
An Updated Review of Pemphigus Diseases

 , ,
, ,
Abstract
:1. Introduction
2. Pathophysiology
3. Clinical Features
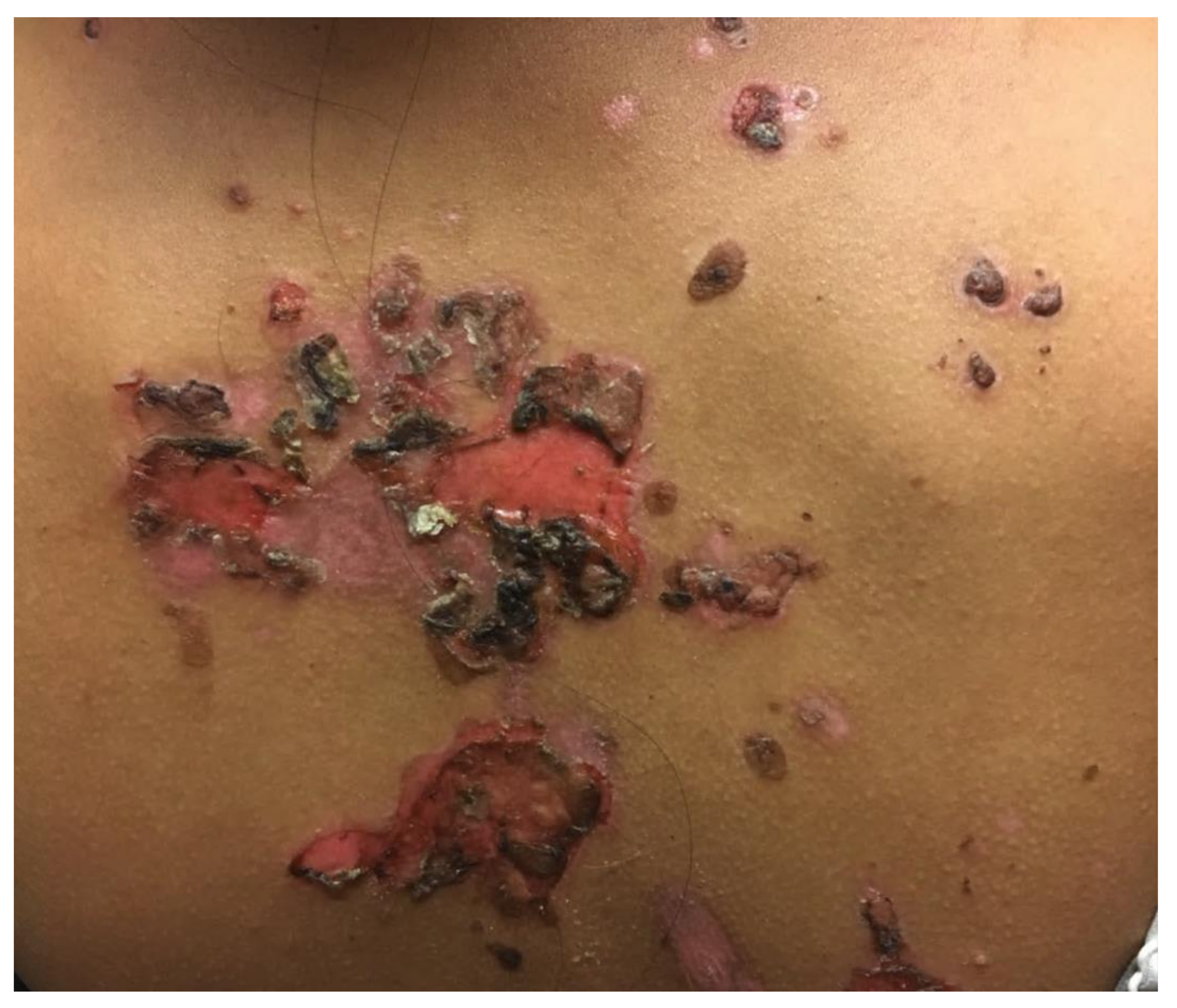
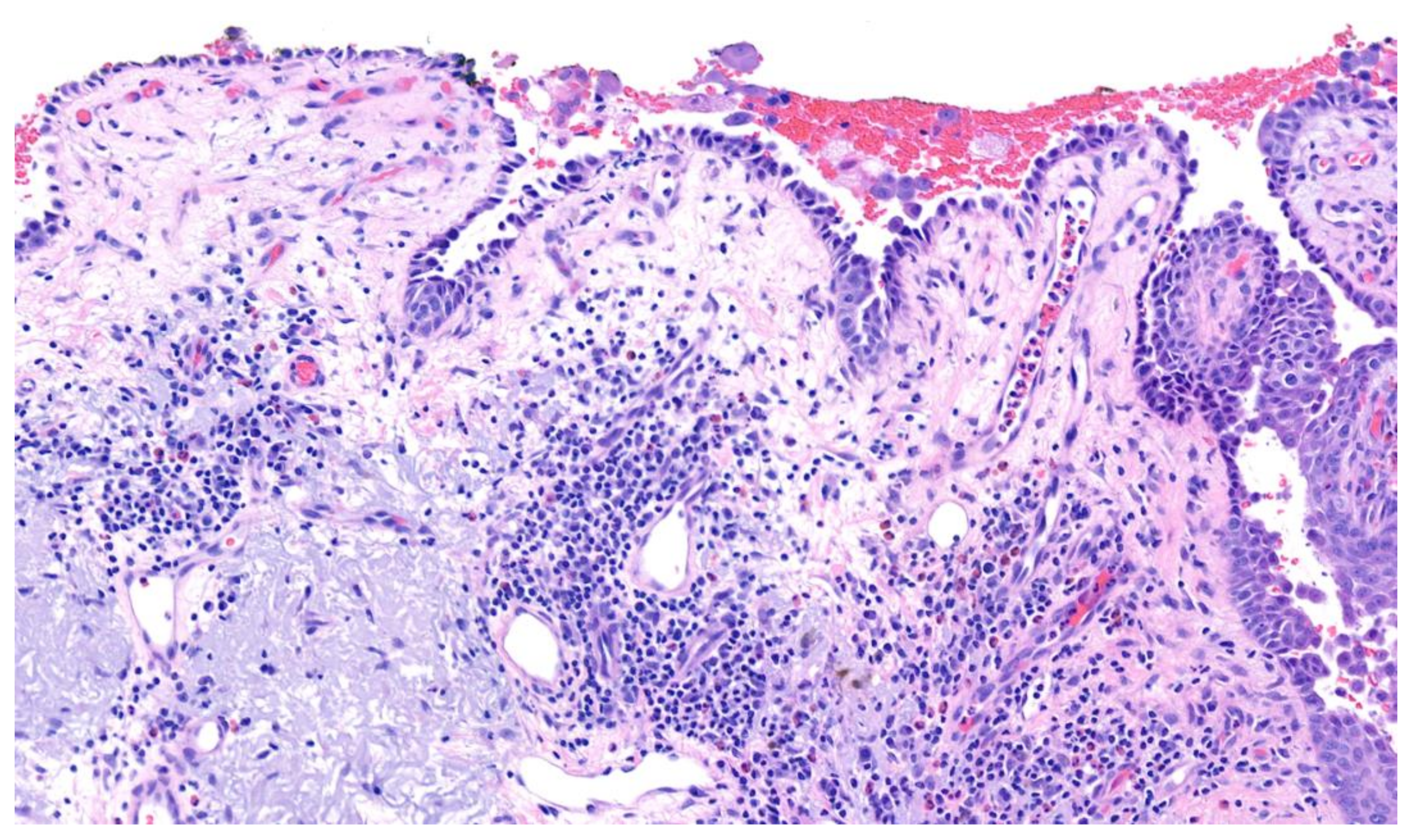
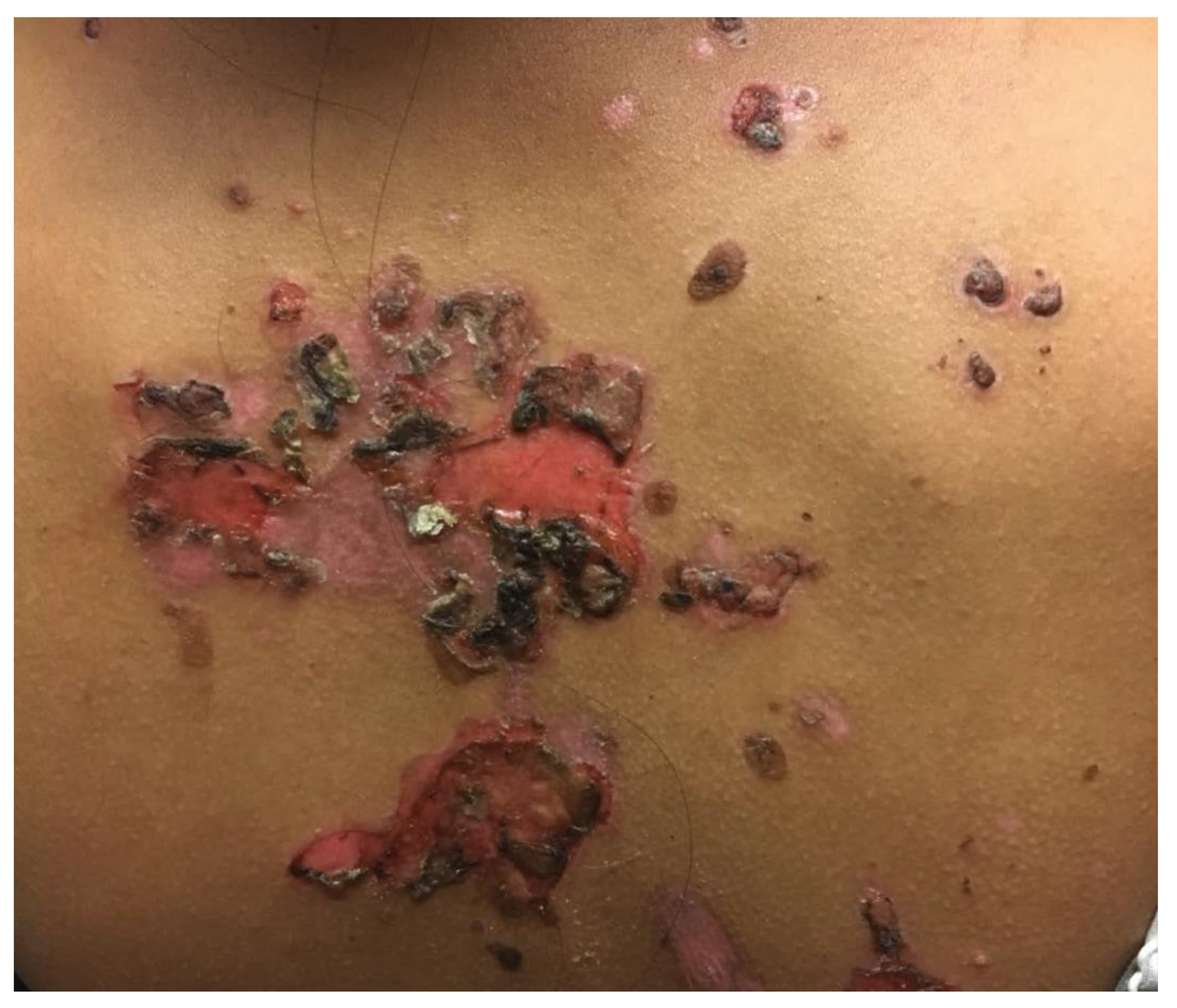
3.1. Pemphigus Vulgaris
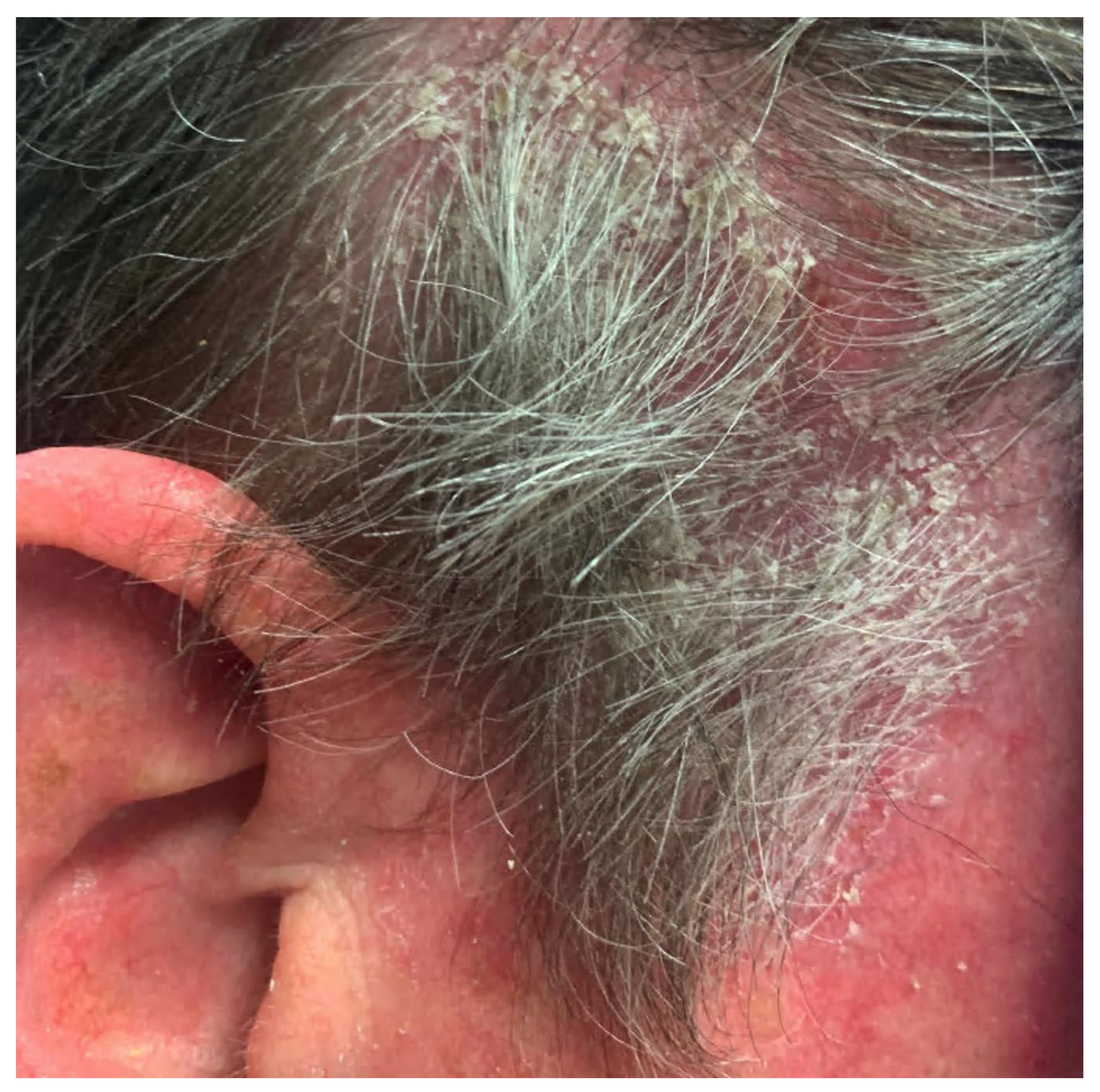
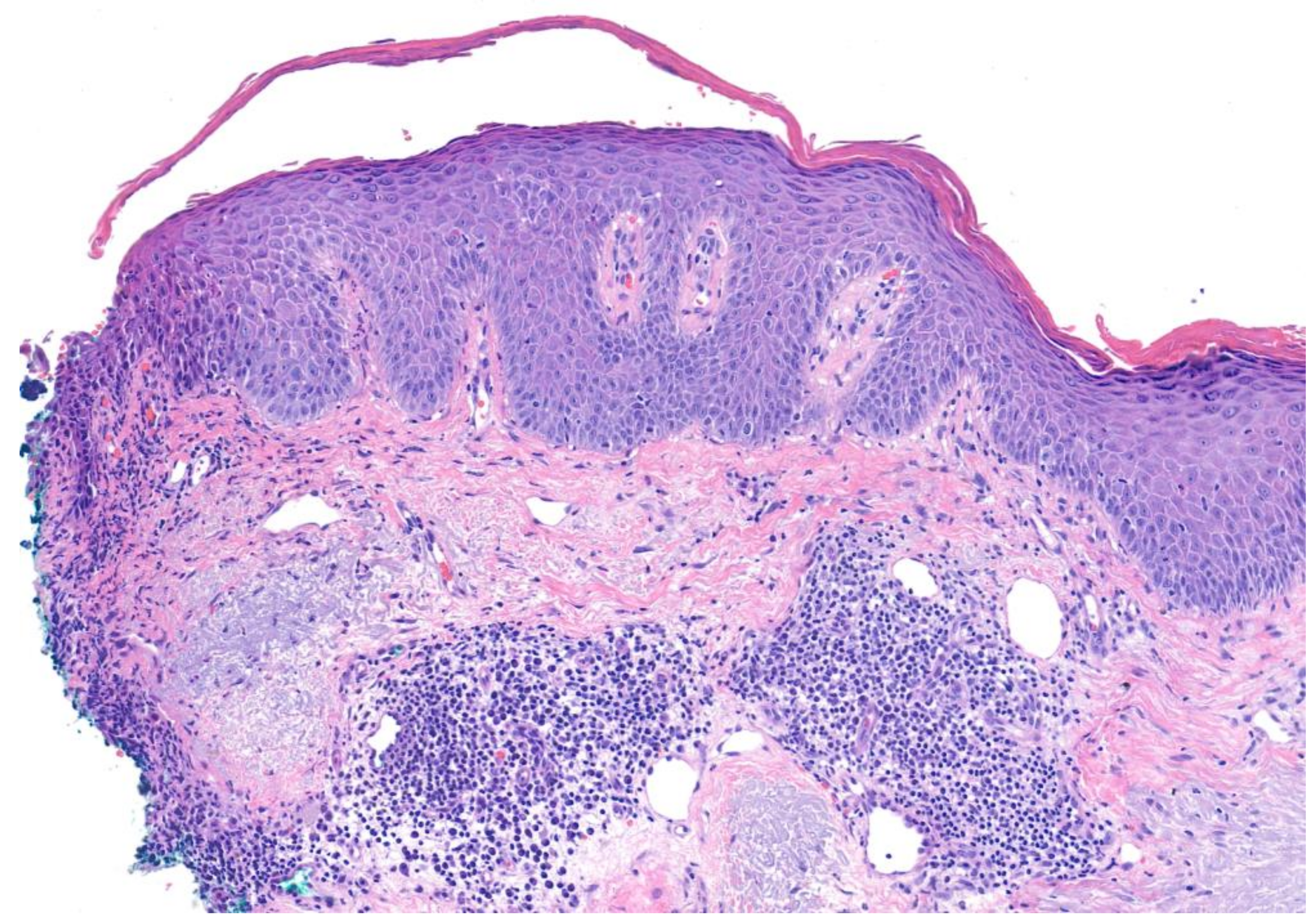
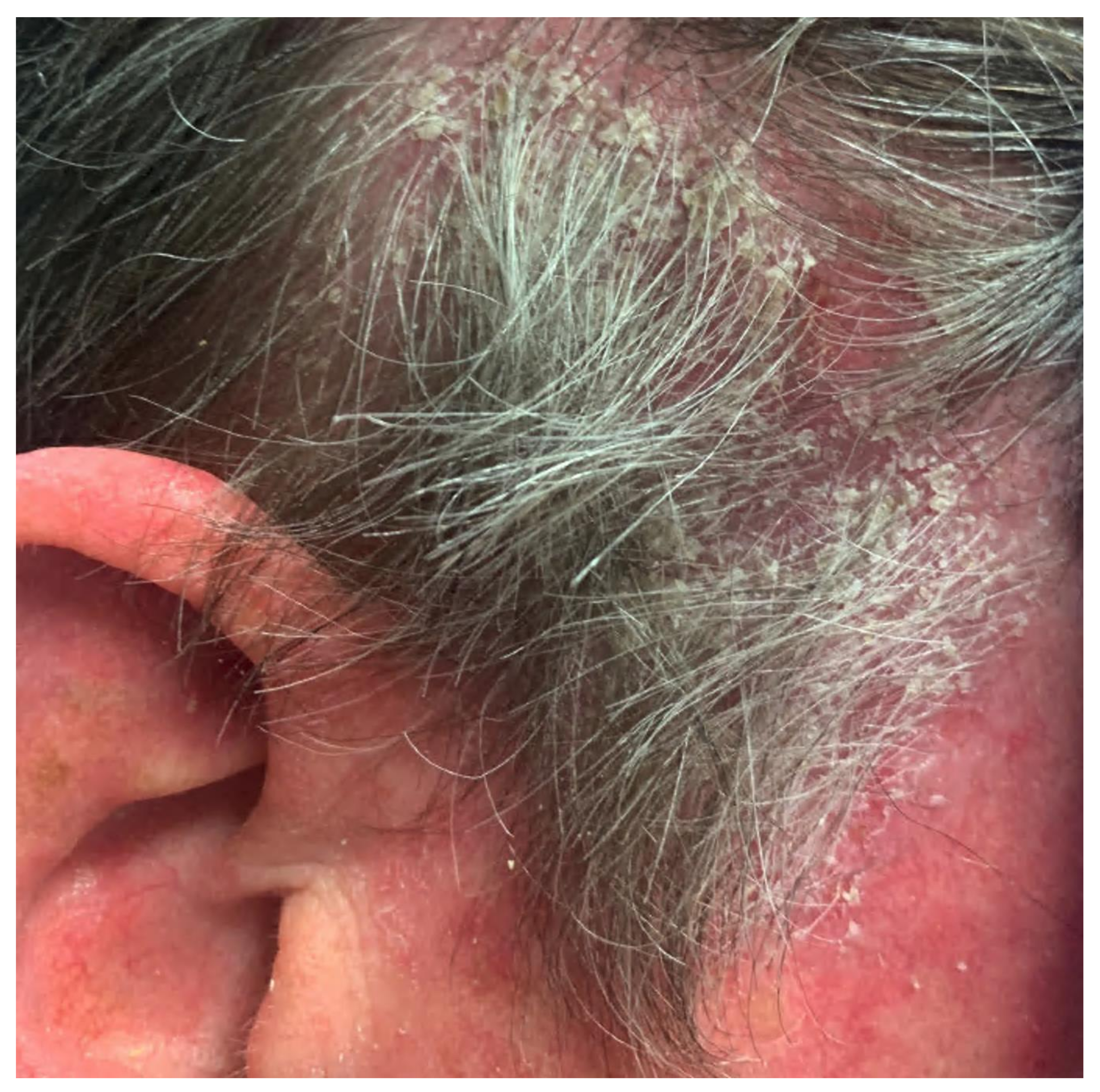
3.2. Pemphigus Foliaceus
3.3. Paraneoplastic Pemphigus
3.4. IgA Pemphigus
4. Diagnosis
5. Management
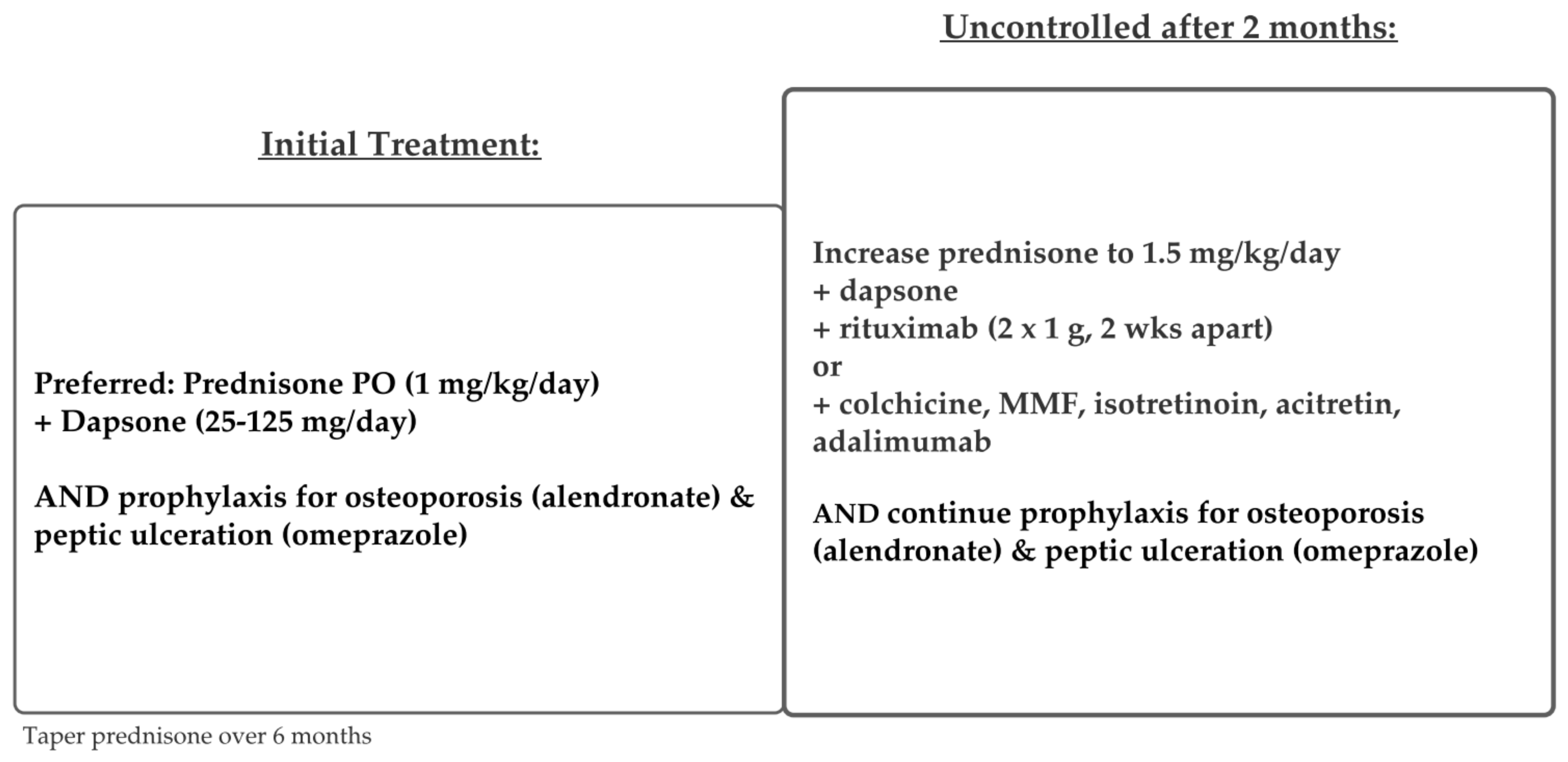
5.1. Pemphigus Vulgaris and Foliaceus
5.2. IgA Pemphigus
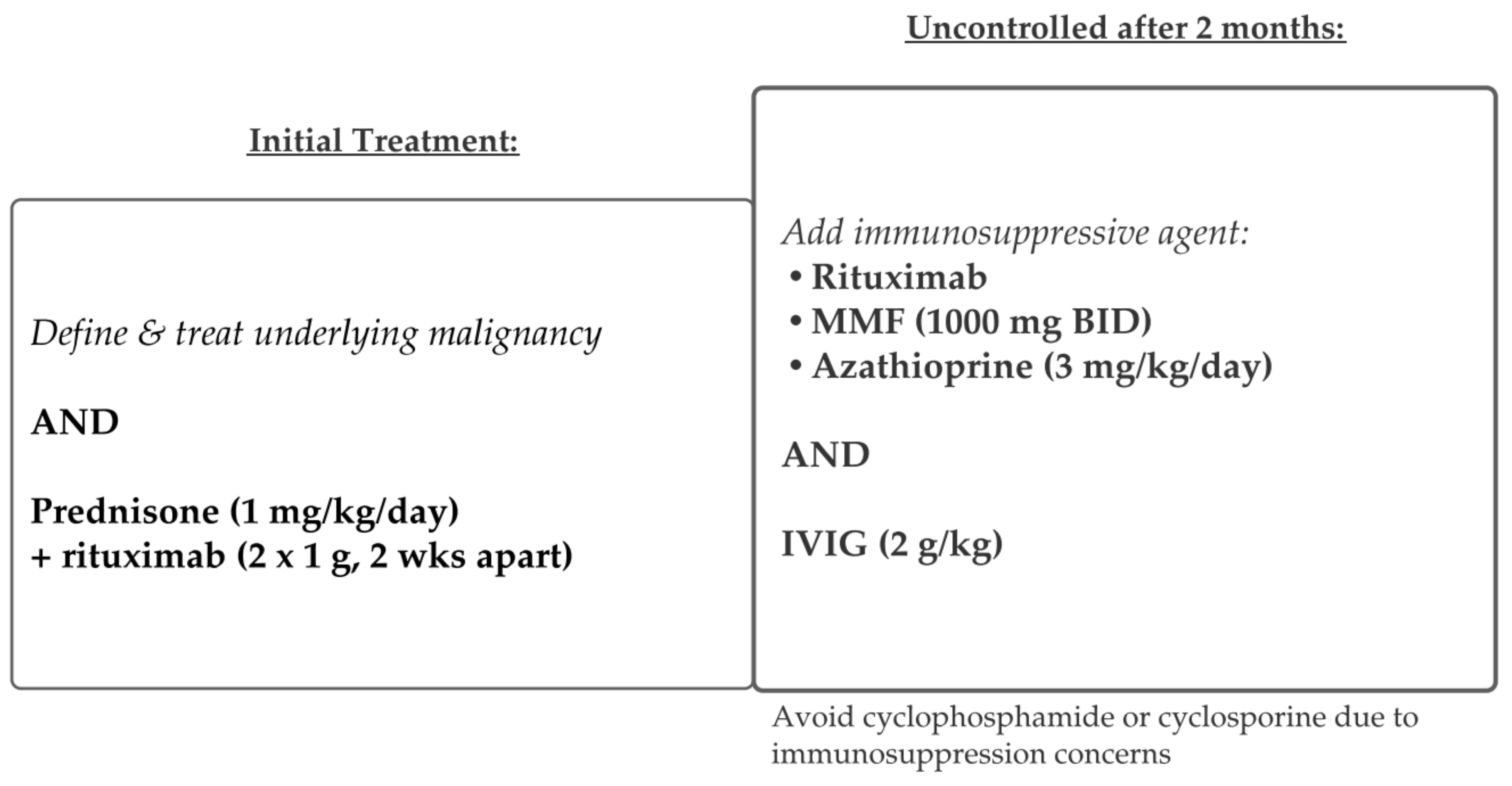
5.3. Paraneoplastic Pemphigus
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Simon, D.G.; Krutchkoff, D.; Kaslow, A.R.; Zarbo, R. Pemphigus in Hartford County, Connecticut, from 1972 to 1977. Arch. Dermatol. 1980, 116, 1035–1037. [Google Scholar] [CrossRef]
- Bastuji-Garin, S.; Souissi, R.; Blum, L.; Turki, H.; Nouira, R.; Jomaa, B.; Zahaf, A.; Ben Osman, A.; Mokhtar, I.; Fazaa, B.; et al. Comparative Epidemiology of Pemphigus in Tunisia and France: Unusual Incidence of Pemphigus Foliaceus in Young Tunisian Women. J. Investig. Dermatol. 1995, 104, 302–305. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.; Wojnarowska, F.; Mehra, N.; Pasricha, J. Pemphigus in Oxford, UK, and New Delhi, India: A Comparative Study of Disease Characteristics and HLA Antigens. Dermatology 1994, 189 (Suppl. 1), 108–110. [Google Scholar] [CrossRef] [PubMed]
- Kridin, K.; Zelber-Sagi, S.; Bergman, R. Pemphigus Vulgaris and Pemphigus Foliaceus: Differences in Epidemiology and Mortality. Acta Derm. Venereol. 2017, 97, 1095–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.R.; Wagner, R.; Khatri, K.; Notani, G.; Awdeh, Z.; Alper, C.A.; Yunis, E.J. Major histocompatibility complex haplotypes and class II genes in non-Jewish patients with pemphigus vulgaris. Proc. Natl. Acad. Sci. USA 1991, 88, 5056–5060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.R.; Yunis, E.J.; Khatri, K.; Wagner, R.; Notani, G.; Awdeh, Z.; Alper, C.A. Major histocompatibility complex haplotype studies in Ashkenazi Jewish patients with pemphigus vulgaris. Proc. Natl. Acad. Sci. USA 1990, 87, 7658–7662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krain, L.S. Increased frequency of HL-A10 in pemphigus vulgaris. Arch. Dermatol. 1973, 108, 803–805. [Google Scholar] [CrossRef] [PubMed]
