
Staphylococcal Scalded Skin Syndrome and Bullous Impetigo


Abstract
:1. Introduction
2. Pathophysiology
3. Clinical Features in Children
4. Clinical Features in Adults
5. Diagnostic Workup
6. Differential Diagnosis
7. Management
1. Introduction
2. Pathophysiology
3. Clinical Features and Diagnostic Workup
4. Differential Diagnosis
5. Management
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liy-Wong, C.; Pope, E.; Weinstein, M.; Lara-Corrales, I. Staphylococcal scalded skin syndrome: An epidemiological and clinical review of 84 cases. Pediatr. Dermatol. 2021, 38, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Handler, M.; Schwartz, R. Staphylococcal scalded skin syndrome: Diagnosis and management in children and adults. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 1418–1423. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.; Shoff, H.W. Staphylococcal Scalded Skin Syndrome. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Leung, A.K.C.; Barankin, B.; Leong, K.F. Staphylococcal-scalded skin syndrome: Evaluation, diagnosis, and management. World J. Pediatr. 2018, 14, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Jordan, K.S. Staphylococcal Scalded Skin Syndrome: A Pediatric Dermatological Emergency. Adv. Emerg. Nurs. J. 2019, 41, 129–134. [Google Scholar] [CrossRef]
- Vernali, S.; Blasiak, R.C.; Morrell, D.S. Demographic characteristics, clinical features, and optimal management of hospitalized patients with staphylococcal scalded skin syndrome. Pediatr. Dermatol. 2021, 38, 825–830. [Google Scholar] [CrossRef] [PubMed]
- Amagai, M.; Matsuyoshi, N.; Wang, Z.H.; Andl, C.; Stanley, J.R. Toxin in bullous impetigo and staphylococcal scalded-skin syndrome targets desmoglein 1. Nat. Med. 2000, 6, 1275–1277. [Google Scholar] [CrossRef]
- Wu, H.; Wang, Z.H.; Yan, A.; Lyle, S.; Fakharzadeh, S.; Wahl, J.K.; Wheelock, M.J.; Ishikawa, H.; Uitto, J.; Amagai, M.; et al. Protection against Pemphigus Foliaceus by Desmoglein 3 in Neonates. N. Engl. J. Med. 2000, 343, 31–35. [Google Scholar] [CrossRef]
- Kanathur, S.; Sarvajnyamurthy, S.; Somaiah, S. Characteristic facies: An index of the disease. Indian J. Dermatol. Venereol. Leprol. 2013, 79, 439–443. [Google Scholar] [CrossRef]
- Staiman, A.; Hsu, D.Y.; I Silverberg, J. Epidemiology of staphylococcal scalded skin syndrome in U.S. children. Br. J. Dermatol. 2018, 178, 704–708. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.K.; Yadav, P.; Mishra, A. A Systemic Review on Staphylococcal Scalded Skin Syndrome (SSSS): A Rare and Critical Disease of Neonates. Open Microbiol. J. 2016, 10, 150–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodiuk-Gad, R.P.; Hung, S.-I.; Valeyrie-Allanore, L.; Shear, N.H. Stevens–Johnson Syndrome and Toxic Epidermal Necrolysis: An Update. Am. J. Clin. Dermatol. 2015, 16, 475–493. [Google Scholar] [CrossRef]
- Szatkowski, J.; Schwartz, R.A. Acute generalized exanthematous pustulosis (AGEP): A review and update. J. Am. Acad. Dermatol. 2015, 73, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Feldmeyer, L.; Heidemeyer, K.; Yawalkar, N. Acute Generalized Exanthematous Pustulosis: Pathogenesis, Genetic Background, Clinical Variants and Therapy. Int. J. Mol. Sci. 2016, 17, 1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman-Adams, H.; Banvard, C.; Juckett, G. Impetigo: Diagnosis and treatment. Am. Fam. Physician 2014, 90, 229–235. [Google Scholar]
- Burnham, J.P.; Kollef, M.H. Understanding toxic shock syndrome. Intensiv. Care Med. 2015, 41, 1707–1710. [Google Scholar] [CrossRef]
- Vuzevski, V.D.; Van Joost, T.; Wagenvoort, J.H.T.; Dey, J.J.M. Cutaneous Pathology in Toxic Shock Syndrome. Int. J. Dermatol. 1989, 28, 94–97. [Google Scholar] [CrossRef]
- Agarwal, S.; Agrawal, D.K. Kawasaki disease: Etiopathogenesis and novel treatment strategies. Expert Rev. Clin. Immunol. 2017, 13, 247–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Gupta, A. Kawasaki disease for dermatologists. Indian Dermatol. Online J. 2016, 7, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Pardo, S.; Perera, T.B. Scarlet Fever. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- McMahon, P. Staphylococcal Scalded Skin Syndrome. 2021. Available online: https://www.uptodate.com/contents/staphylococcal-scalded-skin-syndrome (accessed on 22 October 2021).
