Focused Update on Pulmonary Hypertension in Children—Selected Topics of Interest for the Adult Cardiologist


Abstract
:1. Introduction
2. Definition and Classification
3. Group 1 PH (Table 1)
3.1. Vasoreactivity
3.2. Advanced Treatments for Patients Who Are Not Vasoreactive
End-Stage/Bridging Treatment Strategies
3.3. Pulmonary Hypertension in Fontan Physiology
3.4. Eisenmenger Syndrome
Down Syndrome and PHVD
4. Group 2 PH (Table 1)
4.1. Pulmonary Hypertension Due to Left Heart Disease: The Role of Mechanical Support—The Way to Transplantation
4.2. Pulmonary Hypertension Due to Left Heart Disease and Subsequent Heart Alone Versus Heart and Lung Transplantation
5. Group 3 PH (Table 1)
Bronchopulmonary Dysplasia (BPD)
6. Group 4 PH (Table 1)
Chronic Thromboembolic Pulmonary Hypertension
7. Group 5 PH (Table 1)
7.1. Pulmonary Hypertension in Patients with Scimitar Syndrome
- Increased pulmonary blood flow due to anomalous pulmonary venous connection;
- Left-to-right shunt due to the aorto-pulmonary collateral flow and associated cardiac defects (VSD, PDA, ASD);
- Restriction of the pulmonary vascular bed because of pulmonary hypoplasia with subsequent volume overload of the contralateral lung;
- Pulmonary venous obstruction.
7.2. Segmental Pulmonary Hypertension
7.3. PH in Children with Sickle Cell Disease
8. Summary
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| APAH | associated pulmonary arterial hypertension |
| AVT | acute vasoreactivity testing |
| ASD | atrial septal defect |
| AVSD | atrioventricular septal defect |
| BMPRII | bone morphogenetic protein receptor 2 |
| BNP | brain natriuretic peptide |
| BPA | balloon pulmonary angioplasty |
| BPD | Bronchopulmonary dysplasia |
| CCB | calcium channel blocker |
| CHD | congenital heart disease |
| CI | cardiac index |
| COPD | chronic obstructive pulmonary disease |
| CTEPH | chronic thromboembolic pulmonary hypertension |
| DLTX | Double lung transplantation |
| DPG | diastolic pressure gradient (diastolic PAP—mean PAWP) |
| ECMO | extracorporeal membrane oxygenation |
| ES | Eisenmenger Syndrome |
| ERA | endothelin receptor antagonist |
| FC | functional class |
| HPAH | heritable pulmonary arterial hypertension |
| HLHS | hypoplastic left heart syndrome |
| HLTX | Heart lung transplantation |
| HFrEF | heart failure with reduced ejection fraction |
| IPAH | idiopathic pulmonary arterial hypertension |
| LH | left heart disease |
| LV | left ventricle/ventricular |
| LVEDP | left ventricular end-diastolic pressure |
| MAPCAS | major aorto-pulmonary collateral arteries |
| MCS | mechanical circulatory support |
| NO | nitric oxide |
| NT-proBNP | N-terminal pro-brain natriuretic peptide |
| PPAT | Pulmonary artery acceleration time |
| PAH | pulmonary arterial hypertension |
| PAP | pulmonary arterial pressure |
| PAPm | mean pulmonary arterial pressure |
| PAPs | systolic pulmonary arterial pressure |
| PAWP | pulmonary artery wedge pressure |
| PHVD | Pulmonary hypertensive vascular disease |
| PDA | persistent ductus arteriosus |
| PDE-5i | phosphodiesterase type 5 inhibitor |
| PPHN | persistent pulmonary hypertension of the new-born |
| PLE | pleural effusion |
| PVR | pulmonary vascular resistance |
| 6MWD/6MWT | 6-min walking distance/6-min walking test |
| RAP | Right atrial pressure |
| SCD | sickle cell disease |
| S/D ratio | systolic to diastolic duration ratio |
| SVR | systemic vascular resistance |
| T21 | Trisomy 21 or Down Syndrome |
| TPG | transpulmonary gradient (mean PAP- mean PAWP) |
| TAC | Truncus arteriosus communis |
| TRV | tricuspid regurgitation velocity |
| VAD | ventricular assist device |
| VSD | ventricular septal defect |
| WHO-FC | World Health Organization functional class |
| WU | Wood Units |
| WSPH | World Symposium on Pulmonary hypertension |
References
- Berger, R.M.F.; Beghetti, M.; Humpl, T.; Raskob, G.E.; Ivy, D.D.; Jing, Z.-C.; Bonnet, D.; Schulze-Neick, I.; Barst, R.J. Clinical features of paediatric pulmonary hypertension: A registry study. Lancet 2012, 379, 537–546. [Google Scholar] [CrossRef] [Green Version]
- Van Loon, R.L.E.; Roofthooft, M.T.; Hillege, H.L.; Harkel, A.D.T.; van Osch-Gevers, M.; Delhaas, T.; Kapusta, L.; Strengers, J.L.M.; Rammeloo, L.; Clur, S.-A.B.; et al. Pediatric Pulmonary Hypertension in the Netherlands. Circulation 2011, 124, 1755–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraisse, A.; Jaïs, X.; Schleich, J.-M.; Di Filippo, S.; Maragnes, P.; Beghetti, M.; Gressin, V.; Voisin, M.; Dauphin, C.; Clerson, P.; et al. Characteristics and prospective 2-year follow-up of children with pulmonary arterial hypertension in France. Arch. Cardiovasc. Dis. 2010, 103, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, J.; Żuk, M.; Migdal, A.; Kusa, J.; Skiba, E.; Zygielo, K.; Przetocka, K.; Werynski, P.; Banaszak, P.; Rzeznik-Bieniaszewska, A.; et al. Children and Adolescents with Pulmonary Arterial Hypertension: Baseline and Follow-Up Data from the Polish Registry of Pulmonary Hypertension (BNP-PL). J. Clin. Med. 2020, 9, 1717. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, S.; Gibbs, J.S.R.; Wachter, R.; De Marco, T.; Vonk-Noordegraaf, A.; Vachiéry, J.-L. Left ventricular heart failure and pulmonary hypertension. Eur. Heart J. 2015, 37, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, E.B.; Abman, S.H.; Adatia, I.; Beghetti, M.; Bonnet, D.; Haworth, S.; Ivy, D.D.; Berger, R.M. Paediatric pulmonary arterial hypertension: Updates on definition, classification, diagnostics and management. Eur. Respir. J. 2019, 53, 1801916. [Google Scholar] [CrossRef]
- del Cerro, M.J.; Abman, S.; Diaz, G.; Freudenthal, A.H.; Freudenthal, F.; Harikrishnan, S.; Haworth, S.G.; Ivy, D.; Lopes, A.A.; Raj, J.U.; et al. A Consensus Approach to the Classification of Pediatric Pulmonary Hypertensive Vascular Disease: Report from the PVRI Pediatric Taskforce, Panama 2011. Pulm. Circ. 2011, 1, 286–298. [Google Scholar] [CrossRef] [Green Version]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef]
- Kovacs, G.; Berghold, A.; Scheidl, S.; Olschewski, H. Pulmonary arterial pressure during rest and exercise in healthy subjects: A systematic review. Eur. Respir. J. 2009, 34, 888–894. [Google Scholar] [CrossRef] [Green Version]
- Vanderpool, R.R.; Saul, M.; Nouraie, M. Association Between Hemodynamic Markers of Pulmonary Hypertension and Outcomes in Heart Failure With Preserved Ejection Fraction. JAMA Cardiol. 2018, 3, 298–306. [Google Scholar] [CrossRef]
- Valerio, C.J.; Schreiber, B.E.; Handler, C.E.; Denton, C.P.; Coghlan, J.G. Borderline Mean Pulmonary Artery Pressure in Patients with Systemic Sclerosis: Transpulmonary Gradient Predicts Risk of Developing Pulmonary Hypertension. Arthritis Rheum. 2013, 65, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Kolte, D.; Lakshmanan, S.; Jankowich, M.D.; Brittain, E.L.; Maron, B.A.; Choudhary, G. Mild Pulmonary Hypertension Is Associated with Increased Mortality: A Systematic Review and Meta-Analysis. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weitzenblum, E.; Hirth, C.; Ducolone, A.; Mirhom, R.; Rasaholinjanahary, J.; Ehrhart, M. Prognostic value of pulmonary artery pressure in chronic obstructive pulmonary disease. Thorax 1981, 36, 752–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maron, B.A.; Hess, E.; Maddox, T.M.; Opotowsky, A.R.; Tedford, R.J.; Lahm, T.; Joynt, K.E.; Kass, D.J.; Stephens, T.; Stanislawski, M.A.; et al. Association of Borderline Pulmonary Hypertension With Mortality and Hospitalization in a Large Patient Cohort: Insights From the Veterans Affairs Clinical Assessment, Reporting, and Tracking Program. Circulation 2016, 133, 1240–1248. [Google Scholar] [CrossRef] [Green Version]
- Wiedenroth, C.B.; Olsson, K.M.; Guth, S.; Breithecker, A.; Haas, M.; Kamp, J.-C.; Fuge, J.; Hinrichs, J.B.; Roller, F.; Hamm, C.W.; et al. Balloon pulmonary angioplasty for inoperable patients with chronic thromboembolic disease. Pulm. Circ. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Taboada, D.; Pepke-Zaba, J.; Jenkins, D.P.; Berman, M.; Treacy, C.M.; Cannon, J.E.; Toshner, M.; Dunning, J.J.; Ng, C.; Tsui, S.S.; et al. Outcome of pulmonary endarterectomy in symptomatic chronic thromboembolic disease. Eur. Respir. J. 2014, 44, 1635–1645. [Google Scholar] [CrossRef]
- Hansmann, G.; Koestenberger, M.; Alastalo, T.-P.; Apitz, C.; Austin, E.D.; Bonnet, D.; Budts, W.; D’Alto, M.; Gatzoulis, M.A.; Hasan, B.S.; et al. 2019 updated consensus statement on the diagnosis and treatment of pediatric pulmonary hypertension: The European Pediatric Pulmonary Vascular Disease Network (EPPVDN), endorsed by AEPC, ESPR and ISHLT. J. Heart Lung Transplant. 2019, 38, 879–901. [Google Scholar] [CrossRef] [Green Version]
- Kurland, G.; Deterding, R.R.; Hagood, J.S.; Young, L.R.; Brody, A.S.; Castile, R.G.; Dell, S.D.; Fan, L.L.; Hamvas, A.; Hilman, B.C.; et al. An Official American Thoracic Society Clinical Practice Guideline: Classification, Evaluation, and Management of Childhood Interstitial Lung Disease in Infancy. Am. J. Respir. Crit. Care Med. 2013, 188, 376–394. [Google Scholar] [CrossRef] [Green Version]
- Marín, M.J.D.C.; Sabaté-Rotés, A.; Ogando, A.R.; Soto, A.M.; Jiménez, M.Q.; Camacho, J.L.G.; Sonnenfeld, I.R.; Bonora, A.M.; Brotons, D.C.A.; Galdó, A.M. Assessing Pulmonary Hypertensive Vascular Disease in Childhood. Data from the Spanish Registry. Am. J. Respir. Crit. Care Med. 2014, 190, 1421–1429. [Google Scholar] [CrossRef]