- Mobini, N.; Yunis, E.J.; Alper, C.A.; Yunis, J.J.; Delgado, J.C.; Yunis, D.E.; AliReza, F.A.; Yahya, D.G.; Kamal, B.H.; Peter, K.G.I.; et al. Identical MHC markers in non-Jewish Iranian and Ash-kenazi Jewish patients with pemphigus vulgaris: Possible common central Asian ancestral origin. Hum. Immunol. 1997, 57, 62–67. [Google Scholar] [CrossRef]
- Tallab, T.; Joharji, H.; Bahamdan, K.; Karkashan, E.; Mourad, M.; Ibrahim, K. The incidence of pemphigus in the southern region of Saudi Arabia. Int. J. Dermatol. 2001, 40, 570–572. [Google Scholar] [CrossRef]
- Laskaris, G.; Stoufi, E. Oral pemphigus vulgaris in a 6-year-old girl. Oral Surg. Oral Med. Oral Pathol. 1990, 69, 609–613. [Google Scholar] [CrossRef]
- Hietanen, J.; Salo, O.P. Pemphigus: An epidemiological study of patients treated in Finnish hospitals between 1969 and 1978. Acta Derm. Venereol. 1982, 62, 491–496. [Google Scholar] [PubMed]
- Hübner, F.; König, I.; Holtsche, M.; Zillikens, D.; Linder, R.; Schmidt, E. Prevalence and age distribution of pem-phigus and pemphigoid diseases among paediatric patients in Germany. J. Eur. Acad. Dermatol. Venereol. 2020, 34, 2600–2605. [Google Scholar] [CrossRef] [PubMed]
- Amagai, M.; Kárpáti, S.; Prussick, R.; Klaus-Kovtun, V.; Stanley, J.R. Autoantibodies against the amino-terminal cadherin-like binding domain of pemphigus vulgaris antigen are pathogenic. J. Clin. Investig. 1992, 90, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Sardana, K.; Garg, V.; Agarwal, P. Is there an emergent need to modify the desmoglein compensation theory in pemphigus on the basis of Dsg ELISA data and alternative pathogenic mechanisms? Br. J. Dermatol. 2013, 168, 669–674. [Google Scholar] [CrossRef]
- Jamora, M.J.J.; Jiao, D.; Bystryn, J.-C. Antibodies to desmoglein 1 and 3, and the clinical phenotype of pemphigus vulgaris. J. Am. Acad. Dermatol. 2003, 48, 976–977. [Google Scholar] [CrossRef] [PubMed]
- Aslanova, M.; Yarrarapu, S.N.S.; Zito, P.M. IgA Pemphigus. In StatPearls; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Kridin, K.; Patel, P.M.; Jones, V.A.; Cordova, A.; Amber, K.T. IgA Pemphigus: A Systematic Review. J. Am. Acad. Dermatol. 2020, 82, 1386–1392. [Google Scholar] [CrossRef]