- Bamberger, D.M.; E Boyd, S. Management of Staphylococcus aureus infections. Am. Fam. Physician 2005, 72, 2474–2481. [Google Scholar] [PubMed]
- Wang, Z.; Feig, J.L.; Mannschreck, D.B.; Cohen, B.A. Antibiotic sensitivity and clinical outcomes in staphylococcal scalded skin syndrome. Pediatr. Dermatol. 2019, 37, 222–223. [Google Scholar] [CrossRef]
- Davidson, J.; Polly, S.; Hayes, P.J.; Fisher, K.R.; Talati, A.J.; Patel, T. Recurrent Staphylococcal Scalded Skin Syndrome in an Extremely Low-Birth-Weight Neonate. Am. J. Perinatol. Rep. 2017, 7, e134–e137. [Google Scholar] [CrossRef] [Green Version]
- Preda-Naumescu, A.; Elewski, B.; Mayo, T.T. Common Cutaneous Infections: Patient Presentation, Clinical Course, and Treatment Options. Med. Clin. N. Am. 2021, 105, 783–797. [Google Scholar] [CrossRef]
- Nardi, N.M.; Schaefer, T.J. Impetigo. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Johnson, M.K. Impetigo. Adv. Emerg. Nurs. J. 2020, 42, 262–269. [Google Scholar] [CrossRef]
- Mannschreck, D.; Feig, J.; Selph, J.; Cohen, B. Disseminated bullous impetigo and atopic dermatitis: Case series and literature review. Pediatr. Dermatol. 2020, 37, 103–108. [Google Scholar] [CrossRef]
- Sommer, L.L.; Reboli, A.C.; Heymann, W.R. Chapter 74: Bacterial Diseases. In Dermatology, 4th ed.; Bolognia, J.L., Schaf-fer, J.V., Cerroni, L., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1259–1295. [Google Scholar]
- Gahlawat, G.; Tesfaye, W.; Bushell, M.; Abrha, S.; Peterson, G.M.; Mathew, C.; Sinnollareddy, M.; McMillan, F.; Samarawickrema, I.; Calma, T.; et al. Emerging Treatment Strategies for Impetigo in Endemic and Nonendemic Settings: A Systematic Review. Clin. Ther. 2021, 43, 986–1006. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Children | Adults | |