- Moledina, S.; Hislop, A.A.; Foster, H.; Schulze-Neick, I.; Haworth, S.G. Childhood idiopathic pulmonary arterial hypertension: A national cohort study. Heart 2010, 96, 1401–1406. [Google Scholar] [CrossRef]
- Lammers, A.E.; Apitz, C.; Zartner, P.; Hager, A.; Dubowy, K.-O.; Hansmann, G. Diagnostics, monitoring and outpatient care in children with suspected pulmonary hypertension/paediatric pulmonary hypertensive vascular disease. Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 2016, 102, ii1–ii13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansmann, G.; Apitz, C.; Abdul-Khaliq, H.; Alastalo, T.-P.; Beerbaum, P.; Bonnet, D.; Dubowy, K.-O.; Gorenflo, M.; Hager, A.; Hilgendorff, A.; et al. Executive summary. Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 2016, 102, ii86–ii100. [Google Scholar] [CrossRef] [PubMed]
- Abman, S.H.; Hansmann, G.; Archer, S.L.; Ivy, D.D.; Adatia, I.; Chung, W.K.; Hanna, B.D.; Rosenzweig, E.B.; Raj, J.U.; Cornfield, D.; et al. Pediatric Pulmonary Hypertension. Circulation 2015, 132, 2037–2099. [Google Scholar] [CrossRef] [PubMed]
- Apitz, C.; Abdul-Khaliq, H.; Albini, S.; Beerbaum, P.; Dubowy, K.O.; Gorenflo, M.; Hager, A.; Hansmann, G.; Hilgendorff, A.; Humpl, T.; et al. Neue hämodynamische Definition der pulmonalen Hypertonie. Monatsschrift Kinderheilkd. 2019, 168, 252–256. [Google Scholar] [CrossRef] [Green Version]
- Hoeper, M.; Bogaard, H.J.; Condliffe, R.; Frantz, R.; Khanna, D.; Kurzyna, M.; Langleben, D.; Manes, A.; Satoh, T.; Torres, F.; et al. Definitions and Diagnosis of Pulmonary Hypertension. J. Am. Coll. Cardiol. 2013, 62, D42–D50. [Google Scholar] [CrossRef] [Green Version]
- Sitbon, O.; Humbert, M.; Jaïs, X.; Ioos, V.; Hamid, A.M.; Provencher, S.; Garcia, G.; Parent, F.; Hervé, P.; Simonneau, G. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005, 111, 3105–3111. [Google Scholar] [CrossRef] [Green Version]
- Douwes, J.M.; Humpl, T.; Bonnet, D.; Beghetti, M.; Ivy, D.D.; Berger, R.M.F.; Weintraub, R.; Geiger, R.; Marx, M.; Jing, Z.; et al. Acute Vasodilator Response in Pediatric Pulmonary Arterial Hypertension. J. Am. Coll. Cardiol. 2016, 67, 1312–1323. [Google Scholar] [CrossRef]
- Barst, R.J.; McGoon, M.D.; Elliott, C.G.; Foreman, A.J.; Miller, D.P.; Ivy, D.D. Survival in Childhood Pulmonary Arterial Hypertension. Circulation 2012, 125, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Apitz, C.; Hansmann, G.; Schranz, D. Hemodynamic assessment and acute pulmonary vasoreactivity testing in the evaluation of children with pulmonary vascular disease. Expert consensus statement on the diagnosis and treatment of paediatric pulmonary hypertension. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 2016, 102, ii23–ii29. [Google Scholar] [CrossRef] [Green Version]
- Ivy, D.D.; Abman, S.H.; Barst, R.J.; Berger, R.M.; Bonnet, D.; Fleming, T.R.; Haworth, S.G.; Raj, J.U.; Rosenzweig, E.B.; Neick, I.S.; et al. Pediatric Pulmonary Hypertension. J. Am. Coll. Cardiol. 2013, 62, D117–D126. [Google Scholar] [CrossRef]
- Law, M.A.; Grifka, R.G.; Mullins, C.E.; Nihill, M.R. Atrial septostomy improves survival in select patients with pulmonary hypertension. Am. Heart J. 2007, 153, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Lammers, A.E.; Haworth, S.G.; Diller, G.-P. Atrial septostomy in patients with pulmonary hypertension: Should it be recommended? Expert Rev. Respir. Med. 2011, 5, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Memon, M.M.; Amin, E.; Yamani, N.; Khan, S.U.; Figueredo, V.M.; Deo, S.; Rich, J.D.; Benza, R.L.; Krasuski, R.A. Use of Balloon Atrial Septostomy in Patients With Advanced Pulmonary Arterial Hypertension: A Systematic Review and Meta-Analysis. Chest 2019, 156, 53–63. [Google Scholar] [CrossRef]
- Sandoval, J.; Rothman, A.; Pulido, T. Atrial Septostomy for Pulmonary Hypertension. Clin. Chest Med. 2001, 22, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Latus, H.; Delhaas, T.; Schranz, D.; Apitz, C. Treatment of pulmonary arterial hypertension in children. Nat. Rev. Cardiol. 2015, 12, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Keogh, A.; Mayer, E.; Benza, R.L.; Corris, P.; Dartevelle, P.G.; Frost, A.E.; Kim, N.H.; Lang, I.M.; Pepke-Zaba, J.; Sandoval, J. Interventional and Surgical Modalities of Treatment in Pulmonary Hypertension. J. Am. Coll. Cardiol. 2009, 54, S67–S77. [Google Scholar] [CrossRef] [Green Version]
- Klepetko, W.; Mayer, E.; Sandoval, J.; Trulock, E.P.; Vachiéry, J.-L.; Dartevelle, P.; Pepke-Zaba, J.; Jamieson, S.W.; Lang, I.; Corris, P. Interventional and surgical modalities of treatment for pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, S73–S80. [Google Scholar] [CrossRef]
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Gérald, S.; Andrew, P.; Anton, V.N.; Maurice, B.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS)Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar]
- Baruteau, A.-E.; Belli, E.; Boudjemline, Y.; Laux, D.; Lévy, M.; Simonneau, G.; Carotti, A.; Humbert, M.; Bonnet, D. Palliative Potts shunt for the treatment of children with drug-refractory pulmonary arterial hypertension: Updated data from the first 24 patients. Eur. J. Cardio-Thoracic Surg. 2014, 47, e105–e110. [Google Scholar] [CrossRef]
- Blanc, J.; Vouhe, P.; Bonnet, D. Potts Shunt in Patients with Pulmonary Hypertension. N. Engl. J. Med. 2004, 350, 623. [Google Scholar] [CrossRef] [Green Version]
- Grady, R.M.; Eghtesady, P.; Information, P.E.K.F.C. Potts Shunt and Pediatric Pulmonary Hypertension: What We Have Learned. Ann. Thorac. Surg. 2016, 101, 1539–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esch, J.J.; Shah, P.B.; Cockrill, B.A.; Farber, H.W.; Landzberg, M.J.; Mehra, M.R.; Mullen, M.P.; Opotowsky, A.R.; Waxman, A.B.; Lock, J.; et al. Transcatheter Potts shunt creation in patients with severe pulmonary arterial hypertension: Initial clinical experience. J. Heart Lung Transplant. 2013, 32, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Latus, H.; Apitz, C.; Moysich, A.; Kerst, G.; Jux, C.; Bauer, J.; Schranz, D. Creation of a functional Potts shunt by stenting the persistent arterial duct in newborns and infants with suprasystemic pulmonary hypertension of various etiologies. J. Heart Lung Transplant. 2014, 33, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, V.; Petit, C.J.; Glatz, A.C.; Goldstein, B.H.; Qureshi, A.M. Stenting of the ductus arteriosus for ductal-dependent pulmonary blood flow—current techniques and procedural considerations. Congenital Heart Disease 2019, 14, 110–115. [Google Scholar] [CrossRef] [Green Version]
- Michel-Behnke, I.; Akintuerk, H.; Marquardt, I.; Mueller, M.; Thul, J.; Bauer, J.; Hagel, K.J.; Kreuder, J.; Vogt, P.; Schranz, D. Stenting of the ductus arteriosus and banding of the pulmonary arteries: Basis for various surgical strategies in newborns with multiple left heart obstructive lesions. Heart 2003, 89, 645–650. [Google Scholar] [CrossRef] [Green Version]
- Rosenzweig, E.; Ankola, A.; Krishnan, U.; Middlesworth, W.; Bacha, E.; Bacchetta, M. A novel unidirectional-valved shunt approach for end-stage pulmonary arterial hypertension: Early experience in adolescents and adults. J. Thorac. Cardiovasc. Surg. 2019. [Google Scholar] [CrossRef]
- Salna, M.; van Boxtel, B.; Rosenzweig, E.B.; Bacchetta, M. Modified Potts Shunt in an Adult with Idiopathic Pulmonary Arterial Hypertension. Ann. Am. Thorac. Soc. 2017, 14, 607–609. [Google Scholar] [CrossRef]
- Bartolome, S.; Hoeper, M.; Klepetko, W. Advanced pulmonary arterial hypertension: Mechanical support and lung transplantation. Eur. Respir. Rev. 2017, 26, 170089. [Google Scholar] [CrossRef] [Green Version]
- Hayes, N.; Cherikh, W.S.; Chambers, D.C.; Harhay, M.O.; Khush, K.K.; Lehman, R.R.; Meiser, B.; Rossano, J.W.; Hsich, E.; Potena, L.; et al. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Twenty-second pediatric lung and heart-lung transplantation report-2019; Focus theme: Donor and recipient size match. J. Heart Lung Transplant. 2019, 38, 1015–1027. [Google Scholar] [CrossRef]
- Kirkby, S.; Hayes, N. Pediatric lung transplantation: Indications and outcomes. J. Thorac. Dis. 2014, 6, 1024–1031. [Google Scholar]
- Le Pavec, J.; Hascoet, S.; Fadel, E. Heart-lung transplantation: Current indications, prognosis and specific considerations. J. Thorac. Dis. 2018, 10, 5946–5952. [Google Scholar] [CrossRef] [PubMed]
- Olland, A.; Falcoz, P.-E.; Canuet, M.; Massard, G. Should we perform bilateral-lung or heart-lung transplantation for patients with pulmonary hypertension? Interact. Cardiovasc. Thorac. Surg. 2013, 17, 166–170. [Google Scholar] [CrossRef] [PubMed]
- D’Udekem, Y.; Iyengar, A.J.; Galati, J.C.; Forsdick, V.; Weintraub, R.G.; Wheaton, G.R.; Bullock, A.; Justo, R.N.; Grigg, L.E.; Sholler, G.F.; et al. Redefining Expectations of Long-Term Survival After the Fontan Procedure: Twenty-Five Years of Follow-Up From the Entire Population of Australia and New Zealand. Circulation 2014, 130, S32–S38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenland, P.; Lloyd-Jones, D. Time to End the Mixed—and Often Incorrect—Messages About Prevention and Treatment of Atherosclerotic Cardiovascular Disease⁎⁎Editorials published in the Journal of the American College of Cardiologyreflect the views of the authors and do not necessarily represent the views of JACCor the American College of Cardiology. J. Am. Coll. Cardiol. 2007, 50, 2133–2135. [Google Scholar] [CrossRef] [Green Version]
