An Intergenic rs9275596 Polymorphism on Chr. 6p21 Is Associated with Multiple Sclerosis in Latvians

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Case-Control Study
2.2. DNA Extraction and Genotyping
2.3. Data Management and Analysis
2.4. SNP Functional Analysis in Silico
3. Results
3.1. Polymorphism Discovery and Genetic Diversity
3.2. Eventual Functional Significance of the SNPs’ Allelic Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Simpson, S., Jr.; Blizzard, L.; Otahal, P.; Van der Mei, I.; Taylor, B. Latitude is significantly associated with the prevalence of multiple sclerosis: A meta-analysis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Ahlgren, C.; Oden, A.; Lycke, J. High nationwide prevalence of multiple sclerosis in Sweden. Mult. Scler. 2011, 17, 901–908. [Google Scholar] [CrossRef]
- Compston, A.; Confavreux, C. The distribution of multiple sclerosis. In McAlpine’s Multiple Sclerosis, 4th ed.; Pioli, S., Ed.; Churchill Livingstone/Elsevier: Philadelphia, PA, USA, 2006; pp. 69–180. [Google Scholar]
- Miller, A.F. Multiple sclerosis: Where will be in 2020? Mt. Sinai. J. Med. 2011, 78, 268–279. [Google Scholar]
- Song, G.G.; Choi, S.J.; Ji, J.D.; Lee, Y.H. Genome-wide pathway analysis of a genome-wide association study on multiple sclerosis. Mol. Biol. Rep. 2013, 40, 2557–2564. [Google Scholar] [CrossRef] [PubMed]
- Patsopoulos, N.A.; Bayer Pharma MS Genetics Working Group; Steering Committees of Studies Evaluating IFNβ-1b and a CCR1-Antagonist; ANZgene Consortium; GeneMSA; International Multiple Sclerosis Genetics Consortium; de Bakker, P.I. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann. Neurol. 2011, 70, 897–912. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium (IMSGC). IL12A, MPHOSPH9/CDK2AP1 and RGS1 are novel multiple sclerosis susceptibility loci. Genes Immun. 2010, 11, 397–405. [Google Scholar] [CrossRef] [Green Version]
- International Multiple Sclerosis Genetics Consortium (IMSGC); Hafler, D.A.; Compston, A.; Sawcer, S.; Lander, E.S.; Daly, M.J.; de Jager, P.L.; de Bakker, P.I.; Gabriel, S.B.; Mirel, D.B.; et al. Risk allels for multiple sclerosis identified by a genome wide study. N. Engl. J. Med. 2007, 357, 851–862. [Google Scholar]