|---|---|---|
| Site of primary infection | Conjunctiva, nose, throat, diaper area, umbilical stump, or circumcision wound | Abscess, arteriovenous fistula infection, pneumonia, osteoarthritis, or septic arthritis |
| Common comorbidities | None | Immunodeficiency and renal disease |
| Blood cultures | Negative | Positive |
| Mortality rate | <5% | >60% |
| Disorder | Demographics | Triggers | Clinical Presentation | Histopathology | Clinical Course |
|---|---|---|---|---|---|
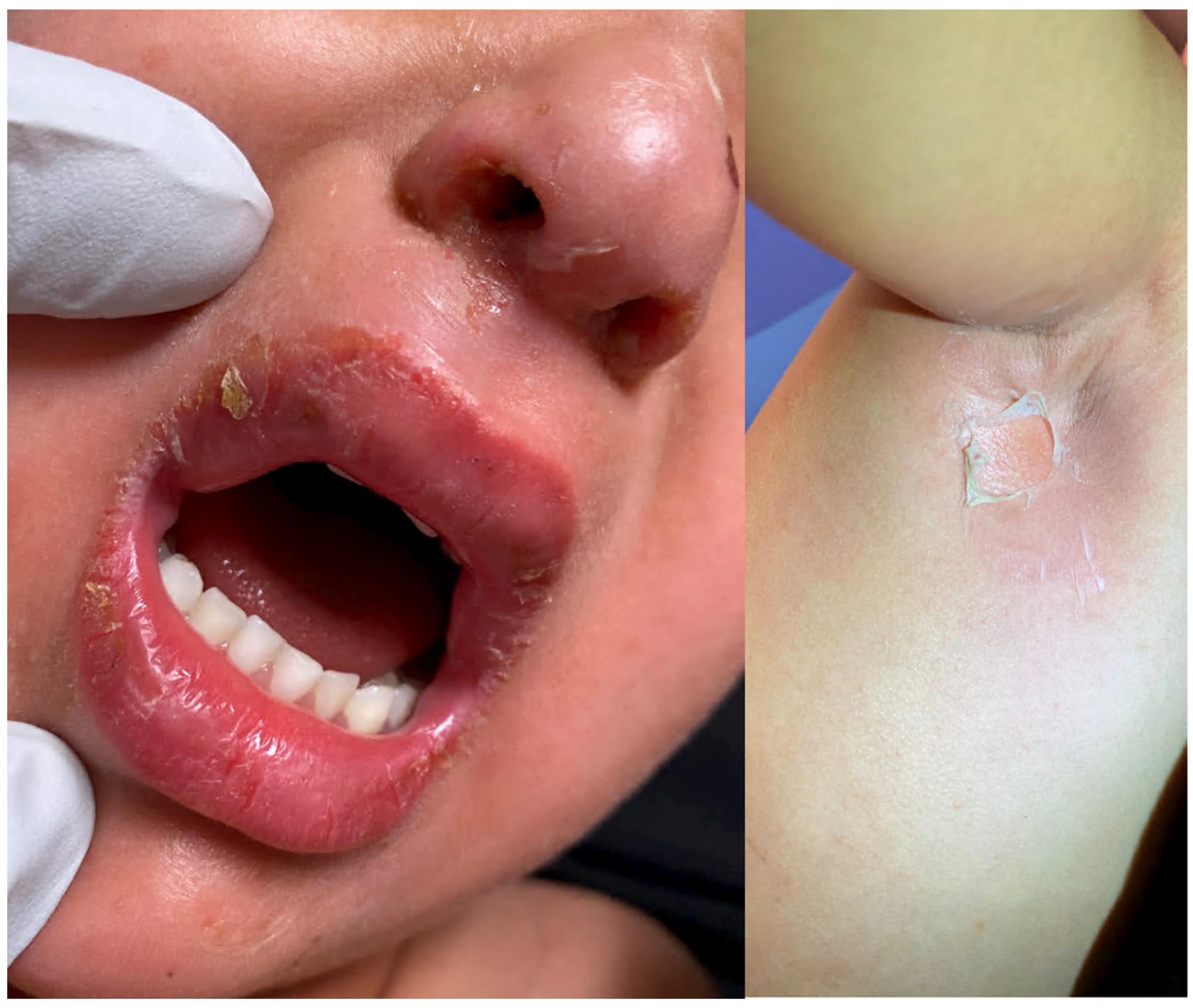
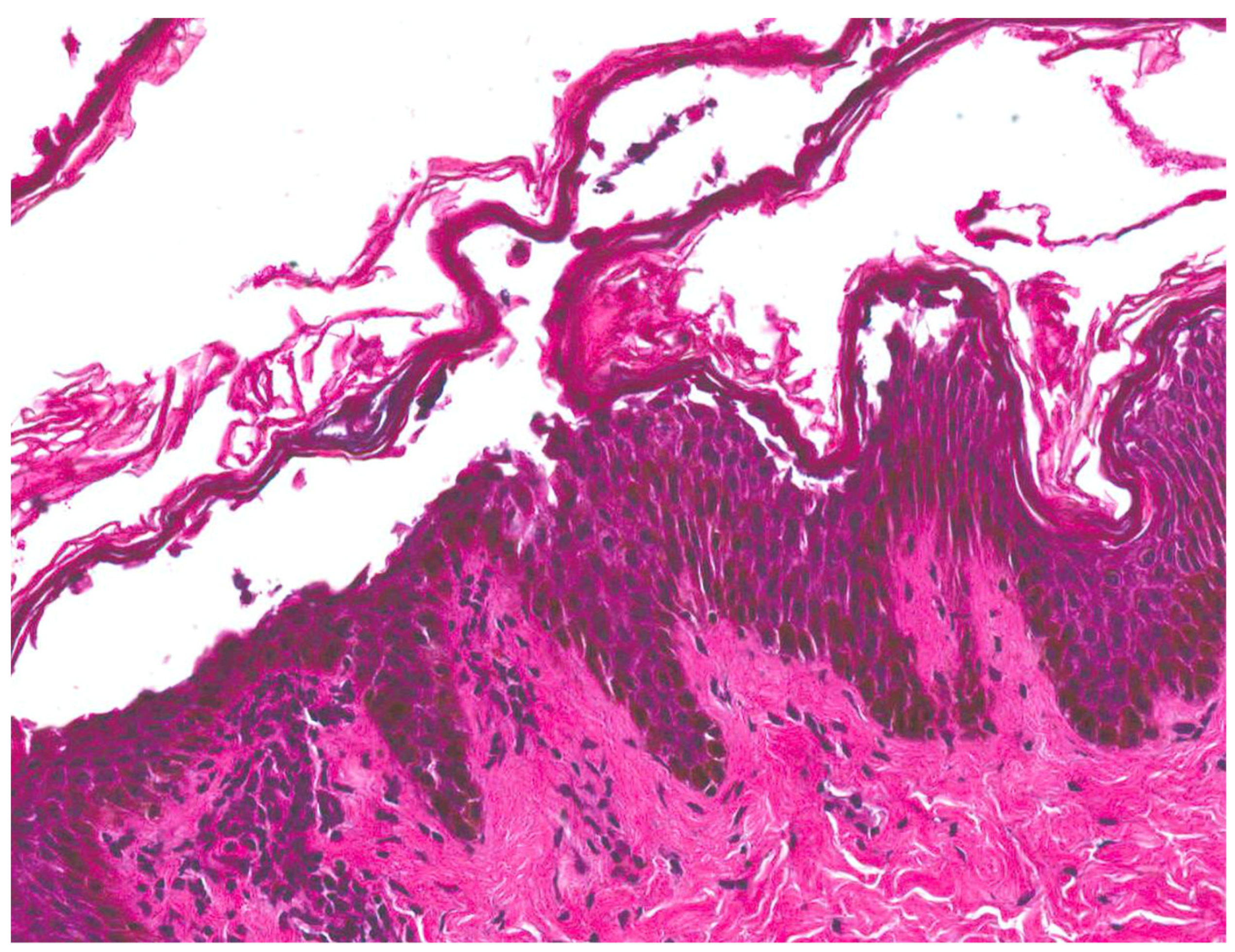
| Staphylococcal scalded skin syndrome (SSSS) [1,2,3,4] | Most commonly seen in children under the age of six | Initial localized infection by S. aureus | Prodrome of irritability, generalized fatigue, and fever followed by the progression of skin lesions. Positive Nikolsky sign | Superficial intraepidermal cleavage beneath the stratum corneum | Within 24–48 h, tender erythematous patches develop. Several hours later, fragile blisters develop within the areas. The blisters progress to form bullae, which rupture easily and then desquamate. |
| Stevens-Johnson syndrome(SJS)/Toxic epidermal necrolysis (TEN) [12] | Most common in older children and adults | 80% of cases are linked to medication intake: sulfa drugs, allopurinol, tetracyclines, anticonvulsants, and NSAIDS | Full thickness epidermal detachment often involving the trunk and proximal extremities; mucosal surfaces affected in >90% of cases. Lesions start as red to dusky macules that coalesce. The necrotic