- Helbing, W.A.; Bosch, E.V.D.; Bogers, A.J.; Helbing, W.A. State of the art of the Fontan strategy for treatment of univentricular heart disease. F1000Research 2018, 7, 935. [Google Scholar] [CrossRef]
- Clift, P.; Celermajer, D.S. Managing adult Fontan patients: Where do we stand? Eur. Respir. Rev. 2016, 25, 438–450. [Google Scholar] [CrossRef] [Green Version]
- Egbe, A.C.; Connolly, H.M.; Miranda, W.R.; Ammash, N.M.; Hagler, N.J.; Veldtman, G.R.; Borlaug, B.A. Hemodynamics of Fontan Failure: The Role of Pulmonary Vascular Disease. Circ. Heart Fail. 2017, 10. [Google Scholar] [CrossRef]
- Hauck, A.; Porta, N.F.M.; Lestrud, S.; Berger, S.; Lestrud, S. The Pulmonary Circulation in the Single Ventricle Patient. Children 2017, 4, 71. [Google Scholar] [CrossRef] [Green Version]
- Gewillig, M.; Goldberg, D.J. Failure of the Fontan Circulation. Heart Fail. Clin. 2014, 10, 105–116. [Google Scholar] [CrossRef]
- Ridderbos, F.-J.S.; Wolff, D.; Timmer, A.; van Melle, J.P.; Ebels, T.; Dickinson, M.G.; Timens, W.; Berger, R.M.F. Adverse pulmonary vascular remodeling in the Fontan circulation. J. Heart Lung Transplant. 2015, 34, 404–413. [Google Scholar] [CrossRef]
- Raj, J.U.; Kääpä, P.; Anderson, J. Effect of pulsatile flow on microvascular resistance in adult rabbit lungs. J. Appl. Physiol. 1992, 72, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Hakim, T.S. Flow-induced release of EDRF in the pulmonary vasculature: Site of release and action. Am. J. Physiol. Circ. Physiol. 1994, 267, H363–H369. [Google Scholar] [CrossRef] [PubMed]
- Henaine, R.; Vergnat, M.; Bacha, E.A.; Baudet, B.; Lambert, V.; Belli, E.; Serraf, A. Effects of lack of pulsatility on pulmonary endothelial function in the Fontan circulation. J. Thorac. Cardiovasc. Surg. 2013, 146, 522–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snarr, B.S.; Paridon, S.M.; Rychik, J.; Goldberg, D.J. Pulmonary vasodilator therapy in the failing Fontan circulation: Rationale and efficacy. Cardiol. Young 2015, 25, 1489–1492. [Google Scholar] [CrossRef] [PubMed]
- Ishida, H.; Kogaki, S.; Takahashi, K.; Ozono, K. Attenuation of bone morphogenetic protein receptor type 2 expression in the pulmonary arteries of patients with failed Fontan circulation. J. Thorac. Cardiovasc. Surg. 2012, 143, e24–e26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishida, H.; Kogaki, S.; Ichimori, H.; Narita, J.; Nawa, N.; Ueno, T.; Takahashi, K.; Kayatani, F.; Kishimoto, H.; Nakayama, M.; et al. Overexpression of endothelin-1 and endothelin receptors in the pulmonary arteries of failed Fontan patients. Int. J. Cardiol. 2012, 159, 34–39. [Google Scholar] [CrossRef]
- Chowdhury, U.K.; Govindappa, R.M.; Das, P.; Ray, R.; Kalaivani, M.; Reddy, S.M. Histomorphometric analysis of intrapulmonary vessels in patients undergoing bidirectional Glenn shunt and total cavopulmonary connection. J. Thorac. Cardiovasc. Surg. 2010, 140, 1251–1256.e14. [Google Scholar] [CrossRef] [Green Version]
- Goldman, A.P.; Delius, R.E.; Deanfield, J.E.; Miller, O.I.; de Leval, M.R.; Sigston, P.E.; Macrae, D.J. Pharmacological control of pulmonary blood flow with inhaled nitric oxide after the fenestrated Fontan operation. Circulation 1996, 94 (Suppl. S9), II44–II48. [Google Scholar]
- Agarwal, H.S.; Churchwell, K.B.; Doyle, T.P.; Christian, K.G.; Drinkwater, D.C.; Byrne, D.W.; Taylor, M.B. Inhaled Nitric Oxide Use in Bidirectional Glenn Anastomosis for Elevated Glenn Pressures. Ann. Thorac. Surg. 2006, 81, 1429–1434. [Google Scholar] [CrossRef]
- Urcelay, G.E.; Borzutzky, A.J.; Becker, P.A.; Castillo, M.E. Nitric Oxide in Pulmonary Arteriovenous Malformations and Fontan Procedure. Ann. Thorac. Surg. 2005, 80, 338–340. [Google Scholar] [CrossRef]
- Khambadkone, S.; Li, J.; de Leval, M.; Cullen, S.; Deanfield, J.; Redington, A. Basal Pulmonary Vascular Resistance and Nitric Oxide Responsiveness Late after Fontan-Type Operation. Circulation 2003, 107, 3204–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latus, H.; Gerstner, B.; Kerst, G.; Moysich, A.; Gummel, K.; Apitz, C.; Bauer, J.; Schranz, D. Effect of Inhaled Nitric Oxide on Blood Flow Dynamics in Patients After the Fontan Procedure Using Cardiovascular Magnetic Resonance Flow Measurements. Pediatr. Cardiol. 2015, 37, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Giardini, A.; Balducci, A.; Specchia, S.; Gargiulo, G.; Bonvicini, M.; Picchio, F.M. Effect of sildenafil on haemodynamic response to exercise and exercise capacity in Fontan patients. Eur. Heart J. 2008, 29, 1681–1687. [Google Scholar] [CrossRef] [PubMed]
- Uzun, O.; Wong, J.K.; Bhole, V.; Stumper, O. Resolution of Protein-Losing Enteropathy and Normalization of Mesenteric Doppler Flow with Sildenafil after Fontan. Ann. Thorac. Surg. 2006, 82, e39–e40. [Google Scholar] [CrossRef] [PubMed]
- Haseyama, K.; Satomi, G.; Yasukochi, S.; Matsui, H.; Harada, Y.; Uchita, S. Pulmonary vasodilation therapy with sildenafil citrate in a patient with plastic bronchitis after the Fontan procedure for hypoplastic left heart syndrome. J. Thorac. Cardiovasc. Surg. 2006, 132, 1232–1233. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, D.J.; French, B.; McBride, M.G.; Marino, B.S.; Mirarchi, N.; Hanna, B.D.; Wernovsky, G.; Paridon, S.M.; Rychik, J. Impact of Oral Sildenafil on Exercise Performance in Children and Young Adults After the Fontan Operation. Circulation 2011, 123, 1185–1193. [Google Scholar] [CrossRef]
- Van De Bruaene, A.; La Gerche, A.; Claessen, G.; De Meester, P.; Devroe, S.; Gillijns, H.; Bogaert, J.; Claus, P.; Heidbuchel, H.; Gewillig, M.; et al. Sildenafil Improves Exercise Hemodynamics in Fontan Patients. Circ. Cardiovasc. Imaging 2014, 7, 265–273. [Google Scholar] [CrossRef] [Green Version]
- Do, P.; Randhawa, I.; Chin, T.; Parsapour, K.; Nussbaum, E. Successful Management of Plastic Bronchitis in a Child Post Fontan: Case Report and Literature Review. Lung 2012, 190, 463–468. [Google Scholar] [CrossRef] [Green Version]
- Butts, R.J.; Chowdhury, S.M.; Baker, G.H.; Bandisode, V.; Savage, A.J.; Atz, A.M. Effect of Sildenafil on Pressure-Volume Loop Measures of Ventricular Function in Fontan Patients. Pediatr. Cardiol. 2015, 37, 184–191. [Google Scholar] [CrossRef]
- Amedro, P.; Gavotto, A.; Abassi, H.; Picot, M.; Matecki, S.; Malekzadeh-Milani, S.; Levy, M.; Ladouceur, M.; Ovaert, C.; Aldebert, P.; et al. Efficacy of phosphodiesterase type 5 inhibitors in univentricular congenital heart disease: The SV-INHIBITION study design. ESC Heart Fail. 2020, 7, 747–756. [Google Scholar] [CrossRef] [Green Version]
- Bowater, S.E.; Weaver, R.A.; Thorne, S.A.; Clift, P. The Safety and Effects of Bosentan in Patients with a Fontan Circulation. Congenit. Heart Dis. 2012, 7, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Hebert, A.; Mikkelsen, U.R.; Thilén, U.; Idorn, L.; Jensen, A.S.; Nagy, E.; Hanseus, K.; Sørensen, K.E.; Søndergaard, L.; Søendergaard, L. Bosentan Improves Exercise Capacity in Adolescents and Adults After Fontan Operation. Circulation 2014, 130, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Lu, R.; Zhang, X.; Zhang, C.; Xiao, S.; Liu, M.; Wang, B.; Dong, N.-G. Efficacy of bosentan in patients after fontan procedures: A double-blind, randomized controlled trial. J. Huazhong Univ. Sci. Technol. 2016, 36, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Schuuring, M.J.; Vis, J.C.; Van Dijk, A.P.J.; Van Melle, J.P.; Vliegen, H.W.; Pieper, P.G.; Sieswerda, G.T.; De Bruin-Bon, R.H.; Mulder, B.J.; Bouma, B.J.; et al. Impact of bosentan on exercise capacity in adults after the Fontan procedure: A randomized controlled trial. Eur. J. Heart Fail. 2013, 15, 690–698. [Google Scholar] [CrossRef]
- Agnoletti, G.; Gala, S.; Ferroni, F.; Bordese, R.; Appendini, L.; Napoleone, C.P.; Bergamasco, L. Endothelin inhibitors lower pulmonary vascular resistance and improve functional capacity in patients with Fontan circulation. J. Thorac. Cardiovasc. Surg. 2017, 153, 1468–1475. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, J.; Tikkanen, A.U.; Clair, M.; Fernandes, S.M.; Graham, D.A.; Milliren, C.E.; Daly, K.P.M.; Mullen, M.P.; Landzberg, M.J. Effect of inhaled iloprost on the exercise function of Fontan patients: A demonstration of concept. Int. J. Cardiol. 2013, 168, 2435–2440. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.H.; Chae, M.H.; Choi, D.Y. Inhaled iloprost for the treatment of patient with Fontan circulation. Korean J. Pediatr. 2014, 57, 461–463. [Google Scholar] [CrossRef] [Green Version]
- Grosse-Wortmann, L.; Al-Otay, A.; Yoo, S.-J. Aortopulmonary Collaterals after Bidirectional Cavopulmonary Connection or Fontan Completion. Circ. Cardiovasc. Imaging 2009, 2, 219–225. [Google Scholar] [CrossRef] [Green Version]
- Kutty, S.; Rathod, R.H.; Danford, D.A.; Celermajer, D.S. Role of imaging in the evaluation of single ventricle with the Fontan palliation. Heart 2015, 102, 174–183. [Google Scholar] [CrossRef]
- Fogel, M.A.; Pawlowski, T.W.; Whitehead, K.K.; Harris, M.A.; Keller, M.S.; Glatz, A.C.; Zhu, W.; Shore, D.; Diaz, L.K.; Rome, J.J. Cardiac Magnetic Resonance and the Need for Routine Cardiac Catheterization in Single Ventricle Patients Prior to Fontan: A Comparison of 3 Groups. J. Am. Coll. Cardiol. 2012, 60, 1094–1102. [Google Scholar] [CrossRef] [Green Version]