- Czernik, A.; Wieczorek, M. Paraneoplastic pemphigus: A short review. Clin. Cosmet. Investig. Dermatol. 2016, 9, 291–295. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-H.; Kuo, C.-F.; Chen, Y.-H.; Yang, Y.-W. Incidence, Mortality, and Causes of Death of Patients with Pemphigus in Taiwan: A Nationwide Population-Based Study. J. Investig. Dermatol. 2012, 132, 92–97. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, M.B.; Porter, S.R.; Smoller, B.R.; Sitaru, C. Oral mucosal manifestations of autoimmune skin diseases. Autoimmun. Rev. 2015, 14, 930–951. [Google Scholar] [CrossRef]
- Popescu, I.A.; Statescu, L.; Vata, D.; Porumb-Andrese, E.; Patrascu, A.I.; Grajdeanu, I.-A.; Solovastru, L.G. Pemphigus vulgaris—Approach and management (Review). Exp. Ther. Med. 2019, 18, 5056–5060. [Google Scholar] [CrossRef] [Green Version]
- Bystryn, J.C.; Steinman, N.M. The adjuvant therapy of pemphigus. An update. Arch. Dermatol. 1996, 132, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Baican, A.; Chiorean, R.; Leucuta, D.C.; Baican, C.; Danescu, S.; Ciuce, D.; Sitaru, C. Prediction of survival for patients with pemphigus vulgaris and pemphigus foliaceus: A retrospective cohort study. Orphanet J. Rare Dis. 2015, 10, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messersmith, L.; Krauland, K. Pemphigus Vegetans. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- James, K.A.; Culton, D.A.; Diaz, L.A. Diagnosis and Clinical Features of Pemphigus Foliaceus. Dermatol. Clin. 2011, 29, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Kasperkiewicz, M.; Ellebrecht, C.T.; Takahashi, H.; Yamagami, J.; Zillikens, D.; Payne, A.S.; Amagai, M. Pemphigus. Nat. Rev. Dis. Prim. 2017, 3, 17026. [Google Scholar] [CrossRef] [Green Version]
- Hans-Filho, G.; Aoki, V.; Bittner, N.R.H.; Bittner, G.C. Fogo selvagem: Endemic pemphigus foliaceus. An. Bras. Dermatol. 2018, 93, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Anhalt, G.J.; Kim, S.; Stanley, J.R.; Korman, N.J.; Jabs, D.A.; Kory, M.; Izumi, H.; Ratrie, H.; Mutasim, D.; Ariss-Abdo, L.; et al. Paraneoplastic Pemphigus. N. Engl. J. Med. 1990, 323, 1729–1735. [Google Scholar] [CrossRef]