epidermis detaches from the dermis and results in bullae formation. Nikolsky and Asboe–Hansen signs are positive | Uninvolved stratum corneum, vacuolar interface dermatitis with necrotic keratinocytes in the epidermis, with progression to full thickness epidermal necrosis | Lesions appear 7–21 days after drug exposure. Progresses for about 4–5 days before entering a plateau phase. Complete healing can take up to several weeks. Mortality rate near 5% for SJS, but up to 50% in TEN |
| Acute generalized exanthematous pustulosis [13,14] | Can occur in any age group. More common in women | Most cases linked to medication intake: aminopenicillins, cephalosporins, clindamycin, macrolide, and calcium channel blockers | Small non-follicular pustules arising in areas of background erythema often initially on face and flexural sites. Mucosal involvement sometimes present. Associated with fever and facial edema | Subcorneal, and/or intraepidermal pustules with eosinophilic and neutrophilic infiltrates and papillary dermal edema | Lesions appear <4 days after drug exposure then heal quickly over a few days with no evidence of scarring as level of split is subcorneal |
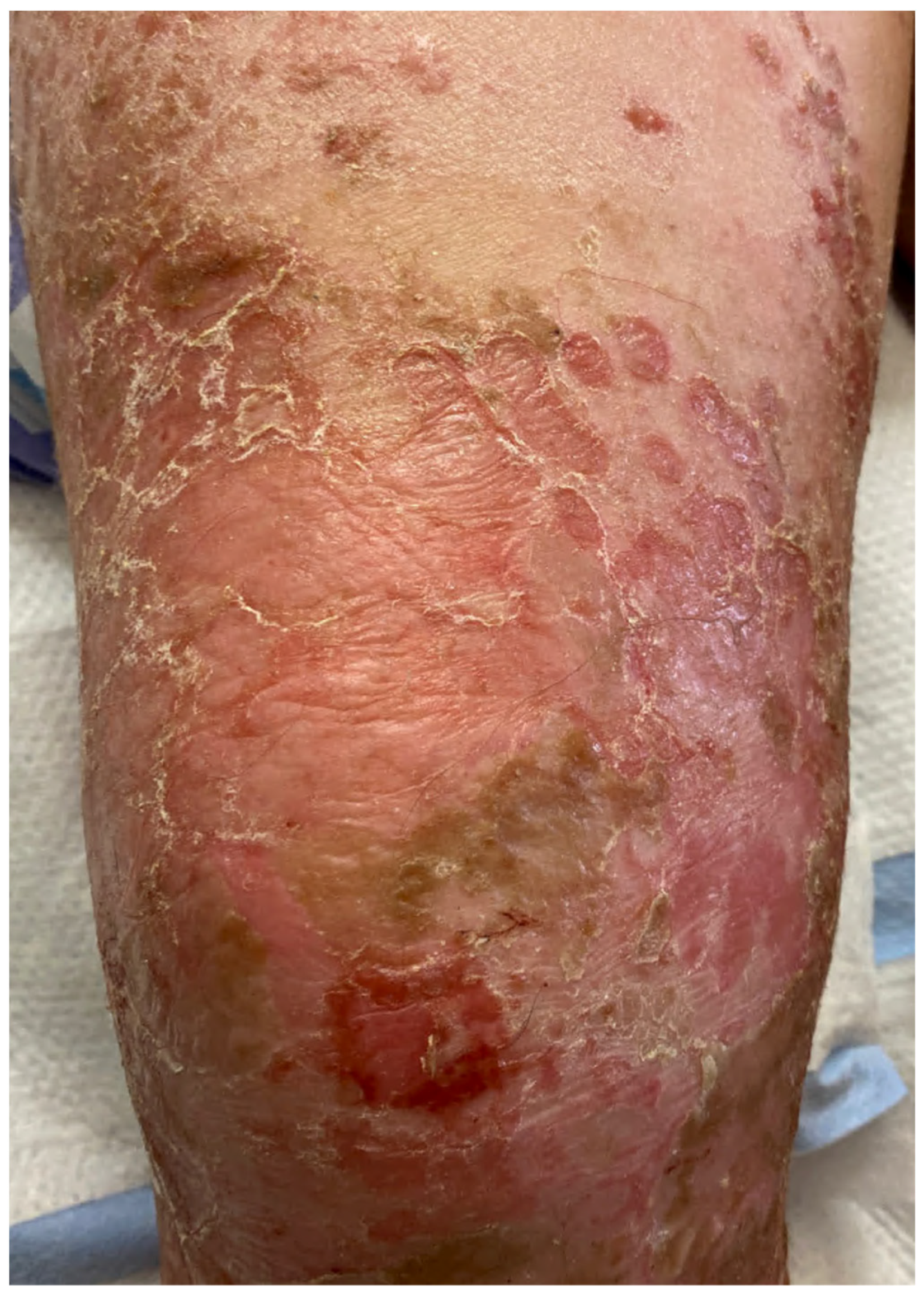
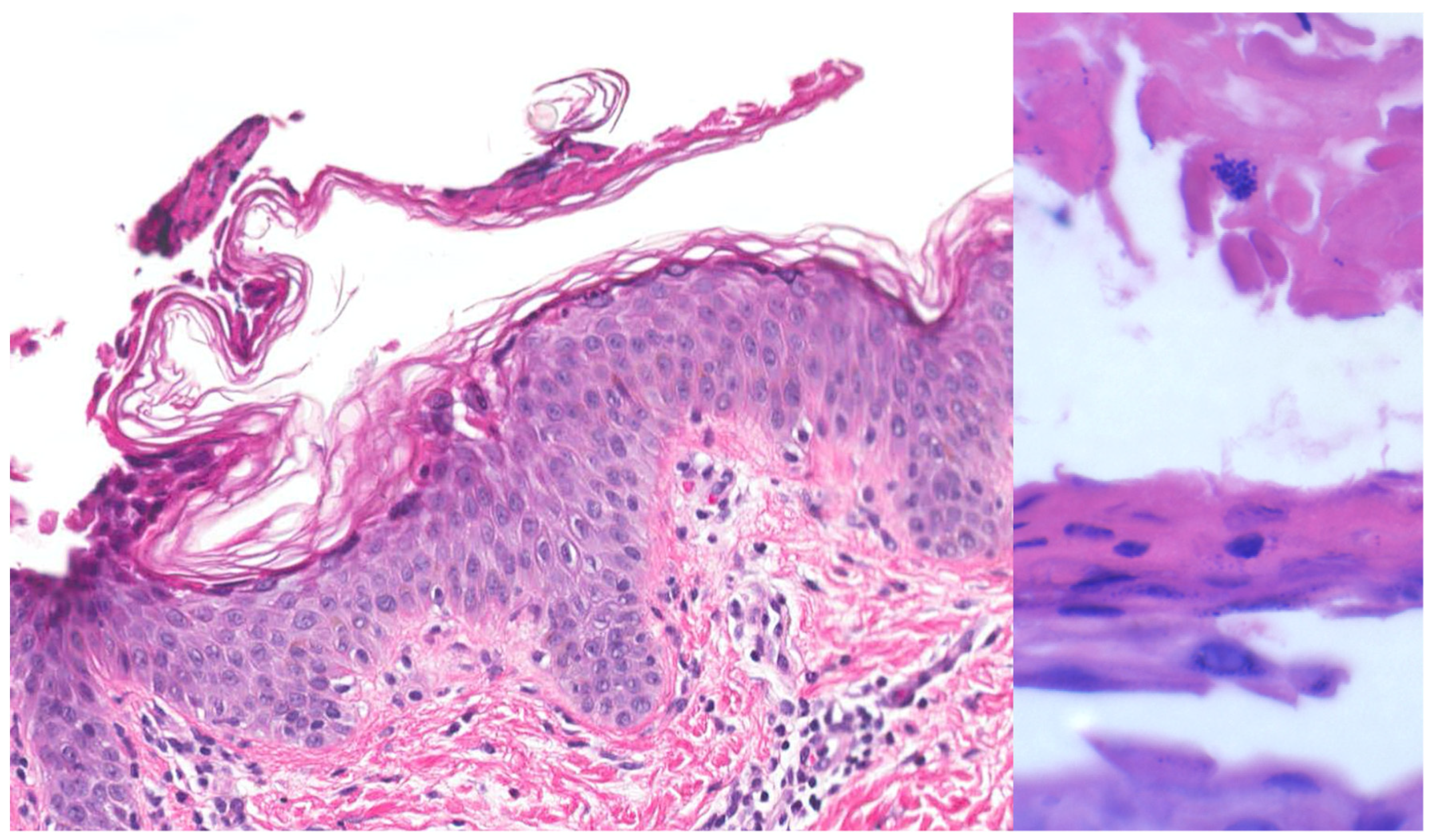
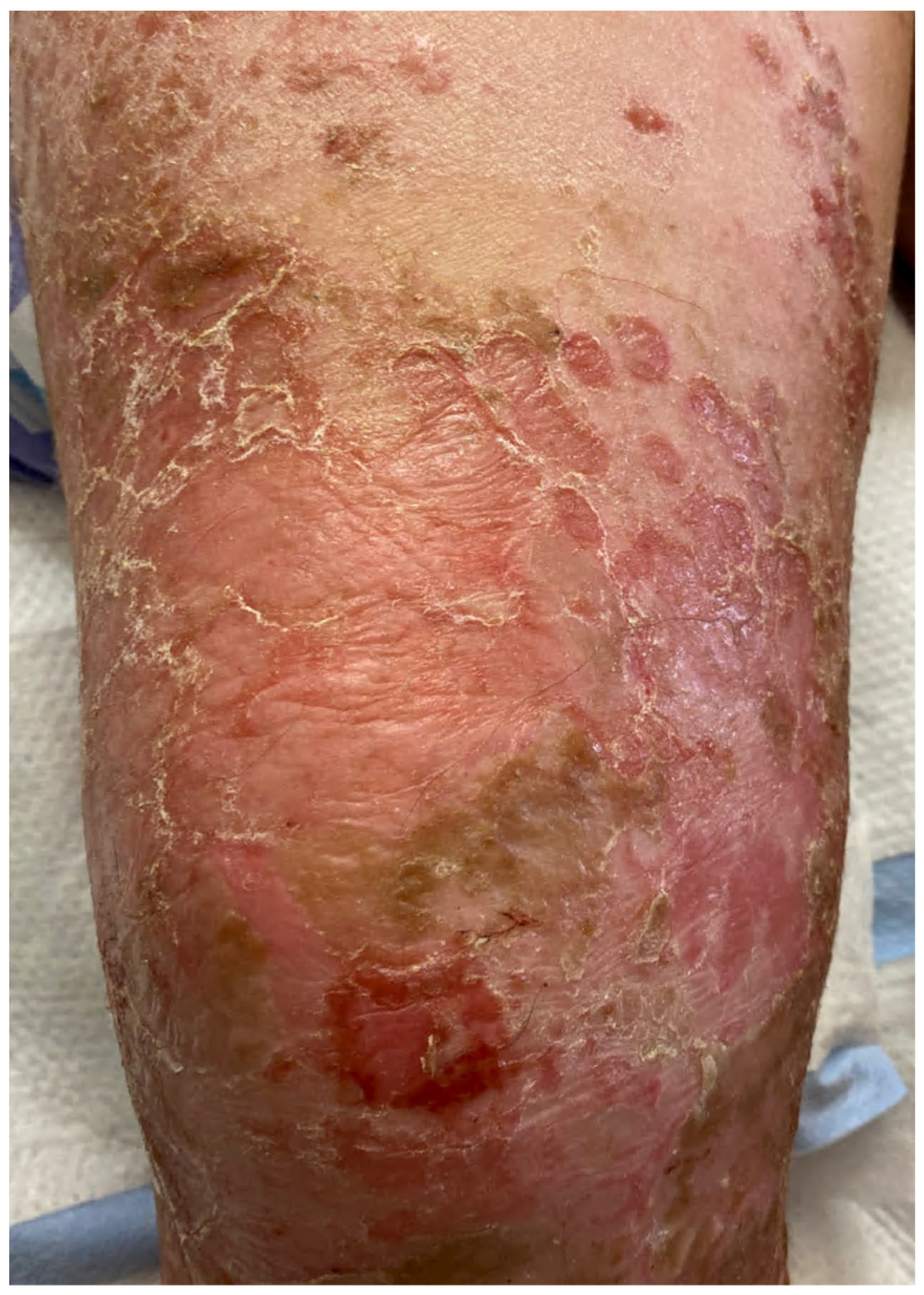
| Bullous impetigo [4,15] | Newborns and infants most commonly affected | Caused by S. aureus production of exfoliative toxins which cleave DSG1 resulting in acantholysis and bullae formation | Small vesicles that grow into tense bullae that rupture and leave behind a narrow rim of scale. The bullae appear in well-demarcated clusters at the initial site of