- De Jager, P.L.; Jia, X.; Wang, P.I.; de Bakker, L.; Ottoboni, L.; Aggarval, N.T.; Piccio, L.; Raychaudhuri, S.; Tran, D.; Aubin, C.; et al. Meta-analysis of genome scans and replication identity CD6, IRF8 and TNFRF1A as new multiple sclerosis susceptibility loci. Nat. Genet. 2009, 41, 776–782. [Google Scholar] [CrossRef] [Green Version]
- Kofler, D.M.; Severson, C.A.; Mousissian, N.; de Jager, P.L.; Hafler, D.A. The CD6 multiple sclerosis susceptibility allele is associated with alterations in CD4+ T cell proliferation. J. Immunol. 2011, 187, 3286–3291. [Google Scholar] [CrossRef] [Green Version]
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.; Patsopoulos, N.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Edkins, S.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar]
- Zhang, K.; Chang, S.; Cui, S.; Guo, L.; Zhang, L.; Wang, J. ICSNPathway: Identify candidate causal SNPs and pathways from genome-wide association study by one analytical framework. Nucleic Acids Res. 2011, 39, W437–W443. [Google Scholar] [CrossRef] [PubMed]
- Shiina, T.; Hosomichi, K.; Inoko, H.; Kulski, J.K. The HLA genomic loci map:expression, interaction, diversity and disease. J. Hum. Genet. 2009, 54, 15–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baranzini, S.E.; Wang, J.; Gibson, R.A.; Galwey, N.; Naegelin, Y.; Barkhof, F.; Radue, E.W.; Lindberg, R.L.; Uitdehaag, B.M.; Johnson, M.R.; et al. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum. Mol. Genet. 2009, 18, 767–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharavi, A.G.; Kiryluk, K.; Choi, M.; Li, Y.; Hou, P.; Xie, J.; Sanna-Cherchi, S.; Men, C.J.; Julian, B.A.; Wyatt, R.J.; et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat. Genet. 2011, 43, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Matzaraki, V.; Kumar, V.; Wijmenga, C.; Zhernakova, A. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol. 2017, 18, 76. [Google Scholar] [CrossRef] [PubMed]
- Hill-Burns, E.M.; Factor, S.A.; Zabetian, C.P.; Thomson, G.; Payami, H. Evidence for more than one Parkinson’s disease-associated variant within the HLA region. PLoS ONE 2011, 6, e27109. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Miranda, C.J.; Braun, L.; Meyer, K.; Frakes, A.E.; Ferraiuolo, L.; Likhite, S.; Bevan, A.K.; Foust, K.D.; McConnell, M.J.; et al. Major histocompatibility compex class I molecules protect motor neurons from astrocyte-induced toxicity in amyotrophic lateral sclerosis (ALS). Nat. Med. 2016, 22, 397–403. [Google Scholar] [CrossRef] [Green Version]