- Kaemmerer, H.; Mebus, S.; Schulze-Neick, I.; Eicken, A.; Trindade, P.T.; Hager, A.; Oechslin, E.; Niwa, K.; Lang, I.; Hess, J. The Adult Patient with Eisenmenger Syndrome: A Medical Update after Dana Point Part I: Epidemiology, Clinical Aspects and Diagnostic Options. Curr. Cardiol. Rev. 2010, 6, 343–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, P. The Eisenmenger Syndrome: II. BMJ 1958, 2, 755–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niwa, K.; Perloff, J.K.; Kaplan, S.; Child, J.S.; Miner, P.D. Eisenmenger syndrome in adults. J. Am. Coll. Cardiol. 1999, 34, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Cantor, W.J.; Harrison, D.; Moussadji, J.S.; Connelly, M.S.; Webb, G.D.; Liu, P.; McLaughlin, P.R.; Siu, S.C. Determinants of survival and length of survival in adults with Eisenmenger syndrome. Am. J. Cardiol. 1999, 84, 677–681. [Google Scholar] [CrossRef]
- Diller, G.-P.; Dimopoulos, K.; Broberg, C.S.; Kaya, M.G.; Naghotra, U.S.; Uebing, A.; Harries, C.; Goktekin, O.; Gibbs, J.S.R.; Gatzoulis, M.A. Presentation, survival prospects, and predictors of death in Eisenmenger syndrome: A combined retrospective and case-control study. Eur. Heart J. 2006, 27, 1737–1742. [Google Scholar] [CrossRef] [Green Version]
- Korten, M.; Helm, P.; Abdul-Khaliq, H.; Baumgartner, H.; Kececioglu, D.; Schlensak, C.; Ulrike, M.M.B.; Gerhard-Paul, D.; Competence Network for Congenital Heart Defects Investigators. Competence Network for Congenital Heart Defects Investigators Eisenmenger syndrome and long-term survival in patients with Down syndrome and congenital heart disease. Heart 2016, 102, 1552–1557. [Google Scholar] [CrossRef]
- Diller, G.-P.; Körten, M.-A.; Bauer, U.M.; Miera, O.; Tutarel, O.; Kaemmerer, H.; Berger, F.; Baumgartner, H. Current therapy and outcome of Eisenmenger syndrome: Data of the German National Register for congenital heart defects. Eur. Heart J. 2016, 37, 1449–1455. [Google Scholar] [CrossRef]
- Duffels, M.; Engelfriet, P.; Berger, R.; Van Loon, R.; Hoendermis, E.; Vriend, J.W.; Van Der Velde, E.T.; Bresser, P.; Mulder, B. Pulmonary arterial hypertension in congenital heart disease: An epidemiologic perspective from a Dutch registry. Int. J. Cardiol. 2007, 120, 198–204. [Google Scholar] [CrossRef]
- Engelfriet, P.M.; Duffels, M.G.J.; Möller, T.; Boersma, E.; Tijssen, J.G.P.; Thaulow, E.; Gatzoulis, M.A.; Mulder, B.J. Pulmonary arterial hypertension in adults born with a heart septal defect: The Euro Heart Survey on adult congenital heart disease. Heart 2006, 93, 682–687. [Google Scholar] [CrossRef] [Green Version]
- Van De Bruaene, A.; Delcroix, M.; Pasquet, A.; De Backer, J.; De Pauw, M.; Naeije, R.; Vachiery, J.-L.; Paelinck, B.; Morissens, M.; Budts, W. The Belgian Eisenmenger syndrome registry: Implications for treatment strategies? Acta Cardiol. 2009, 64, 447–453. [Google Scholar] [CrossRef]
- Li, L.; Jick, S.; Breitenstein, S.; Hernandez, G.; Michel, A.; Vizcaya, D. Pulmonary arterial hypertension in the USA: An epidemiological study in a large insured pediatric population. Pulm. Circ. 2017, 7, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Haworth, S.G.; Hislop, A.A. Treatment and survival in children with pulmonary arterial hypertension: The UK Pulmonary Hypertension Service for Children 2001–2006. Heart 2009, 95, 312–317. [Google Scholar] [CrossRef]
- Frank, D.B.; Hanna, B.D. Pulmonary arterial hypertension associated with congenital heart disease and Eisenmenger syndrome: Current practice in pediatrics. Minerva Pediatr. 2015, 67, 169–185. [Google Scholar]
- D’Alto, M.; Diller, G.-P. Pulmonary hypertension in adults with congenital heart disease and Eisenmenger syndrome: Current advanced management strategies. Heart 2014, 100, 1322–1328. [Google Scholar] [CrossRef]
- Oechslin, E. Management of adults with cyanotic congenital heart disease. Heart 2014, 101, 485–494. [Google Scholar] [CrossRef]
- Chaix, M.-A.; Gatzoulis, M.A.; Diller, G.-P.; Khairy, P.; Oechslin, E. Eisenmenger Syndrome: A Multisystem Disorder-Do Not Destabilize the Balanced but Fragile Physiology. Can. J. Cardiol. 2019, 35, 1664–1674. [Google Scholar] [CrossRef]
- Sandoval, J.; Aguirre, J.S.; Pulido, T.; Martínez-Guerra, M.L.; Santos, E.; Alvarado, P.; Rosas, M.; Bautista, E. Nocturnal Oxygen Therapy in Patients with the Eisenmenger Syndrome. Am. J. Respir. Crit. Care Med. 2001, 164, 1682–1687. [Google Scholar] [CrossRef] [Green Version]
- Daliento, L.; Somerville, J.; Presbitero, P.; Menti, L.; Brach-Prever, S.; Rizzoli, G.; Stone, S. Eisenmenger syndrome. Factors relating to deterioration and death. Eur. Heart J. 1998, 19, 1845–1855. [Google Scholar] [CrossRef] [Green Version]
- Opotowsky, A.R.; Landzberg, M.J.; Beghetti, M. The Exceptional and Far-Flung Manifestations of Heart Failure in Eisenmenger Syndrome. Heart Fail. Clin. 2014, 10, 91–104. [Google Scholar] [CrossRef]
- Galiè, N. Bosentan Therapy in Patients with Eisenmenger Syndrome: A Multicenter, Double-Blind, Randomized, Placebo-Controlled Study. Circulation 2006, 114, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Apostolopoulou, S.C.; Manginas, A.; Cokkinos, D.V.; Rammos, S. Long-term oral bosentan treatment in patients with pulmonary arterial hypertension related to congenital heart disease: A 2-year study. Heart 2006, 93, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Hislop, A.A.; Moledina, S.; Foster, H.; Schulze-Neick, I.; Haworth, S.G. Long-term efficacy of bosentan in treatment of pulmonary arterial hypertension in children. Eur. Respir. J. 2010, 38, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-N.; Jiang, X.; Zhang, R.; Li, X.-L.; Wu, B.; Zhao, Q.-H.; Wang, Y.; Dai, L.-Z.; Pan, L.; Gomberg-Maitland, M.; et al. Oral sildenafil treatment for Eisenmenger syndrome: A prospective, open-label, multicentre study. Heart 2011, 97, 1876–1881. [Google Scholar] [CrossRef] [PubMed]
- Humpl, T.; Reyes, J.; Holtby, H.; Stephens, D.; Adatia, I. Beneficial Effect of Oral Sildenafil Therapy on Childhood Pulmonary Arterial Hypertension. Twelve-Month Clinical Trial of a Single-Drug, Open-Label, Pilot Study. ACC Curr. J. Rev. 2005, 14, 57. [Google Scholar] [CrossRef]
- Barst, R.J.; Ivy, D.D.; Gaitan, G.; Szatmari, A.; Rudzinski, A.; Garcia, A.E.; Sastry, B.K.S.; Pulido, T.; Layton, G.R.; Serdarevic-Pehar, M.; et al. A Randomized, Double-Blind, Placebo-Controlled, Dose-Ranging Study of Oral Sildenafil Citrate in Treatment-Naive Children with Pulmonary Arterial Hypertension. Circulation 2012, 125, 324–334. [Google Scholar] [CrossRef]
- Arnott, C.; Strange, G.; Bullock, A.; Kirby, A.C.; O’Donnell, C.; Radford, D.J.; Grigg, L.E.; Celermajer, D.S. Pulmonary vasodilator therapy is associated with greater survival in Eisenmenger syndrome. Heart 2017, 104, 732–737. [Google Scholar] [CrossRef]
- Hascoet, S.; Fournier, E.; Jaïs, X.; Le Gloan, L.; Dauphin, C.; Houeijeh, A.; Godart, F.; Iriart, X.; Richard, A.; Radojevic, J.; et al. Outcome of adults with Eisenmenger syndrome treated with drugs specific to pulmonary arterial hypertension: A French multicentre study. Arch. Cardiovasc. Dis. 2017, 110, 303–316. [Google Scholar] [CrossRef]
- Gatzoulis, M.A.; Landzberg, M.; Beghetti, M.; Berger, R.M.; Efficace, M.; Gesang, S.; He, J.; Papadakis, K.; Pulido, T.; Galiè, N.; et al. Evaluation of Macitentan in Patients With Eisenmenger Syndrome. Circulation 2018, 139, 51–63. [Google Scholar] [CrossRef]
- Rosenzweig, E.B.; Kerstein, D.; Barst, R.J. Long-term prostacyclin for pulmonary hypertension with associated congenital heart defects. Circulation 1999, 99, 1858–1865. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, S.M.; Newburger, J.W.; Lang, P.; Pearson, D.D.; Feinstein, J.A.; Gauvreau, K.; Landzberg, M.J. Usefulness of epoprostenol therapy in the severely ill adolescent/adult with Eisenmenger physiology. Am. J. Cardiol. 2003, 91, 632–635. [Google Scholar] [CrossRef]
- Skoro-Sajer, N.; Gerges, C.; Bálint, O.H.; Kőhalmi, D.; Kaldararova, M.; Šimková, I.; Jakowitsch, J.; Gabriel, H.; Baumgartner, H.; Gerges, M.; et al. Subcutaneous treprostinil in congenital heart disease-related pulmonary arterial hypertension. Heart 2018, 104, 1195–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, H.S.; Kim, G.B.; Song, M.K.; Bang, J.S.; Lee, S.Y.; Bae, E.J.; Noh, C.I. Eisenmenger Syndrome in Adults: Treatment Pattern and Prognostic Factors in the Advanced Pulmonary Vasodilator Era. Pediatr. Cardiol. 2018, 40, 23–28. [Google Scholar] [CrossRef]
- Benza, R.L.; Miller, D.P.; Barst, R.J.; Badesch, D.B.; Frost, A.E.; McGoon, M.D. An Evaluation of Long-term Survival from Time of Diagnosis in Pulmonary Arterial Hypertension from the REVEAL Registry. Chest 2012, 142, 448–456. [Google Scholar] [CrossRef]
- Hjortshøj, C.M.S.; Kempny, A.; Jensen, A.S.; Sørensen, K.; Nagy, E.; Dellborg, M.; Bengt, J.; Virginija, R.; Gu, H.; Alexander, R.O.; et al. Past and current cause-specific mortality in Eisenmenger syndrome. Eur. Heart J. 2017, 38, 2060–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempny, A.; Hjortshøj, C.S.; Gu, H.; Li, W.; Opotowsky, A.R.; Landzberg, M.J.; Jensen, A.S.; Søndergaard, L.; Estensen, M.-E.; Thilén, U.; et al. Predictors of Death in Contemporary Adult Patients with Eisenmenger Syndrome. Circulation 2017, 135, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D. Molecular genetic analysis of Down syndrome. Qual. Life Res. 2009, 126, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, F.K.; Alford, K.A.; Tybulewicz, V.L.; Fisher, E.M. Down syndrome—Recent progress and future prospects. Hum. Mol. Genet. 2009, 18, R75–R83. [Google Scholar] [CrossRef] [Green Version]