infection. Systemic involvement is rare. Nikolsky sign is negative and culture of bullae or erosions is positive | Loss of cell adhesion in the superficial epidermis (granular layer) resulting in a subcorneal blister; mixed dermal inflammatory infiltrate, crusting, epidermal hyperplasia, and lesional cocci in clusters | Usually resolves within 3–6 weeks. However, high risk patients may develop SSSS due to dissemination of exfoliative toxin |
| Toxic shock syndrome (TSS) [16,17] | Usually a result of vaginal or surgical wound colonization | Typically caused by S. aureus (produces toxic shock syndrome toxin-1) or S. pyogenes | Fever >102 F, desquamative rash, hypotension, involvement of 3 or more organ systems. Symptoms often include- myalgias, vomiting, diarrhea, headache, pharyngitis. Rash is less common when caused by S. pyogenes | Necrolysis of keratinocytes in the epidermis with superficial dermal neutrophilic and lymphocytic infiltrate | When caused by S. aureus, begins with fever, confusion, and fatigue. Erythema starts on the trunk, spreads to the extremities, and desquamates 1–2 weeks later. Sequelae can include Beau lines, nail shedding, and telogen effluvium |
| Kawasaki disease [18,19] | Mostly affects children below the age of five | Exact etiology is unknown but thought to be due to an infectious trigger | Diagnostic criteria: fever at least 5 days and 4 of the following: Bilateral conjunctival injection, oropharyngeal changes, cervical lymphadenopathy, edema/erythema/desquamation of hands/feet, and polymorphous rash | Dilation of small vessels in the papillary dermis with infiltration of CD4+ T cells and macrophages in the dermis and epidermis | If left untreated, can lead to coronary aneurysms and sudden death. |