- Imrell, K.; Greiner, E.; Hillert, J.; Masterman, T. HLA-DRB115 and cerebrospinal-fluid-specific oligoclonal immunoglobulin G bands lower age at attainment of important disease milestones in multiple sclerosis. J. Neuroimmunol. 2009, 210, 128–130. [Google Scholar] [CrossRef]
- Okuda, D.; Srinivasan, R.; Oksenberg, J.; Goodin, D.; Baranzini, S.; Beheshtian, A.; Waubant, E.; Zamvil, S.S.; Leppert, D.; Qualley, P.; et al. Genotype–phenotype correlations in multiple sclerosis: HLA genes influence disease severity inferred by 1HMR spectroscopy and MRI measures. Brain 2009, 132, 250–259. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, K. A genome-wide association study identifies variants in the HLA-DP locus associated with chronic hepatitis B in Asians. Nat. Genet. 2009, 41, 591–595. [Google Scholar]
- Zhou, X.J.; Qi, Y.Y.; Hou, P.; Lv, J.C.; Shi, S.F.; Liu, L.J.; Zhao, N.; Zhang, H. Cumulative effects of variants identified by genome-wide association studies in IgA nephropathy. Sci. Rep. 2014, 4, 4904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas-Villarraga, A.; Amaya-Amaya, J.; Rodriguez-Rodriguez, A.; Mantilla, R.D.; Anaya, J.M. Introducing polyautoimmunity: Secondary autoimmune diseases no longer exist. Autoimmune Dis. 2012, 2012, 254319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic criteria for multiple sclerosis: 2010 Revisions to the McDonald criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.M. Genetic association studies: Design, analysis and interpretation. Briefings Bioinform. 2002, 3, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Uthoff, S.M.S.; Hunt, L.E.; Grant, B.S.; Young, V.; Eichenberger, M.R.; Cobbs, G.A.; Galandiuk, S. T-Cell Receptor γ: A Microsatellite Marker for Colorectal Cancer. Ann. Surg. Oncol. 2002, 9, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Cartharius, K.; Frech, K.; Grote, K.; Klocke, B.; Haltmeier, M.; Klingenhoff, A.; Frisch, M.; Bayerlein, M.; Werner, T. MatInspector and beyond: Promoter analysis based on transcription factor binding sites. Bioinformatics 2005, 21, 2933–2942. [Google Scholar] [CrossRef] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Morgan, D.; Raff, M.; Roberts, K.; Walter, P. Molecular Biology of the Cell, 6th ed.; Garland Science: New York, NY, USA; Abingdon, UK, 2014; 1464p. [Google Scholar]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Beroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucl. Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Search and Contextual Analysis of Transfer RNA Genes. Nucl. Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucl. Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Deyneko, I.V.; Kel, A.E.; Kel-Margoulis, O.V.; Deineko, E.V.; Wingender, E.; Weiss, S. MatrixCatch-a novel tool for the recognition of composite regulatory elements in promoters. BMC Bioinform. 2013, 14, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksenberg, J.R.; Barcellos, L.F.; Cree, B.A.; Baranzini, S.E.; Bugawan, T.L.; Khan, O.; Lincoln, R.R.; Swerdlin, A.; Mignot, E.; Lin, L.; et al. Mapping multiple sclerosis susceptibility to the HLA-DR locus in African Americans. Am. J. Hum. Genet. 2004, 74, 160–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.H.; Pappas, D.; De Jager, P.L.; Pelletier, D.; de Bakker, P.I.W.; Kappos, L.; Polman, C.H.; Chibnik, L.B.; Hafler, D.A.; Matthews, P.M.; et al. Australian and New Zealand Multiple Sclerosis Genetics Consortium. Modeling the cumulative genetic risk for multiple sclerosis from genome-wide association data. Genome Med. 2011, 3, 3. [Google Scholar] [CrossRef]
- Purnamawati, K.; Ong, J.A.; Deshpande, S.; Tan, W.K.; Masurkar, N.; Low, J.K.; Drum, C.L. The Importance of Sex Stratification in Autoimmune Disease Biomarker Research: A Systematic Review. Front. Immunol. 2018, 9, 1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chanda, P.; Zhang, A.; Sucheston, L.; Ramanathan, M. A two-stage search strategy for detecting multiple loci associated with rheumatoid arthritis. BMC Proc. 2009, 3 (Suppl. 7), S72. [Google Scholar] [CrossRef] [Green Version]
- Alarcón-Riquelme, M.E. A RUNX trio with a taste for autoimmunity. Nat. Genet. 2003, 35, 299–300. [Google Scholar] [CrossRef]
- Yamada, R.; Tokuhiro, S.; Chang, X.; Yamamoto, K. Review SLC22A4 and RUNX1: Identification of RA susceptible genes. J. Mol. Med. 2004, 82, 558–564. [Google Scholar] [CrossRef]