- Saji, T. Clinical characteristics of pulmonary arterial hypertension associated with Down syndrome. Pediatr. Int. 2014, 56, 297–303. [Google Scholar] [CrossRef]
- Cua, C.; Blankenship, A.; North, A.L.; Hayes, J.; Nelin, L.D. Increased Incidence of Idiopathic Persistent Pulmonary Hypertension in Down Syndrome Neonates. Pediatr. Cardiol. 2007, 28, 250–254. [Google Scholar] [CrossRef]
- Bush, D.; Abman, S.H.; Galambos, C. Prominent Intrapulmonary Bronchopulmonary Anastomoses and Abnormal Lung Development in Infants and Children with Down Syndrome. J. Pediatr. 2017, 180, 156–162.e1. [Google Scholar] [CrossRef]
- Galambos, C.; Minic, A.D.; Bush, D.; Nguyen, D.; Dodson, B.; Seedorf, G.; Abman, S.H. Increased Lung Expression of Anti-Angiogenic Factors in Down Syndrome: Potential Role in Abnormal Lung Vascular Growth and the Risk for Pulmonary Hypertension. PLoS ONE 2016, 11, e0159005. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, O.; Dominguez, C.; Ruiz, A.; Ribera, I.; Reig, J.A.; Cabero, L.; Carreras, E.; Llurba, E. Angiogenic Gene Expression in Down Syndrome Fetal Hearts. Fetal Diagn. Ther. 2015, 40, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Nugent, A.W.; Daubeney, P.E.; Chondros, P.; Carlin, J.B.; Cheung, M.; Wilkinson, L.C.; Andrew, M.D.; Stephen, G.K.; Chow, C.W.; James, L.W.; et al. The epidemiology of childhood cardiomyopathy in Australia. N. Engl. J. Med. 2003, 348, 1639–1646. [Google Scholar] [CrossRef] [PubMed]
- Lipshultz, S.E.; Sleeper, L.A.; Towbin, J.A.; Lowe, A.M.; Orav, E.J.; Cox, G.F.; Lurie, P.R.; McCoy, K.L.; McDonald, M.A.; Messere, J.E.; et al. The incidence of pediatric cardiomyopathy in two regions of the United States. N. Engl. J. Med. 2003, 348, 1647–1655. [Google Scholar] [CrossRef] [Green Version]
- Massin, M.M.; Astadicko, I.; Dessy, H. Epidemiology of Heart Failure in a Tertiary Pediatric Center. Clin. Cardiol. 2008, 31, 388–391. [Google Scholar] [CrossRef]
- Sommers, C.; Nagel, B.H.P.; Neudorf, U.; Schmaltz, A.A. Congestive heart failure in childhood. Herz 2005, 30, 652–662. [Google Scholar] [CrossRef]
- Kirk, R.; Dipchand, A.I.; Rosenthal, D.N.; Addonizio, L.; Burch, M.; Chrisant, M.; Dubin, A.; Everitt, M.; Gajarski, R.; Mertens, L.; et al. The International Society for Heart and Lung Transplantation Guidelines for the management of pediatric heart failure: Executive summary. J. Heart Lung Transplant. 2014, 33, 888–909. [Google Scholar] [CrossRef]
- Rao, S.D.; Adusumalli, S.; Mazurek, J.A. Pulmonary Hypertension in Heart Failure Patients. Card. Fail. Rev. 2020, 6. [Google Scholar] [CrossRef] [Green Version]
- Vachiéry, J.-L.; Delcroix, M.; Al-Hiti, H.; Efficace, M.; Hutyra, M.; Lack, G.; Papadakis, K.; Rubin, L.J. Macitentan in pulmonary hypertension due to left ventricular dysfunction. Eur. Respir. J. 2018, 51, 1701886. [Google Scholar] [CrossRef] [Green Version]
- Guimaraes, L.; Del Val, D.; Bergeron, S.; O’Connor, K.; Bernier, M.; Rodés-Cabau, J. Interatrial Shunting for Treating Acute and Chronic Left Heart Failure. Eur. Cardiol. Rev. 2020, 15, e18. [Google Scholar] [CrossRef]
- Bauer, A.; Khalil, M.; Schmidt, D.; Bauer, J.; Esmaeili, A.; Apitz, C.; Voelkel, N.F.; Schranz, D. Creation of a restrictive atrial communication in pulmonary arterial hypertension (PAH): Effective palliation of syncope and end-stage heart failure. Pulm. Circ. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akintürk, H.; Sen-Hild, B.; Yörüker, U.; Müller, M.; Thul, J.; Jux, C.; Schranz, D. Reverse Potts-Shunt for Bridging to Transplant, Recovery or Long-term Palliation. Thorac. Cardiovasc. Surg. 2018, 66, 71. [Google Scholar] [CrossRef]
- Schranz, D.; Akintuerk, H.; Voelkel, N.F. ‘End-stage’ heart failure therapy: Potential lessons from congenital heart disease: From pulmonary artery banding and interatrial communication to parallel circulation. Heart 2016, 103, 262–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodès-Cabau, J.; Bernier, M.; Amat-Santos, I.J.; Ben Gal, T.; Nombela-Franco, L.; Del Blanco, B.G.; Kerner, A.; Bergeron, S.; Del Trigo, M.; Pibarot, P.; et al. Interatrial Shunting for Heart Failure. JACC Cardiovasc. Interv. 2018, 11, 2300–2310. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.J.; Feldman, T.; Ricciardi, M.J.; Kahwash, R.; Lilly, S.; Litwin, S.; Nielsen, C.D.; Van Der Harst, P.; Hoendermis, E.; Penicka, M.; et al. One-Year Safety and Clinical Outcomes of a Transcatheter Interatrial Shunt Device for the Treatment of Heart Failure With Preserved Ejection Fraction in the Reduce Elevated Left Atrial Pressure in Patients With Heart Failure (REDUCE LAP-HF I) Trial: A Randomized Clinical Trial. JAMA Cardiol. 2018, 3, 968–977. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, R.; Schranz, D. Creation of a restrictive atrial left-to-right shunt: A novel treatment for heart failure. Heart Fail. Rev. 2018, 23, 841–847. [Google Scholar] [CrossRef]
- Lin, Y.-N.; Chen, Y.-H.; Wang, H.-J.; Hung, J.-S.; Chang, K.-C.; Lo, P.-H. Atrial Septostomy for Left Atrial Decompression during Extracorporeal Membrane Oxygenation by Inoue Balloon Catheter. Circ. J. 2017, 81, 1419–1423. [Google Scholar] [CrossRef] [Green Version]
- Selim, A.M.; Wadhwani, L.; Burdorf, A.; Raichlin, E.; Lowes, B.D.; Zolty, R. Left Ventricular Assist Devices in Pulmonary Hypertension Group 2 With Significantly Elevated Pulmonary Vascular Resistance: A Bridge to Cure. Heart Lung Circ. 2019, 28, 946–952. [Google Scholar] [CrossRef]
- Benden, C.; Edwards, L.B.; Kucheryavaya, A.Y.; Christie, J.D.; Dipchand, A.I.; Dobbels, F.; Jason, D.C.; Fabienne, D.; Axel, O.R.; Josef, S.; et al. The Registry of the International Society for Heart and Lung Transplantation: Fifteenth pediatric lung and heart-lung transplantation report—2012. J. Heart Lung Transplant. 2012, 31, 1087–1095. [Google Scholar] [CrossRef] [Green Version]
- Miera, O.; Potapov, E.V.; Redlin, M.; Stepanenko, A.; Berger, F.; Hetzer, R.; Hübler, M. First Experiences with the HeartWare Ventricular Assist System in Children. Ann. Thorac. Surg. 2011, 91, 1256–1260. [Google Scholar] [CrossRef]
- Thangappan, K.; Morales, D.L.S.; Vu, Q.; Lehenbauer, D.; Villa, C.; Wittekind, S.; Russel, H.; Angela, L.; Farhan, Z. Impact of Mechanical Circulatory Support on Pediatric Heart Transplant Candidates with Elevated Pulmonary Vascular Resistance. Artif. Organs. 2020. [Google Scholar] [CrossRef] [PubMed]
- Richmond, M.E.; Law, Y.M.; Das, B.B.; Everitt, M.D.; Kukreja, M.; Naftel, D.C.; Kemna, M.S.; Henderson, H.T.; Beddows, K.; Fricker, F.J.; et al. Elevated pre-transplant pulmonary vascular resistance is not associated with mortality in children without congenital heart disease: A multicenter study. J. Heart Lung Transplant. 2015, 34, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Fayyaz, A.U.; Edwards, W.D.; Maleszewski, J.J.; Konik, E.A.; Dubrock, H.M.; Borlaug, B.A.; Frantz, R.P.; Jenkins, S.M.; Redfield, M.M. Global Pulmonary Vascular Remodeling in Pulmonary Hypertension Associated With Heart Failure and Preserved or Reduced Ejection Fraction. Circulation 2018, 137, 1796–1810. [Google Scholar] [CrossRef] [PubMed]
- Zafar, F.; Castleberry, C.; Khan, M.S.; Mehta, V.; Bryant, R.; Lorts, A.; Wilmot, I.; Jefferies, J.L.; Chin, C.; Morales, D.L. Pediatric heart transplant waiting list mortality in the era of ventricular assist devices. J. Heart Lung Transplant. 2015, 34, 82–88. [Google Scholar] [CrossRef]
- Hosseinpour, A.-R.; Cullen, S.; Tsang, V.T. Transplantation for adults with congenital heart disease? Eur. J. Cardio-Thoracic Surg. 2006, 30, 508–514. [Google Scholar] [CrossRef]
- Rossano, J.W.; Cherikh, W.S.; Chambers, D.C.; Goldfarb, S.; Khush, K.; Kucheryavaya, A.Y.; Levvey, B.; Lund, L.; Meiser, B.; Yusen, R.D.; et al. The Registry of the International Society for Heart and Lung Transplantation: Twentieth Pediatric Heart Transplantation Report—2017; Focus Theme: Allograft ischemic time. J. Heart Lung Transplant. 2017, 36, 1060–1069. [Google Scholar] [CrossRef] [Green Version]
- Dipchand, A.I. Current state of pediatric cardiac transplantation. Ann. Cardiovasc. Surg. 2018, 7, 31. [Google Scholar]
- Rivinius, R.; Helmschrott, M.; Ruhparwar, A.; Schmack, B.; Darche, F.F.; Thomas, D.; Bruckner, T.; Doesch, A.O.; Katus, H.A.; Ehlermann, P. Elevated pre-transplant pulmonary vascular resistance is associated with early post-transplant atrial fibrillation and mortality. ESC Heart Fail. 2020, 7, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Everitt, M.D.; Boyle, G.J.; Schechtman, K.B.; Zheng, J.; Bullock, E.A.; Kaza, A.K.; Dipchand, A.I.; Naftel, D.C.; Kirklin, J.K.; Canter, C.E. Early survival after heart transplant in young infants is lowest after failed single-ventricle palliation: A multi-institutional study. J. Heart Lung Transplant. 2012, 31, 509–516. [Google Scholar] [CrossRef]