- Pile, H.D.; Yarrarapu, S.N.S.; Crane, J.S. Drug Induced Pemphigus. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Kaplan, I. Neoplasms associated with paraneoplastic pemphigus: A review with emphasis on non-hematologic malignancy and oral mucosal manifestations. Oral Oncol. 2004, 40, 553–562. [Google Scholar] [CrossRef]
- Nousari, H.C.; Deterding, R.; Wojtczack, H.; Uitto, J.; Aho, S.; Hashimoto, T.; Anhalt, G.J. The Mechanism of Respiratory Failure in Paraneoplastic Pemphigus. N. Engl. J. Med. 1999, 340, 1406–1410. [Google Scholar] [CrossRef]
- Nikolskaia, O.; Nousari, C.; Anhalt, G. Paraneoplastic pemphigus in association with Castleman’s disease. Br. J. Dermatol. 2003, 149, 1143–1151. [Google Scholar] [CrossRef]
- Maldonado, F.; Pittelkow, M.R.; Ryu, J.H. Constrictive bronchiolitis associated with paraneoplastic autoimmune multi-organ syndrome. Respirology 2009, 14, 129–133. [Google Scholar] [CrossRef]
- Ohzono, A.; Sogame, R.; Li, X.; Teye, K.; Tsuchisaka, A.; Numata, S.; Koga, H.; Kawakami, T.; Tsuruta, D.; Ishii, N.; et al. Clinical and immunological findings in 104 cases of paraneoplastic pemphigus. Br. J. Dermatol. 2015, 173, 1447–1452. [Google Scholar] [CrossRef]
- Sehgal, V.N.; Srivastava, G. Paraneoplastic pemphigus/paraneoplastic autoimmune multiorgan syndrome. Int. J. Dermatol. 2009, 48, 162–169. [Google Scholar] [CrossRef]
- Anan, T.; Shimizu, F.; Hatano, Y.; Okamoto, O.; Katagiri, K.; Fujiwara, S. Paraneoplastic pemphigus associated with corneal perforation and cutaneous alternariosis: A case report and review of cases treated with rituximab. J. Dermatol. 2011, 38, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Svoboda, S.A.; Huang, S.; Liu, X.; Hsu, S.; Motaparthi, K. Paraneoplastic pemphigus: Revised diagnostic criteria based on literature analysis. J. Cutan. Pathol. 2021, 48, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Hohwy, T.; Bang, K.; Steiniche, T.; Peterslund, N.A.; d’Amore, F. Alemtuzumab-induced remission of both severe para-neoplastic pemphigus and leukaemic bone marrow infiltration in a case of treatment-resistant B-cell chronic lymphocytic leukaemia. Eur. J. Haematol. 2004, 73, 206–209. [Google Scholar] [CrossRef]