- Wang, W.; Li, G.; Hong, D.; Zou, Y.; Fei, D.; Wang, L. Replication of genome-wide association study identified seven susceptibility genes, affirming the effect of rs2856717 on renal function and poor outcome of IgA nephropathy. Nephrology 2017, 22, 811–817. [Google Scholar] [CrossRef]
- Qin, X.; Wang, C.; Lu, G.; Peng, M.; Cheng, G.; Zhu, H.; Cao, Y.; Liu, J.; Li, Y.; Cai, H.; et al. Risk alleles for IgA nephropathy-associated SNPs conferred completely opposite effects to idiopathic membranous nephropathy in Chinese Han. Immunol. Res. 2017, 65, 1059–1064. [Google Scholar] [CrossRef] [Green Version]
- Hong, X.; Hao, K.; Ladd-Acosta, C.; Hansen, K.D.; Tsai, H.; Liu, X.; Xu, X.; Thornton, T.A.; Caruso, D.; Keet, C.A.; et al. Genome-wide Association Study Identifies Peanut Allergy-Specific Loci and Evidence of Epigenetic Mediation in U.S. Children. Nat. Commun. 2015, 6, 6304. [Google Scholar] [CrossRef]
- Lemaire, S.; Fontrodona, N.; Aubé, F.; Claude, J.B.; Polvèche, H.; Modolo, L.; Bourgeois, C.F.; Mortreux, F.; Auboeuf, D. Characterizing the interplay between gene nucleotide composition bias and splicing. Genome Biol. 2019, 20, 259. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.C.; Zhurkin, V.B.; Louis, J.M.; Cornilescu, G.; Clore, G.M. Structural basis for SRY-dependent 46-X,Y sex reversal: Modulation of DNA bending by a naturally occurring point mutation. J. Mol. Biol. 2001, 312, 481–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, T.; Uchida, H.; Ohta, A.; Horiuchi, H. Involvement of protein kinase C in the suppression of apoptosis and in polarity establishment in Aspergillus nidulans under conditions of heat stress. PLoS ONE 2012, 7, e50503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, R.; Kim, G.D.; Radomska, H.S.; Lekstrom-Himes, J.; Smith, L.T.; Antonson, P.; Tenen, D.G.; Xanthopoulos, K.G. CCAAT/enhancer binding protein epsilon is preferentially up-regulated during granulocytic differentiation and its functional versatility is determined by alternative use of promoters and differential splicing. Proc. Natl. Acad. Sci. USA 1997, 94, 6462–6467. [Google Scholar] [CrossRef] [Green Version]
- Villard, J.; Peretti, M.; Masternak, K.; Barras, E.; Caretti, G.; Mantovani, R.; Reith, W. A functionally essential domain of RFX5 mediates activation of major histocompatibility complex class II promoters by promoting cooperative binding between RFX and NF-Y. Mol. Cell Biol. 2000, 20, 3364–3376. [Google Scholar] [CrossRef]