- Alsoufi, B.; Mahle, W.T.; Manlhiot, C.; Deshpande, S.; Kogon, B.; McCrindle, B.W.; Kanter, K.; Information, P.E.K.F.C. Outcomes of heart transplantation in children with hypoplastic left heart syndrome previously palliated with the Norwood procedure. J. Thorac. Cardiovasc. Surg. 2016, 151, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Kanter, K.R.; Mahle, W.T.; Vincent, R.; Berg, A.M.; Kogon, B.; Kirshbom, P.M. Heart Transplantation in Children With a Fontan Procedure. Ann. Thorac. Surg. 2011, 91, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, K.A.; Addonizio, L.J.; Kichuk-Chrisant, M.R.; Galantowicz, M.E.; Lamour, J.M.; Quaegebeur, J.M.; Daphne, T.H. Cardiac transplantation after the Fontan or Glenn procedure. J. Am. Coll. Cardiol. 2004, 16, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.B.; Campbell, D.N.; Ivy, D.; Boucek, M.M.; Sondheimer, H.M.; Pietra, B.; Bibhuti, B.D.; Joseph, R.C. Evidence of pulmonary vascular disease after heart transplantation for Fontan circulation failure. J. Thorac. Cardiovasc. Surg. 2004, 128, 693–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormick, A.D.; Schumacher, K.R. Transplantation of the failing Fontan. Transl. Pediatr. 2019, 8, 290–301. [Google Scholar] [CrossRef]
- Kimberling, M.T.; Balzer, D.T.; Hirsch, R.; Mendeloff, E.; Huddleston, C.B.; Canter, C.E. Cardiac transplantation for pediatric restrictive cardiomyopathy: Presentation, evaluation, and short-term outcome. J. Heart Lung Transplant. 2002, 21, 455–459. [Google Scholar] [CrossRef]
- Hughes, M.L.; Kleinert, S.; Keogh, A.M.; Macdonald, P.; Wilkinson, J.L.; Weintraub, R.G. Pulmonary vascular resistance and reactivity in children with end-stage cardiomyopathy. J. Heart Lung Transplant. 2000, 19, 701–704. [Google Scholar] [CrossRef]
- Murtuza, B.; Fenton, M.; Burch, M.; Gupta, A.; Muthialu, N.; Elliott, M.J.; Hsia, T.-Y.; Tsang, V.T.; Kostolny, M. Pediatric Heart Transplantation for Congenital and Restrictive Cardiomyopathy. Ann. Thorac. Surg. 2013, 95, 1675–1684. [Google Scholar] [CrossRef]
- Mehra, M.R.; Canter, C.E.; Hannan, M.M.; Semigran, M.J.; Uber, P.A.; Baran, D.A.; Danziger-Isakov, L.; Kirklin, J.K.; Kirk, R.; Kushwaha, S.S.; et al. The 2016 International Society for Heart Lung Transplantation listing criteria for heart transplantation: A 10-year update. J. Heart Lung Transplant. 2016, 35, 1–23. [Google Scholar] [CrossRef]
- Mourani, P.M.; Abman, S.H. Pulmonary Hypertension and Vascular Abnormalities in Bronchopulmonary Dysplasia. Clin. Perinatol. 2015, 42, 839–855. [Google Scholar] [CrossRef] [Green Version]
- Varghese, N.; Rios, D. Pulmonary Hypertension Associated with Bronchopulmonary Dysplasia: A Review. Pediatr. Allergy Immunol. Pulmonol. 2019, 32, 140–148. [Google Scholar] [CrossRef] [Green Version]
- Jakkula, M.; Le Cras, T.D.; Gebb, S.; Hirth, K.P.; Tuder, R.M.; Voelkel, N.F.; Abman, S.H. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am. J. Physiol. Cell. Mol. Physiol. 2000, 279, L600–L607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, R.J.; Weesner, K.M.; Bucher, J.R. Oxygen-Induced Alterations in Lung Vascular Development in the Newborn Rat. Pediatr. Res. 1983, 17, 368–375. [Google Scholar] [CrossRef] [Green Version]
- Cornfield, D.N. Developmental Regulation of Oxygen Sensing and Ion Channels in the Pulmonary Vasculature. Vaccine Des. 2009, 661, 201–220. [Google Scholar] [CrossRef]
- El-Saie, A.; Shivanna, B. Novel Strategies to Reduce Pulmonary Hypertension in Infants with Bronchopulmonary Dysplasia. Front. Pediatr. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Sahni, M.; Bhandari, V. Bronchopulmonary dysplasia-associated pulmonary hypertension. Pediatr. Med. 2019, 2, 4. [Google Scholar] [CrossRef]
- Allen, J.; Zwerdling, R.; Ehrenkranz, R.; Gaultier, C.; Geggel, R.; Greenough, A.; Kleinman, R.; Klijanowicz, A.; Martinez, F.; Ozdemir, A.; et al. Statement on the Care of the Child with Chronic Lung Disease of Infancy and Childhood. Am. J. Respir. Crit. Care Med. 2003, 168, 356–396. [Google Scholar] [CrossRef] [Green Version]
- Khemani, E.; McElhinney, D.B.; Rhein, L.; Andrade, O.; Lacro, R.V.; Thomas, K.C.; Mullen, M.P. Pulmonary Artery Hypertension in Formerly Premature Infants With Bronchopulmonary Dysplasia: Clinical Features and Outcomes in the Surfactant Era. Pediatrics 2007, 120, 1260–1269. [Google Scholar] [CrossRef]
- Arjaans, S.; Zwart, E.A.H.; Ploegstra, M.-J.; Bos, A.F.; Kooi, E.M.W.; Hillege, H.L.; Berger, R.M. Identification of gaps in the current knowledge on pulmonary hypertension in extremely preterm infants: A systematic review and meta-analysis. Paediatr. Périnat. Epidemiol. 2018, 32, 258–267. [Google Scholar] [CrossRef]
- Arjaans, S.; Haarman, M.G.; Roofthooft, M.T.R.; Fries, M.W.F.; Kooi, E.M.W.; Bos, A.F.; Berger, R.M.F. Fate of pulmonary hypertension associated with bronchopulmonary dysplasia beyond 36 weeks postmenstrual age. Arch. Dis. Child. Fetal Neonatal Ed. 2020. [Google Scholar] [CrossRef]
- Al-Ghanem, G.; Shah, P.; Thomas, S.; Banfield, L.; El Helou, S.; Fusch, C.; Mukerji, A. Bronchopulmonary dysplasia and pulmonary hypertension: A meta-analysis. J. Perinatol. 2017, 37, 414–419. [Google Scholar] [CrossRef]
- Kadmon, G.; Schiller, O.; Dagan, T.; Bruckheimer, E.; Birk, E.; Schonfeld, T. Pulmonary hypertension specific treatment in infants with bronchopulmonary dysplasia. Pediatr. Pulmonol. 2016, 52, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Mourani, P.M.; Sontag, M.K.; Ivy, D.D.; Abman, S.H. Effects of Long-Term Sildenafil Treatment for Pulmonary Hypertension in Infants with Chronic Lung Disease. J. Pediatr. 2009, 154, 379–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansmann, G.; Sallmon, H.; Roehr, C.C.; Kourembanas, S.; Austin, E.D.; Koestenberger, M.; for the European Pediatric Pulmonary Vascular Disease Network (EPPVDN). Pulmonary hypertension in bronchopulmonary dysplasia. Pediatr. Res. 2020, 1–11. [Google Scholar] [CrossRef]
- Koroglu, O.A.; Yalaz, M.; Levent, E.; Akisü, M.; Kultursay, N. Cardiovascular Consequences of Bronchopulmonary Dysplasia in Prematurely Born Preschool Children. Neonatology 2013, 104, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Zivanovic, S.; Pushparajah, K.; Calvert, S.; Marlow, N.; Razavi, R.; Peacock, J.L.; Greenough, A. Pulmonary Artery Pressures in School-Age Children Born Prematurely. J. Pediatr. 2017, 191, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Bonadies, L.; Zaramella, P.; Porzionato, A.; Perilongo, G.; Muraca, M.; Baraldi, E. Present and Future of Bronchopulmonary Dysplasia. J. Clin. Med. 2020, 9, 1539. [Google Scholar] [CrossRef]
- Moschino, L.; Stocchero, M.; Filippone, M.; Carraro, S.; Baraldi, E. Longitudinal Assessment of Lung Function in Survivors of Bronchopulmonary Dysplasia from Birth to Adulthood. The Padova BPD Study. Am. J. Respir. Crit. Care Med. 2018, 198, 134–137. [Google Scholar] [CrossRef]
- Thébaud, B.; Goss, K.N.; Laughon, M.; Whitsett, J.A.; Abman, S.H.; Steinhorn, R.H.; Aschner, J.L.; Davis, P.G.; McGrath-Morrow, S.A.; Soll, R.F.; et al. Bronchopulmonary dysplasia. Nat. Rev. Dis. Prim. 2019, 5, 78. [Google Scholar] [CrossRef]
- Bonderman, D.; Wilkens, H.; Wakounig, S.; Schäfers, H.-J.; Jansa, P.; Lindner, J.; Simkova, I.; Martischnig, A.M.; Dudczak, J.; Sadushi, R.; et al. Risk factors for chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2008, 33, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.H.; Lang, I.M. Risk factors for chronic thromboembolic pulmonary hypertension. Eur. Respir. Rev. 2012, 21, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Madani, M.M.; Wittine, L.M.; Auger, W.R.; Fedullo, P.F.; Kerr, K.M.; Kim, N.H.; Test, V.J.; Kriett, J.M.; Jamieson, S.W. Chronic thromboembolic pulmonary hypertension in pediatric patients. J. Thorac. Cardiovasc. Surg. 2011, 141, 624–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, M.; Boyer-Neumann, C.; Parent, F.; Eschwège, V.; Jaillet, H.; Meyer, D.; Simonneau, G. Thrombotic risk factors in pulmonary hypertension. Eur. Respir. J. 2000, 15, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Bonderman, D.; Turecek, P.L.; Jakowitsch, J.; Weltermann, A.; Adlbrecht, C.; Schneider, B.; Kneussl, M.; Rubin, L.J.; Kyrle, P.A.; Klepetko, W.; et al. High prevalence of elevated clotting factor VIII in chronic thromboembolic pulmonary hypertension. Thromb. Haemost. 2003, 90, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Verbelen, T.; Cools, B.; Fejzic, Z.; Eynde, R.V.D.; Maleux, G.; Delcroix, M.; Meyns, B. Pulmonary endarterectomy in a 12-year-old boy with multiple comorbidities. Pulm. Circ. 2019, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.-C.; Wu, E.-T.; Chen, S.-J.; Lu, F.L.; Huang, S.-C.; Wang, J.-K.; Chang, C.-I.; Wu, M.-H. Scimitar syndrome: Incidence, treatment, and prognosis. Eur. J. Nucl. Med. Mol. Imaging 2007, 167, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Schramel, F.M.N.H.; Westermann, C.J.J.; Knaepen, P.J.; Bosch, J.M.M.V.D. The scimitar syndrome: Clinical spectrum and surgical treatment. Eur. Respir. J. 1995, 8, 196–201. [Google Scholar] [CrossRef] [Green Version]
- Haworth, S.G.; Sauer, U.; Bühlmeyer, K. Pulmonary hypertension in scimitar syndrome in infancy. Heart 1983, 50, 182–189. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.-A.; Burrows, P.E.; Benson, L.N.; Rabinovitch, M.; Freedom, R.M. Scimitar syndrome in infancy. J. Am. Coll. Cardiol. 1993, 22, 873–882. [Google Scholar] [CrossRef]
- Al Rukban, H.; Al Ghaihab, M.; Tamimi, O.; Al-Saleh, S. Clinical spectrum of infantile scimitar syndrome: A tertiary center experience. Ann. Pediatr. Cardiol. 2014, 7, 29–33. [Google Scholar] [CrossRef]
- Diaz-Frias, J.; Widrich, J. Scimitar Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. Available online: http://www.ncbi.nlm.nih.gov/books/NBK546602/ (accessed on 6 June 2020).