| Scarlet Fever [20] | School-aged children and teenagers are most commonly affected | Streptococcus pyogenes producing streptococcal pyrogenic exotoxins types A, B and C, often after tonsillitis or pharyngitis | Preceded by sore throat, fever, headache, malaise, vomiting. Blanchable erythema of neck, chest, axillae. Papular, non-blanching rash that spares the palms and soles and is classically described as a “sandpaper” rash. Petechial streaks in folds (Pastia lines), flushing of cheeks | Spongiosis and parakeratosis with perivascular neutrophilic infiltrates | Rash usually develops 2–3 days after initial infection and can desquamate up to 2 weeks later. Patients can return to normal activity once afebrile for 24 h |
| Disorder | Demographics | Triggers | Clinical Presentation | Histopathology | Clinical Course |
|---|---|---|---|---|---|
| Bullous impetigo [4,15] | Newborns and infants most commonly affected | Caused by exfoliative toxins produced by S. aureus, which cleave DSG1, resulting in acantholysis and bullae formation | Small vesicles that grow into tense bullae in well-demarcated clusters at the initial site of infection. Bullae rupture and leave behind a narrow rim of scale. Systemic involvement is rare. Nikolsky sign is negative, and culture of bullae or erosions is positive. | Loss of cell adhesion in the superficial epidermis (granular layer) resulting in a subcorneal blister; mixed dermal inflammatory infiltrate, crusting, epidermal hyperplasia, and lesional cocci in clusters. | Usually resolves within 3–6 weeks. However, high-risk patients may develop SSSS, due to dissemination of exfoliative toxin. |