- Lee, S.E.; Kim, S.C. Paraneoplastic pemphigus. Dermatol. Sin. 2010, 28, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Paolino, G.; Didona, D.; Magliulo, G.; Iannella, G.; Didona, B.; Mercuri, S.R.; Moliterni, E.; Donati, M.; Ciofalo, A.; Granata, G.; et al. Paraneoplastic Pemphigus: Insight into the Autoimmune Pathogenesis, Clinical Features and Therapy. Int. J. Mol. Sci. 2017, 18, 2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leger, S.; Picard, D.; Ingen-Housz-Oro, S.; Arnault, J.-P.; Aubin, F.; Carsuzaa, F.; Chaumentin, G.; Chevrant-Breton, J.; Chosidow, O.; Crickx, B.; et al. Prognostic Factors of Paraneoplastic Pemphigus. Arch. Dermatol. 2012, 148, 1165–1172. [Google Scholar] [CrossRef] [Green Version]
- Tsuruta, D.; Ishii, N.; Hamada, T.; Ohyama, B.; Fukuda, S.; Koga, H.; Imamura, K.; Kobayashi, H.; Karashima, T.; Nakama, T.; et al. IgA pemphigus. Clin. Dermatol. 2011, 29, 437–442. [Google Scholar] [CrossRef]
- Chorzelski, T.P.; Beutner, E.H.; Kowalewski, C.; Olszewska, M.; Maciejowska, E.; Seferowicz, E.; Kumar, V.; Jablonska, S. IgA pemphigus foliaceus with a clinical presentation of pemphigus herpetiformis. J. Am. Acad. Dermatol. 1991, 24, 839–844. [Google Scholar] [CrossRef]
- Giurdanella, F.; Diercks, G.; Jonkman, M.; Pas, H. Laboratory diagnosis of pemphigus: Direct immunofluorescence remains the gold standard. Br. J. Dermatol. 2016, 175, 185–186. [Google Scholar] [CrossRef]
- Gregoriou, S.; Efthymiou, O.; Stefanaki, C.; Rigopoulos, D. Management of pemphigus vulgaris: Challenges and solutions. Clin. Cosmet. Investig. Dermatol. 2015, 8, 521–527. [Google Scholar] [CrossRef] [Green Version]
- Joly, P.; Maho-Vaillant, M.; Prost-Squarcioni, C.; Hebert, V.; Houivet, E.; Calbo, S.; Golinski, M.L.; Labeille, B.; Picard-Dahan, C.; Carle, P.; et al. First-Line Rituximab Combined with Short-Term Prednisone Versus Prednisone Alone for the Treatment of Pem-phigus (Ritux 3): A Prospective, Multicentre, Parallel-Group, Open-Label Randomised Trial. Lancet 2017, 389, 2031–2040. [Google Scholar] [CrossRef]
- Cholera, M.; Chainani-Wu, N. Management of Pemphigus Vulgaris. Adv. Ther. 2016, 33, 910–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joly, P.; Horwath, B.; Patsatsi, A.; Uzun, S.; Bech, R.; Beissert, S.; Bergman, R.; Bernard, P.; Borradori, L.; Caproni, M.; et al. Updated S2K guidelines on the management of pemphigus vulgaris and foliaceus initiated by the european academy of dermatology and venereology (EADV). J. Eur. Acad. Dermatol. Venereol. 2020, 34, 1900–1913. [Google Scholar] [CrossRef]
