glioma Cells Induced by Enhanced IL-6 Signaling. Medicina 2020, 56, 625. https://doi.org/10.3390/medicina56110625
Chu I-R, Pan R-L, Yang C-S. Differentiation of Tumorigenic C6 Glioma Cells Induced by Enhanced IL-6 Signaling. Medicina. 2020; 56(11):625. https://doi.org/10.3390/medicina56110625
Chicago/Turabian StyleChu, Inn-Ray, Rong-Long Pan, and Chung-Shi Yang. 2020. "Differentiation of Tumorigenic C6 Glioma Cells Induced by Enhanced IL-6 Signaling" Medicina 56, no. 11: 625. https://doi.org/10.3390/medicina56110625
APA StyleChu, I.-R., Pan, R.-L., & Yang, C.-S. (2020). Differentiation of Tumorigenic C6 Glioma Cells Induced by Enhanced IL-6 Signaling. Medicina, 56(11), 625. https://doi.org/10.3390/medicina56110625