| Psoriasis [29] | Can occur at any age, but has bimodal peak of 20–30 and 50–60 years of age | Cutaneous injury (koebnerization), infections (HIV, Streptococcus), stress, medications, hypocalcemia | Salmon pink plaques with silvery scale on extensor surfaces | Stratum corneum with alternating neutrophils and parakeratosis, regular acanthosis, tortuous blood vessels in dermal papillae | Chronic condition that can wax and wane in severity. Usually requires treatment for remission |
| Linear IgA bullous dermatosis [29] | Can occur in children and adults | Infections, medications, malignancies | Tense bullae often in herpetiform or annular arrangement (crown of jewels) overlying erythema | Subepidermal blister with neutrophils. Immunofluorescence shows linear IgA deposits along the dermoepidermal junction | Responds quickly to dapsone. If untreated, typically lasts several years then resolves on its own |
| Contact dermatitis [29] | Children and adults | Common allergens: poison ivy, nickel, fragrances, neomycin | Well-demarcated pink scaly patches and thin plaques, variable overlying vesicles, bullae, or crusting | Spongiotic dermatitis (spongiosis, lymphocyte exocytosis, parakeratosis), often with eosinophils and Langerhans cell vesicles | Rash persists until causative agent is removed. Responds to steroids (oral or topical). |
| Superficial pustular folliculitis [29] | Children and adults | Occluded areas, beard | Folliculocentric pustules and papules with collarettes of scale often on the head, neck, trunk, buttocks, legs | Suppurative folliculitis and mixed perifollicular infiltrate including neutrophils and lymphocytes | Lesions come and go; individual lesions will often spontaneously resolve. Response to topical and oral antibiotics. |