- Boulard, C.; Lehembre, S.D.; Picard-Dahan, C.; Kern, J.; Zambruno, G.; Feliciani, C.; Marinovic, B.; Vabres, P.; Borradori, L.; Prost-Squarcioni, C.; et al. Calculation of cut-off values based on the Autoimmune Bullous Skin Disorder Intensity Score (ABSIS) and Pemphigus Disease Area Index (PDAI) pemphigus scoring systems for defining moderate, significant and extensive types of pemphigus. Br. J. Dermatol. 2016, 175, 142–149. [Google Scholar] [CrossRef]
- Harman, K.E.; Brown, D.; Exton, L.S.; Groves, R.W.; Hampton, P.J.; Mustapa, M.F.M.; Setterfield, J.; Yesudian, P.D.; McHenry, P.; Gibbon, K.; et al. British Association of Dermatologists’ guidelines for the management of pemphigus vulgaris 2017. Br. J. Dermatol. 2017, 177, 1170–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniel, B.S.; Hertl, M.; Werth, V.P.; Eming, R.; Murrell, D.F. Severity score indexes for blistering diseases. Clin. Dermatol. 2012, 30, 108–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lernia, V.; Casanova, D.M.; Goldust, M.; Ricci, C. Pemphigus Vulgaris and Bullous Pemphigoid: Update on Diagnosis and Treatment. Dermatol. Pract. Concept. 2020, 10, e2020050. [Google Scholar] [CrossRef]
- Ingen-Housz-Oro, S.; Valeyrie-Allanore, L.; Cosnes, A.; Ortonne, N.; Hue, S.; Paul, M.; Wolkenstein, P.; Chosidow, O. First-line Treatment of Pemphigus Vulgaris with a Combination of Rituximab and High-Potency Topical Corticosteroids. JAMA Dermatol. 2015, 151, 200–203. [Google Scholar] [CrossRef]
- Kushner, C.J.; Wang, S.; Tovanabutra, N.; Tsai, D.E.; Werth, V.P.; Payne, A.S. Factors Associated with Complete Remission After Rituximab Therapy for Pemphigus. JAMA Dermatol. 2019, 155, 1404–1409. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.; Ingen-Housz-Oro, S.; Chosidow, O.; Antonicelli, F.; Bernard, P. Rituximab as Single Long-term Maintenance Therapy in Patients with Difficult-to-Treat Pemphigus. JAMA Dermatol. 2018, 154, 363. [Google Scholar] [CrossRef] [PubMed]
- Genentech Press Releases. Available online: https://www.gene.com/media/press-releases/14727/2018-06-07/fda-approves-genentechs-rituxan-rituxima (accessed on 7 June 2018).