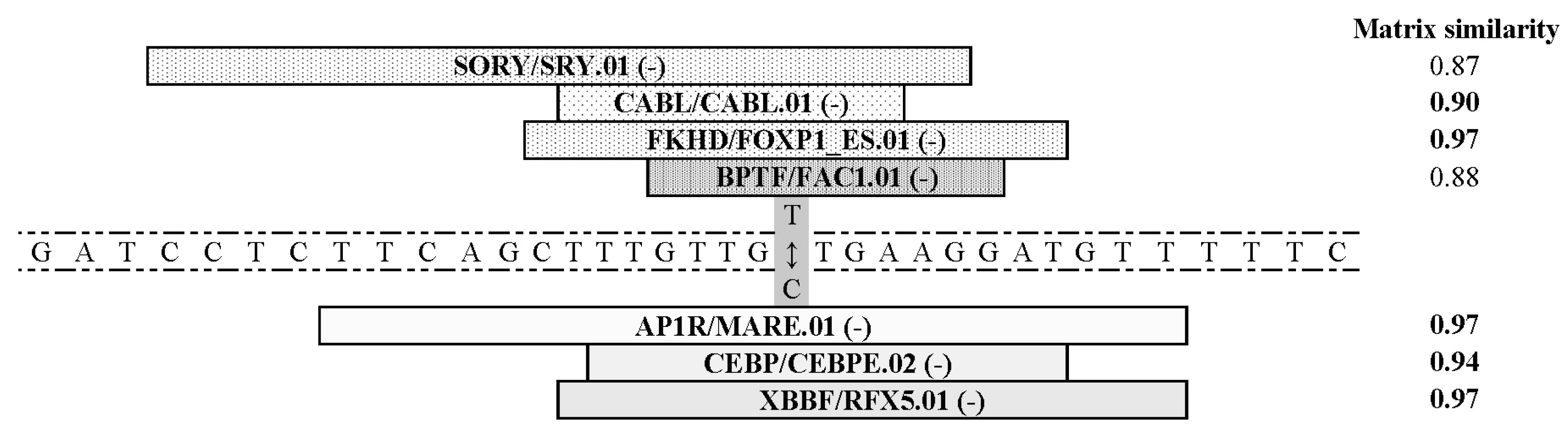
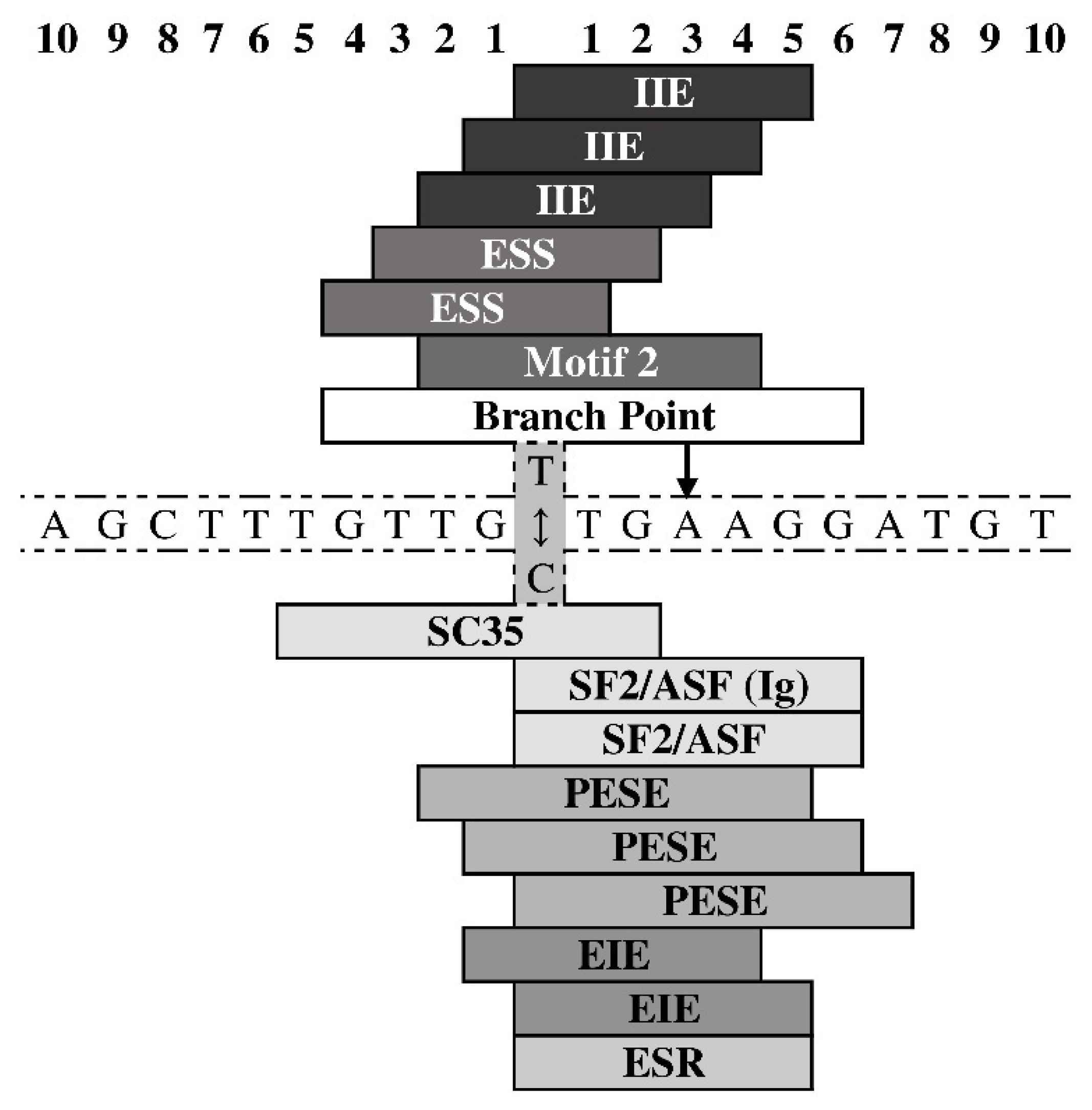
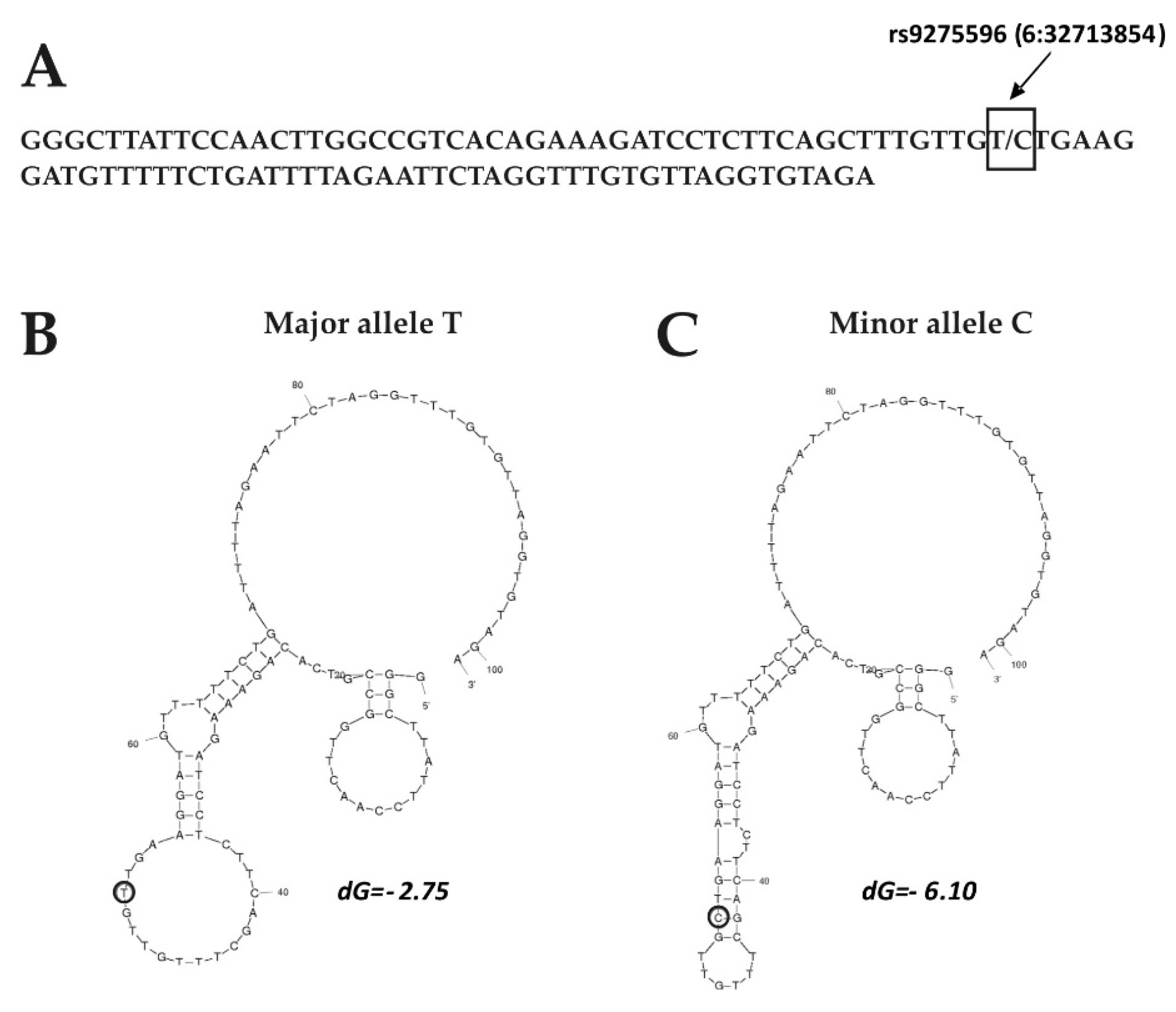


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MAF or Genotype | Distribution of Allele and Genotypes | Data on Association | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Groups | MS * | Controls ^ | Genetic Model | Group | p | OR [95% CI] | |||
| n | % | n | % | ||||||
| C ♦ | Total | 223 | 40.84 | 127 | 30.53 | C vs. T | Total | 0.00099 ** | 1.57 [1.20–2.06] |
| Females | 156 | 40.84 | 71 | 29.10 | Females | 0.0029 * | 1.68 [1.19–2.37] | ||
| Males | 67 | 40.85 | 56 | 32.56 | Males | n.s. | — | ||
| TT | Total | 96 | 35.16 | 103 | 49.52 | TC + CC vs. TT | Total | 0.0015 * | 1.81 [1.25–2.63] |
| Females | 63 | 32.98 | 64 | 52.46 | Females | 0.00062 ** | 2.24 [1.41–3.57] | ||
| Males | 33 | 40.24 | 39 | 45.35 | Males | n.s. | — | ||
| TC | Total | 131 | 47.99 | 83 | 39.90 | TC vs. TT + CC | Total | n.s. (0.08) | — |
| Females | 100 | 52.36 | 45 | 36.89 | Females | 0.0074 * | 1.88 [1.18–2.99] | ||
| Males | 31 | 37.80 | 38 | 44.19 | Males | n.s. | — | ||
| CC | Total | 46 | 16.85 | 22 | 10.58 | CC vs. TT + TC | Total | 0.050 * | 1.71 [0.99–2.95] |
| Females | 28 | 14.66 | 13 | 10.66 | Females | n.s. | — | ||
| Males | 18 | 21.95 | 9 | 10.47 | Males | 0.043 * | 2.41 [1.01–5.72] | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paramonova, N.; Trapina, I.; Dokane, K.; Kalnina, J.; Sjakste, T.; Sjakste, N. An Intergenic rs9275596 Polymorphism on Chr. 6p21 Is Associated with Multiple Sclerosis in Latvians. Medicina 2020, 56, 154. https://doi.org/10.3390/medicina56040154
Paramonova N, Trapina I, Dokane K, Kalnina J, Sjakste T, Sjakste N. An Intergenic rs9275596 Polymorphism on Chr. 6p21 Is Associated with Multiple Sclerosis in Latvians. Medicina. 2020; 56(4):154. https://doi.org/10.3390/medicina56040154
Chicago/Turabian StyleParamonova, Natalia, Ilva Trapina, Kristine Dokane, Jolanta Kalnina, Tatjana Sjakste, and Nikolajs Sjakste. 2020. "An Intergenic rs9275596 Polymorphism on Chr. 6p21 Is Associated with Multiple Sclerosis in Latvians" Medicina 56, no. 4: 154. https://doi.org/10.3390/medicina56040154