- Dupuis, C.; Charaf, L.A.; Brevière, G.-M.; Abou, P. “Infantile” form of the scimitar syndrome with pulmonary hypertension. Am. J. Cardiol. 1993, 71, 1326–1330. [Google Scholar] [CrossRef]
- Dupuis, C.; Charaf, L.A.; Brevière, G.-M.; Abou, P.; Remy-Jardin, M.; Helmius, G. The “adult” form of the scimitar syndrome. Am. J. Cardiol. 1992, 70, 502–507. [Google Scholar] [CrossRef]
- Midyat, L.; Demir, E.; Aşkın, M.; Gulen, F.; Ülger, Z.; Tanac, R.; Bayraktaroğlu, S. Eponym. Eur. J. Nucl. Med. Mol. Imaging 2010, 169, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Vida, V.L.; Guariento, A.; Milanesi, O.; Gregori, D.; Stellin, G.; Zucchetta, F.; Zanotto, L.; Padalino, M.A.; Castaldi, B.; Bosiznik, S.; et al. The natural history and surgical outcome of patients with scimitar syndrome: A multi-centre European study. Eur. Heart J. 2017, 39, 1002–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, R.L.; Moore, P.; Teitel, D.; Hawgood, S.; McQuitty, J.; Fineman, J.R. Abnormal vascular tone in infants and children with lung hypoplasia: Findings from cardiac catheterization and the response to chronic therapy. Pediatric Crit. Care Med. 2006, 7, 589–594. [Google Scholar] [CrossRef]
- Hilgendorff, A.; Apitz, C.; Bonnet, D.; Hansmann, G. Pulmonary hypertension associated with acute or chronic lung diseases in the preterm and term neonate and infant. The European Paediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart 2016, 102, ii49–ii56. [Google Scholar] [CrossRef] [Green Version]
- Von Schnakenburg, C.; Peuster, M.; Norozi, K.; Roebl, M.; Maibohm, M.; Wessel, A.; Fink, C. Acute pulmonary edema caused by epoprostenol infusion in a child with scimitar syndrome and pulmonary hypertension. Pediatr. Crit. Care Med. 2003, 4, 111–114. [Google Scholar] [CrossRef]
- Haworth, S.G.; Macartney, F.J. Growth and development of pulmonary circulation in pulmonary atresia with ventricular septal defect and major aortopulmonary collateral arteries. Heart 1980, 44, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Liao, P.-K.; Edwards, W.D.; Julsrud, P.R.; Puga, F.J.; Danielson, G.K.; Feldt, R.H. Pulmonary blood supply in patients with pulmonary atresia and ventricular septal defect. J. Am. Coll. Cardiol. 1985, 6, 1343–1350. [Google Scholar] [CrossRef] [Green Version]
- Haworth, S.G.; Rees, P.G.; Taylor, J.F.; Macartney, F.J.; de Leval, M.; Stark, J. Pulmonary atresia with ventricular septal defect and major aortopulmonary collateral arteries. Effect of systemic pulmonary anastomosis. Br. Heart J. 1981, 45, 133–141. [Google Scholar] [CrossRef] [Green Version]
- Hofbeck, M.; Singer, H. Pulmonary atresia with ventricular septal defect: Palliative operations of primarily inoperable forms. Z. Kardiol. 1983, 72, 622–632. [Google Scholar]
- Reddy, V.M.; McElhinney, D.B.; Amin, Z.; Moore, P.; Parry, A.J.; Teitel, D.F.; Hanley, F.L. Early and intermediate outcomes after repair of pulmonary atresia with ventricular septal defect and major aortopulmonary collateral arteries: Experience with 85 patients. Circulation 2000, 101, 1826–1832. [Google Scholar] [CrossRef] [PubMed]
- Schouvey, S.; Dragulescu, A.; Ghez, O.; Amedro, P.; Kreitmann, B.; Chetaille, P.; Fraisse, A.; Metras, D. Rehabilitation of hypoplastic pulmonary arteries in pulmonary atresia with ventricular septal defect. Medium term results. Arch. Mal. Vaiss. 2007, 100, 422. [Google Scholar]
- Rabinovitch, M.; Haworth, S.G.; Vance, Z.; Vawter, G.; Castaneda, A.R.; Nadas, A.S.; Reid, L.M. Early pulmonary vascular changes in congenital heart disease studied in biopsy tissue. Hum. Pathol. 1980, 11, 499–509. [Google Scholar] [PubMed]
- Haworth, S.G. Collateral arteries in pulmonary atresia with ventricular septal defect. A precarious blood supply. Heart 1980, 44, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Lofland, G.K. Pulmonary atresia, ventricular septal defect, and multiple aorta pulmonary collateral arteries. Semin. Thorac. Cardiovasc. Surgery Pediatr. Card. Surg. Annu. 2004, 7, 85–94. [Google Scholar] [CrossRef]
- DeGiovanni, J. Timing, frequency, and results of catheter intervention following recruitment of major aortopulmonary collaterals in patients with pulmonary atresia and ventricular septal defect. J. Interv. Cardiol. 2004, 17, 47–52. [Google Scholar] [CrossRef]
- Dimopoulos, K.; Diller, G.; Opotowsky, A.R.; D’Alto, M.; Gu, H.; Giannakoulas, G.; Budts, W.; Broberg, C.S.; Veldtman, G.; Swan, L.; et al. Definition and Management of Segmental Pulmonary Hypertension. J. Am. Heart Assoc. 2018, 7, e008587. [Google Scholar] [CrossRef] [Green Version]
- Lim, Z.S.; Vettukattill, J.J.; Salmon, A.P.; Veldtman, G.R. Sildenafil therapy in complex pulmonary atresia with pulmonary arterial hypertension. Int. J. Cardiol. 2008, 129, 339–343. [Google Scholar] [CrossRef]
- Yamamura, K.; Nagata, H.; Ikeda, K.; Ihara, K.; Hara, T. Efficacy of bosentan therapy for segmental pulmonary artery hypertension due to major aortopulmonary collateral arteries in children. Int. J. Cardiol. 2012, 161, e1–e3. [Google Scholar] [CrossRef]
- Grant, E.K.; Berger, J.T. Use of Pulmonary Hypertension Medications in Patients with Tetralogy of Fallot with Pulmonary Atresia and Multiple Aortopulmonary Collaterals. Pediatr. Cardiol. 2015, 37, 304–312. [Google Scholar] [CrossRef]
- Pinto, V.M.; Balocco, M.; Quintino, S.; Forni, G.L. Sickle cell disease: A review for the internist. Intern. Emerg. Med. 2019, 14, 1051–1064. [Google Scholar] [CrossRef] [PubMed]
- Gordeuk, V.R.; Castro, O.L.; Machado, R.F. Pathophysiology and treatment of pulmonary hypertension in sickle cell disease. Blood 2016, 127, 820–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potoka, K.P.; Gladwin, M.T. Vasculopathy and pulmonary hypertension in sickle cell disease. Am. J. Physiol. Cell. Mol. Physiol. 2015, 308, L314–L324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parent, F.; Bachir, D.; Inamo, J.; Lionnet, F.; Driss, F.; Loko, G.; Habibi, A.; Bennani, S.; Savale, L.; Adnot, S.; et al. A Hemodynamic Study of Pulmonary Hypertension in Sickle Cell Disease. N. Engl. J. Med. 2011, 365, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, G.H.H.; Souza, R.; Salemi, V.M.C.; Jardim, C.V.P.; Gualandro, S.F.M. Pulmonary hypertension diagnosed by right heart catheterisation in sickle cell disease. Eur. Respir. J. 2011, 39, 112–118. [Google Scholar] [CrossRef] [Green Version]
- Gladwin, M.T. Cardiovascular complications in patients with sickle cell disease. Hematology 2017, 2017, 423–430. [Google Scholar] [CrossRef] [Green Version]
- Castro, O.; Hoque, M.; Brown, B.D. Pulmonary hypertension in sickle cell disease: Cardiac catheterization results and survival. Blood 2003, 101, 1257–1261. [Google Scholar] [CrossRef] [Green Version]
- Ambrusko, S.J.; Gunawardena, S.; Sakara, A.; Windsor, B.; Lanford, L.; Michelson, P.; Krishnamurti, L. Elevation of tricuspid regurgitant jet velocity, a marker for pulmonary hypertension in children with sickle cell disease. Pediatr. Blood Cancer 2006, 47, 907–913. [Google Scholar] [CrossRef]
- Chaudry, R.A.; Cikes, M.; Karu, T.; Hutchinson, C.; Ball, S.; Sutherland, G.; Rosenthal, M.; Bush, A.; Dm, S.C. Paediatric sickle cell disease: Pulmonary hypertension but normal vascular resistance. Arch. Dis. Child. 2010, 96, 131–136. [Google Scholar] [CrossRef]
- Pashankar, F.D.; Carbonella, J.; Bazzy-Asaad, A.; Friedman, A. Longitudinal follow up of elevated pulmonary artery pressures in children with sickle cell disease. Br. J. Haematol. 2009, 144, 736–741. [Google Scholar] [CrossRef]
- Lee, M.T.; Small, T.; Khan, M.A.; Rosenzweig, E.B.; Barst, R.J.; Brittenham, G.M. Doppler-defined pulmonary hypertension and the risk of death in children with sickle cell disease followed for a mean of three years. Br. J. Haematol. 2009, 146, 437–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagar, R.W.; Michlitsch, J.G.; Gardner, J.; Vichinsky, E.; Morris, C.R. Clinical differences between children and adults with pulmonary hypertension and sickle cell disease. Br. J. Haematol. 2007, 140, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Agha, H.; El Tagui, M.; El Ghamrawy, M.; Hady, M.A. The 6-min walk test: An independent correlate of elevated tricuspid regurgitant jet velocity in children and young adult sickle cell patients. Ann. Hematol. 2014, 93. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.; Van Hell, A.J.; Visser, M.; Nijveldt, R.J.; Van Leeuwen, P.A.M.; Teerlink, T. Role of the human erythrocyte in generation and storage of asymmetric dimethylarginine. Am. J. Physiol. Circ. Physiol. 2012, 302, H1762–H1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagener, F.A.; Abraham, N.G.; Van Kooyk, Y.; De Witte, T.; Figdor, C.G. Heme-induced cell adhesion in the pathogenesis of sickle-cell disease and inflammation. Trends Pharmacol. Sci. 2001, 22, 52–54. [Google Scholar] [CrossRef]