- Rituxan Prices, Coupons & Patient Assistance Programs. Available online: https://www.drugs.com/price-guide/rituxan (accessed on 24 June 2021).
- Mimouni, D.; Anhalt, G.J.; Cummins, D.L.; Kouba, D.J.; Thorne, J.E.; Nousari, H.C. Treatment of Pemphigus Vulgaris and Pemphigus Foliaceus with Mycophenolate Mofetil. Arch. Dermatol. 2003, 139, 739–742. [Google Scholar] [CrossRef] [Green Version]
- Chams-Davatchi, C.; Esmaili, N.; Daneshpazhooh, M.; Valikhani, M.; Balighi, K.; Hallaji, Z.; Barzegari, M.; Akhyani, M.; Ghodsi, S.Z.; Seirafi, H.; et al. Randomized controlled open-label trial of four treatment regimens for pemphigus vulgaris. J. Am. Acad. Dermatol. 2007, 57, 622–628. [Google Scholar] [CrossRef]
- Chams-Davatchi, C.; Mortazavizadeh, A.; Daneshpazhooh, M.; Davatchi, F.; Balighi, K.; Esmaili, N.; Akhyani, M.; Hallaji, Z.; Seirafi, H.; Mortazavi, H. Randomized double blind trial of prednisolone and azathioprine, vs. prednisolone and placebo, in the treatment of pemphigus vulgaris. J. Eur. Acad. Dermatol. Venereol. 2012, 27, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Olszewska, M.; Kolacinska-Strasz, Z.; Sulej, J.; Labecka, H.; Cwikla, J.; Natorska, U.; Blaszczyk, M. Efficacy and safety of cyclophosphamide, azathioprine, and cyclosporine (ciclosporin) as adjuvant drugs in pemphigus vulgaris. Am. J. Clin. Dermatol. 2007, 8, 85–92. [Google Scholar] [CrossRef]
- Vinall, C.; Stevens, L.; McArdle, P. Pemphigus vulgaris: A multidisciplinary approach to management. Case Rep. 2013, 2013, bcr2013200479. [Google Scholar] [CrossRef]
- Sadaksharam, J. Current Therapeutic Modalities of Immunobullous Lesions. Contemp. Clin. Dent. 2019, 10, 407–409. [Google Scholar]
- Lehman, J.S.; Kalaaji, A.N. Role of primary prophylaxis for pneumocystis pneumonia in patients treated with systemic corticosteroids or other immunosuppressive agents for immune-mediated dermatologic conditions. J. Am. Acad. Dermatol. 2010, 63, 815–823. [Google Scholar] [CrossRef]
- Amber, K.T.; Lamberts, A.; Solimani, F.; Agnoletti, A.F.; Didona, D.; Euverman, I.; Cozzani, E.; Yueh, L.H.; Di Zenzo, G.; Leshem, Y.A.; et al. Determining the Incidence of Pneumocystis Pneumonia in Patients with Autoimmune Blistering Diseases Not Receiving Routine Prophylaxis. JAMA Dermatol. 2017, 153, 1137–1141. [Google Scholar] [CrossRef] [Green Version]
- Hirata, Y.; Abe, R.; Kikuchi, K.; Hamasaka, A.; Shinkuma, S.; Nomura, T.; Nishie, W.; Arita, K.; Shimizu, H. Intraepidermal neutrophilic IgA pemphigus successfully treated with dapsone. Eur. J. Dermatol. 2012, 22, 282–283. [Google Scholar] [CrossRef] [PubMed]
- Howell, S.M.; Bessinger, G.T.; Altman, C.E.; Belnap, C.M. Rapid response of IgA pemphigus of the subcorneal pustular dermatosis subtype to treatment with adalimumab and mycophenolate mofetil. J. Am. Acad. Dermatol. 2005, 53, 540–542. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Presentation | H&E | DIF | IIF | Serology | |
|---|---|---|---|---|---|
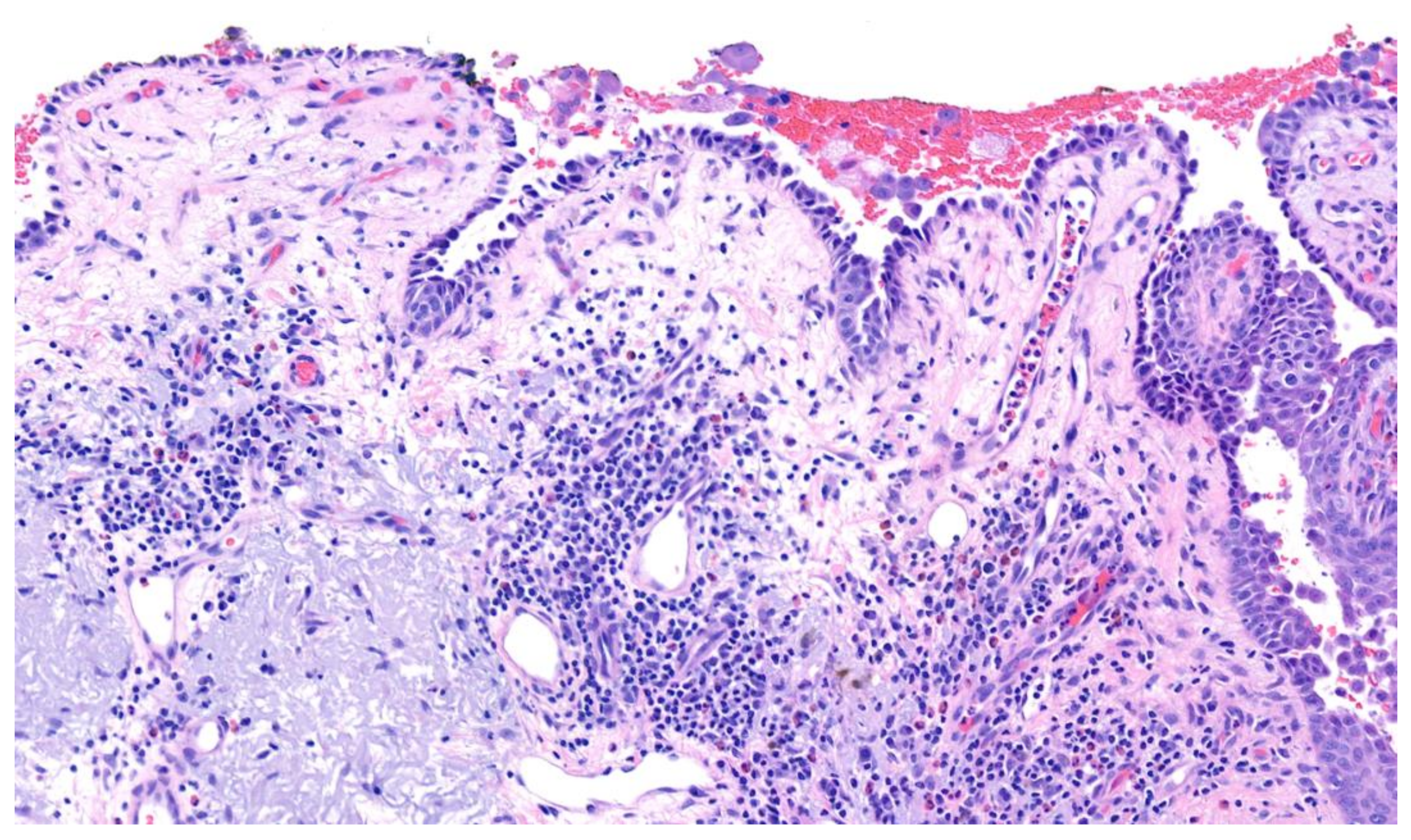