| Pemphigus foliaceus [29] | Usually seen in adults 45–65 years old | Autoantibodies targeting desmoglein 1. Medications (captopril, penicillamine). Possible environmental triggers can cause Fogo Selvagem, a form of pemphigus foliaceus endemic to Brazil | Erythematous “puff pastry-like” crusted erosions often in seborrheic distribution, with a positive Nikolsky sign and no mucosal involvement | Subcorneal split in granular layer with acantholysis and scattered eosinophils +/− neutrophils. DIF shows intercellular IgG and C3 deposition primarily in the upper half of epidermis | Treatment with rituximab achieves complete remission in 90% of patients within two years |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brazel, M.; Desai, A.; Are, A.; Motaparthi, K. Staphylococcal Scalded Skin Syndrome and Bullous Impetigo. Medicina 2021, 57, 1157. https://doi.org/10.3390/medicina57111157
Brazel M, Desai A, Are A, Motaparthi K. Staphylococcal Scalded Skin Syndrome and Bullous Impetigo. Medicina. 2021; 57(11):1157. https://doi.org/10.3390/medicina57111157
Chicago/Turabian StyleBrazel, Morgan, Anand Desai, Abhirup Are, and Kiran Motaparthi. 2021. "Staphylococcal Scalded Skin Syndrome and Bullous Impetigo" Medicina 57, no. 11: 1157. https://doi.org/10.3390/medicina57111157
APA StyleBrazel, M., Desai, A., Are, A., & Motaparthi, K. (2021). Staphylococcal Scalded Skin Syndrome and Bullous Impetigo. Medicina, 57(11), 1157. https://doi.org/10.3390/medicina57111157