- Buehler, P.W.; Baek, J.H.; Lisk, C.; Connor, I.; Sullivan, T.; Kominsky, U.; Majka, S.; Stenmark, K.R.; Nozik-Grayck, E.; Bonaventura, J.; et al. Free hemoglobin induction of pulmonary vascular disease: Evidence for an inflammatory mechanism. Am. J. Physiol. Cell. Mol. Physiol. 2012, 303, L312–L326. [Google Scholar] [CrossRef] [Green Version]
- Ataga, K.I.; Moore, C.G.; Jones, S.; Olajide, O.; Strayhorn, D.; Hinderliter, A.; Orringer, E.P. Pulmonary hypertension in patients with sickle cell disease: A longitudinal study. Br. J. Haematol. 2006, 134, 109–115. [Google Scholar] [CrossRef]
- Hu, W.; Jin, R.; Zhang, J.; You, T.; Peng, Z.; Ge, X.; Bronson, R.T.; Halperin, J.A.; Loscalzo, J.; Qin, X. The critical roles of platelet activation and reduced NO bioavailability in fatal pulmonary arterial hypertension in a murine hemolysis model. Blood 2010, 116, 1613–1622. [Google Scholar] [CrossRef] [Green Version]
- Anthi, A.; Machado, R.F.; Jison, M.L.; Taveira-DaSilva, A.M.; Rubin, L.J.; Hunter, L.; Hunter, C.J.; Coles, W.; Nichols, J.; Avila, N.A.; et al. Hemodynamic and Functional Assessment of Patients with Sickle Cell Disease and Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2007, 175, 1272–1279. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Boil. 1992, 12, 5447–5454. [Google Scholar] [CrossRef] [Green Version]
- Ebert, B.L.; Bunn, H.F. Regulation of the erythropoietin gene. Blood J. Am. Soc. Hematol. 1999, 94, 1864–1877. [Google Scholar]
- Niss, O.; Quinn, C.T.; Lane, A.; Daily, J.; Khoury, P.R.; Bakeer, N.; Kimball, T.R.; Towbin, J.A.; Malik, P.; Taylor, M.D. Cardiomyopathy With Restrictive Physiology in Sickle Cell Disease. JACC Cardiovasc. Imaging 2016, 9, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Mehari, A.; Alam, S.; Tian, X.; Cuttica, M.J.; Barnett, C.F.; Miles, G.; Xu, D.; Seamon, C.; Adams-Graves, P.; Castro, O.L.; et al. Hemodynamic Predictors of Mortality in Adults with Sickle Cell Disease. Am. J. Respir. Crit. Care Med. 2013, 187, 840–847. [Google Scholar] [CrossRef]
- Klings, E.S.; Machado, R.F.; Barst, R.J.; Morris, C.R.; Mubarak, K.K.; Gordeuk, V.R.; Kato, G.J.; Ataga, K.I.; Gibbs, J.S.; Castro, O.; et al. An Official American Thoracic Society Clinical Practice Guideline: Diagnosis, Risk Stratification, and Management of Pulmonary Hypertension of Sickle Cell Disease. Am. J. Respir. Crit. Care Med. 2014, 189, 727–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerath, A.; Murphy, P.; Madonik, M.; Barth, D.; Granton, J.; De Perrot, M. Pulmonary endarterectomy in sickle cell haemoglobin C disease. Eur. Respir. J. 2011, 38, 735–737. [Google Scholar] [CrossRef] [PubMed]
- Yung, G.L.; Channick, R.N.; Fedullo, P.F.; Auger, W.R.; Kerr, K.M.; Jamieson, S.W.; Kapelanski, D.P.; Moser, K.M. Successful Pulmonary Thromboendarterectomy in Two Patients with Sickle Cell Disease. Am. J. Respir. Crit. Care Med. 1998, 157, 1690–1693. [Google Scholar] [CrossRef]
- Freeman, A.T.; Ataga, K.I. Pulmonary endarterectomy as treatment for chronic thromboembolic pulmonary hypertension in sickle cell disease. Am. J. Hematol. 2015, 90, E223–E224. [Google Scholar] [CrossRef]
- Machado, R.F.; Martyr, S.; Kato, G.J.; Barst, R.J.; Anthi, A.; Robinson, M.R.; Hunter, L.; Coles, W.; Nichols, J.; Hunter, C.J.; et al. Sildenafil therapy in patients with sickle cell disease and pulmonary hypertension. Br. J. Haematol. 2005, 130, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Derchi, G.; Forni, G.L.; Formisano, F.; Cappellini, M.D.; Galanello, R.; D’Ascola, G.; Bina, P.; Magnano, C.; Lamagna, M. Efficacy and safety of sildenafil in the treatment of severe pulmonary hypertension in patients with hemoglobinopathies. Haematology 2005, 90, 452–458. [Google Scholar]
- Minniti, C.P.; Machado, R.F.; Coles, W.A.; Sachdev, V.; Gladwin, M.T.; Kato, G.J. Endothelin receptor antagonists for pulmonary hypertension in adult patients with sickle cell disease. Br. J. Haematol. 2009, 147, 737–743. [Google Scholar] [CrossRef] [Green Version]
- Barst, R.J.; Mubarak, K.K.; Machado, R.F.; Ataga, K.I.; Benza, R.L.; Castro, O.; Naeije, R.; Sood, N.; Swerdlow, P.S.; Hildesheim, M.; et al. Exercise capacity and haemodynamics in patients with sickle cell disease with pulmonary hypertension treated with bosentan: Results of the ASSET studies. Br. J. Haematol. 2010, 149, 426–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, R.F.; Barst, R.J.; Yovetich, N.A.; Hassell, K.L.; Kato, G.J.; Gordeuk, V.R.; Gibbs, J.S.R.; Little, J.A.; Schraufnagel, D.; Krishnamurti, L.; et al. Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood 2011, 118, 855–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
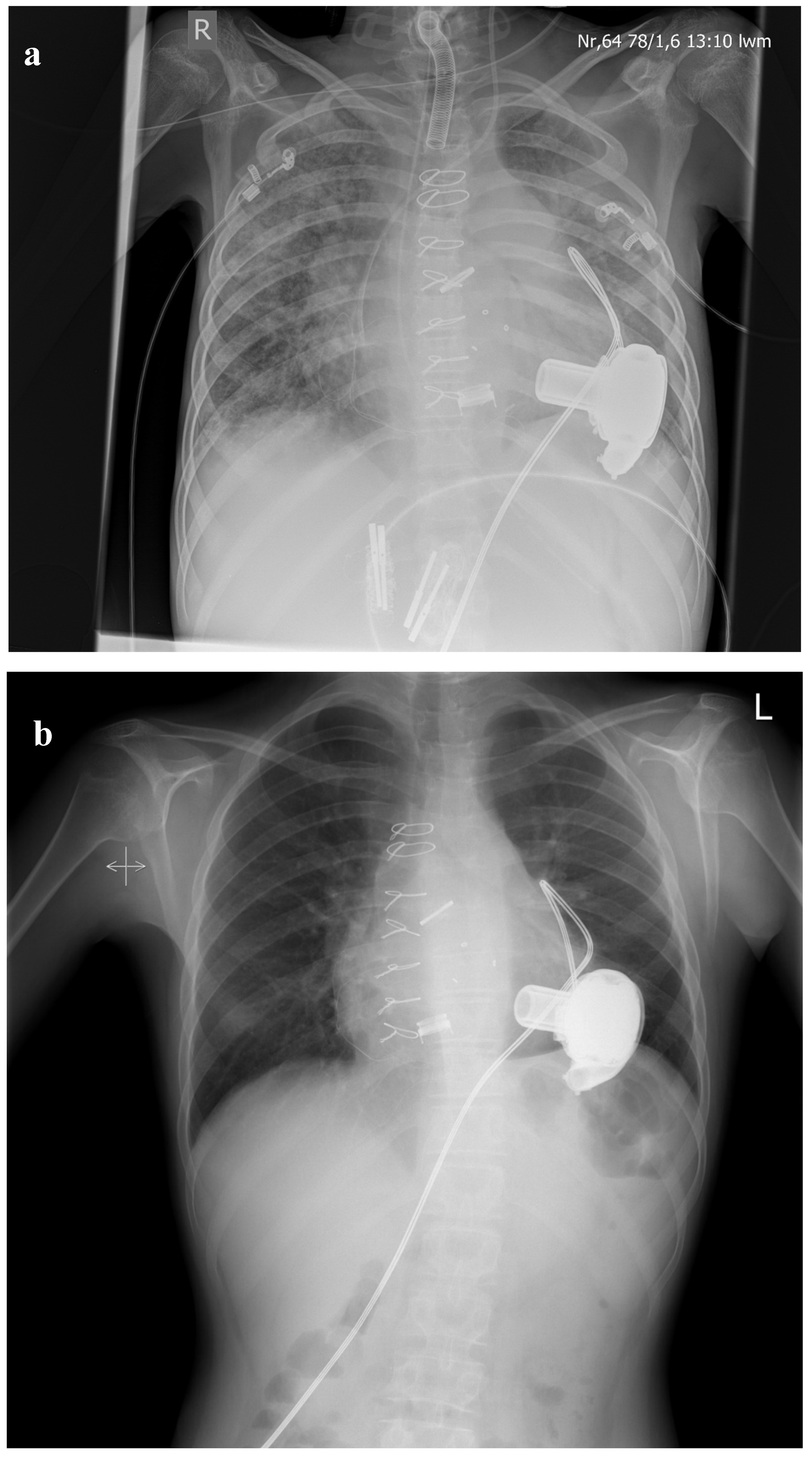

{kind=link}
{kind=link}
| 2019 Hemodynamic Definition of Paediatric Pulmonary Hypertension, Proposed by the European Paediatric Pulmonary Vascular Disease Network [17] | ERS/ESC Updated Clinical Classification of Pulmonary Hypertension (PH) [8] | |||
|---|---|---|---|---|
| Pulmonary Hypertension (PH) mPAP > 20 mmHg—beyond the age of 3 months at sea level | ||||
| Precapillary PH (PAH), or pulmonary hypertensive vascular disease (PHVD) | mPAP > 20 mmHg PAWP or LVEDP ≤ 15 mmHg PVR index ≥ 3 WU × m2 (DPG ≥ 7 mmHg) | 1. Pulmonary Arterial Hypertension 1.1. Idiopathic PAH 1.2. Heritable PAH 1.3. Drug- and toxin-induced PAH 1.4. PAH associated with: 1.4.1. Connective tissue disease 1.4.2. HIV infection 1.4.3. Portal hypertension 1.4.4. Congenital heart disease, including ES - CHD with biventricular circulation mPAP > 20 mmHg and PVR Index ≥ 3 WU × m2 - CHD with univentricular circulation (i.e., Fontan) mean TPG > 6 mmHg or PVR index > 3 WU × m2 1.4.5. Schistosomiasis 1.5. PAH long-term responders to calcium channel blockers 1.6. PAH with overt features of venous/capillaries (PVOD/PCH) involvement 1.7. Persistent PH of the new-born syndrome 5. PH with unclear and/or multifactorial mechanisms 5.1. Hematological disorders 5.2. Systemic and metabolic disorders 5.3. Others 5.4. Complex congenital heart disease | 3. PH due to lung diseases and/or hypoxia 3.1. Obstructive lung disease 3.2. Restrictive lung disease 3.3. Other lung disease with mixed restrictive/obstructive pattern 3.4. Hypoxia without lung disease 3.5. Developmental lung disorders | 4. PH due to pulmonary artery obstructions 4.1. Chronic thromboembolic PH 4.2. Other pulmonary artery obstructions |
| Isolated postcapillary PH | mPAP > 20 mmHg PAWP or LVEDP > 15 mmHg PVR Index < 3 WU × m2 | 2. PH due to left heart disease 2.1. PH due to heart failure with preserved left ventricular ejection fraction 2.2. PH due to heart failure with reduced left ventricular ejection fraction 2.3. Valvular heart disease 2.4. Congenital/acquired cardiovascular conditions leading to post-capillary PH | ||
| Combined pre- and postcapillary PH | mPAP > 20 mmHg PAWP oder LVEDP > 15 mmHg PVR Index ≥ 3 WU × m2 (DPG ≥ 7 mmHg) | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albinni, S.; Marx, M.; Lang, I.M. Focused Update on Pulmonary Hypertension in Children—Selected Topics of Interest for the Adult Cardiologist. Medicina 2020, 56, 420. https://doi.org/10.3390/medicina56090420
Albinni S, Marx M, Lang IM. Focused Update on Pulmonary Hypertension in Children—Selected Topics of Interest for the Adult Cardiologist. Medicina. 2020; 56(9):420. https://doi.org/10.3390/medicina56090420
Chicago/Turabian StyleAlbinni, Sulaima, Manfred Marx, and Irene M. Lang. 2020. "Focused Update on Pulmonary Hypertension in Children—Selected Topics of Interest for the Adult Cardiologist" Medicina 56, no. 9: 420. https://doi.org/10.3390/medicina56090420