| Pemphigus vulgaris | Painful blisters and erosions predominating in oropharyngeal mucous membranes; symptoms can include dysphagia, vocal hoarseness, vaginal irritation, painful sexual intercourse; palms and soles are spared | Suprabasilar acantholysis with retention of basal keratinocytes along the basement membrane (“tombstoning”) | Intercellular deposition of immunoglobulin G (IgG) | Intercellular deposition of immunoglobulin G (IgG) circulating antibodies Utilize monkey esophagus substrate | Presence of antibodies against both desmoglein 1 and desmoglein 3 or antibodies against desmoglein 3 only |
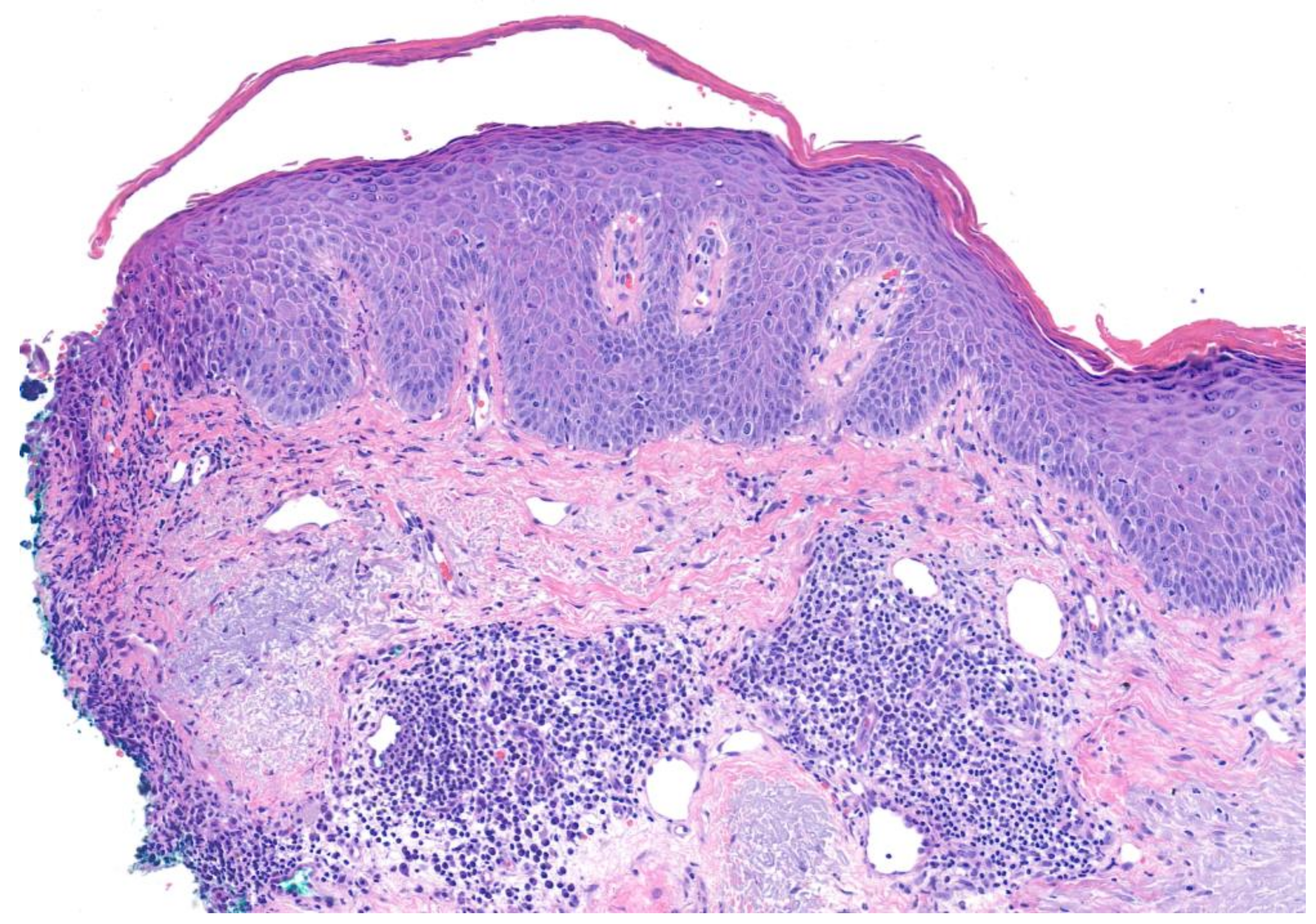
| Pemphigus foliaceus | Painful blisters and erosions without mucosal membrane involvement; cutaneous involvement primarily concentrated in seborrheic areas (scalp, face, upper trunk) | Acantholysis within upper epidermis, adjacent or within the granular layer, leading to a subcorneal cleft If significant eosinophils are present, consider drug-induced pemphigus | Intercellular deposition of immunoglobulin G (IgG) Negative DIF studies are not uncommon in drug-induced pemphigus | Intercellular deposition of immunoglobulin G (IgG) circulating antibodies Utilize guinea pig esophagus substrate | Presence of antibodies against desmoglein 1 only |
| Paraneoplastic pemphigus | Severe mucosal involvement and polymorphous skin lesions with associated underlying malignancy or neoplasm (e.g. Non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, Castleman’s disease) | Suprabasilar acantholysis, keratinocyte necrosis, and interface change | Intercellular deposition of immunoglobulin G (IgG) Negative DIF studies are not uncommon in paraneoplastic pemphigus | Intercellular deposition of immunoglobulin G (IgG) circulating antibodies Utilize rat bladder substrate | Presence of antibodies against plakin proteins |
| IgA pemphigus | Tense bullae that transition into clear fluid-filled blisters; cutaneous involvement of vesicles (81%), pustules (75%), and erythematous annular plaques (64%) primarily seen in flexural areas of proximal extremities and trunk; mucous membranes usually spared | Subcorneal pustular dermatosis type: subcorneal vesiculopustules with minimal acantholysis Intraepidermal neutrophilic dermatosis type: intraepidermal vesiculopustules with variable acantholysis | Intercellular deposition of immunoglobulin A (IgA) | Intercellular deposition of immunoglobulin A (IgA) circulating antibodies | Subcorneal pustular dermatosis type: presence of antibodies against desmocollin 1 Intraepidermal neutrophilic dermatosis type: presence of IgA antibodies against desmoglein 1 and 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malik, A.M.; Tupchong, S.; Huang, S.; Are, A.; Hsu, S.; Motaparthi, K. An Updated Review of Pemphigus Diseases. Medicina 2021, 57, 1080. https://doi.org/10.3390/medicina57101080
Malik AM, Tupchong S, Huang S, Are A, Hsu S, Motaparthi K. An Updated Review of Pemphigus Diseases. Medicina. 2021; 57(10):1080. https://doi.org/10.3390/medicina57101080
Chicago/Turabian StyleMalik, Ali M., Sarah Tupchong, Simo Huang, Abhirup Are, Sylvia Hsu, and Kiran Motaparthi. 2021. "An Updated Review of Pemphigus Diseases" Medicina 57, no. 10: 1080. https://doi.org/10.3390/medicina57101080
APA StyleMalik, A. M., Tupchong, S., Huang, S., Are, A., Hsu, S., & Motaparthi, K. (2021). An Updated Review of Pemphigus Diseases. Medicina, 57(10), 1080. https://doi.org/10.3390/medicina57101080