
Rare Occurrence of Microsatellite Instability in Gastrointestinal Stromal Tumors


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and DNA Extraction
2.2. Microsatellite Instability Analysis
2.3. Somatic Mutation Profile Analysis
2.4. Statistical Analysis
3. Results
3.1. Clinicopathologic Findings of the Patients under Study
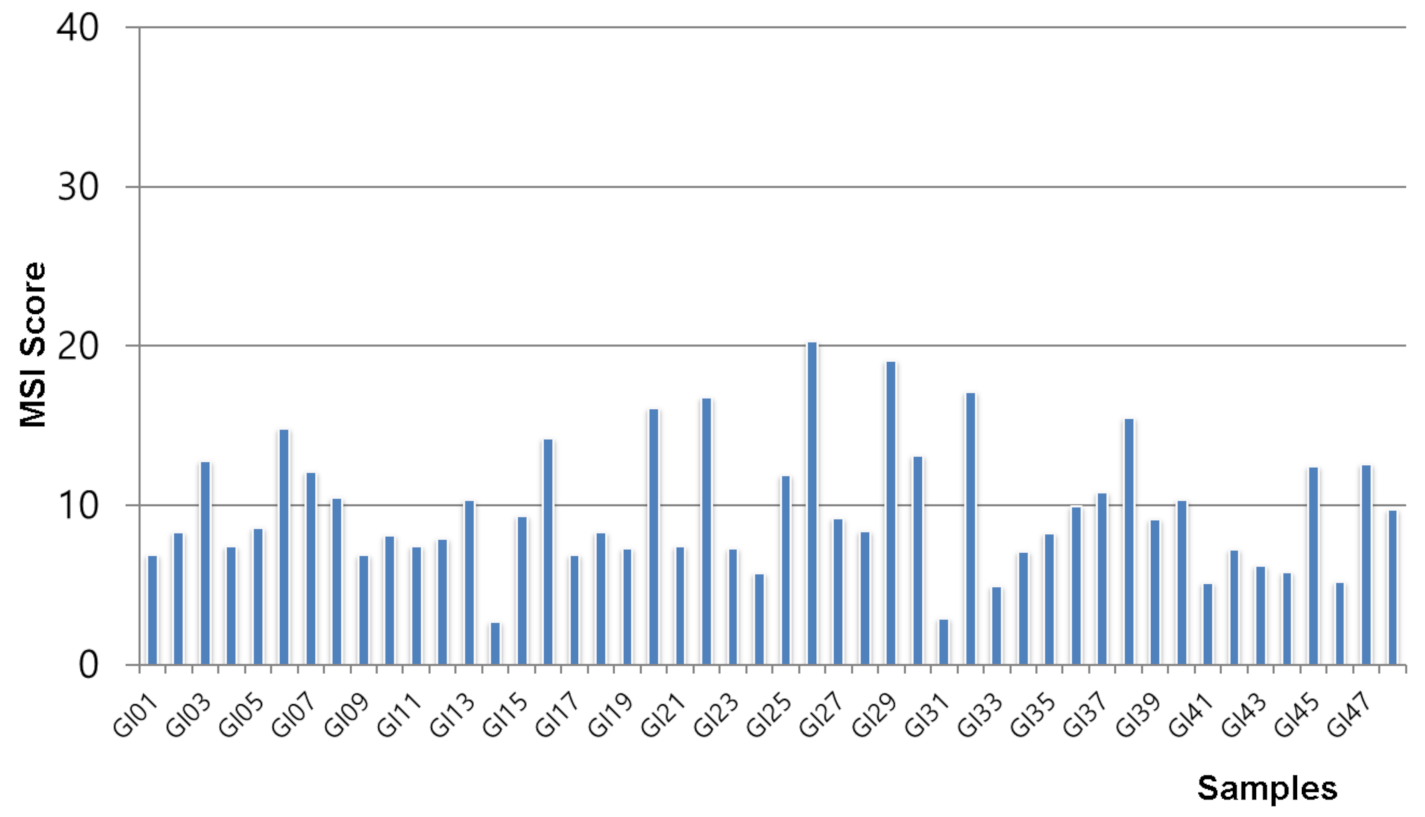
3.2. Microsatellite Instability Status of Gastrointestinal Stromal Tumors
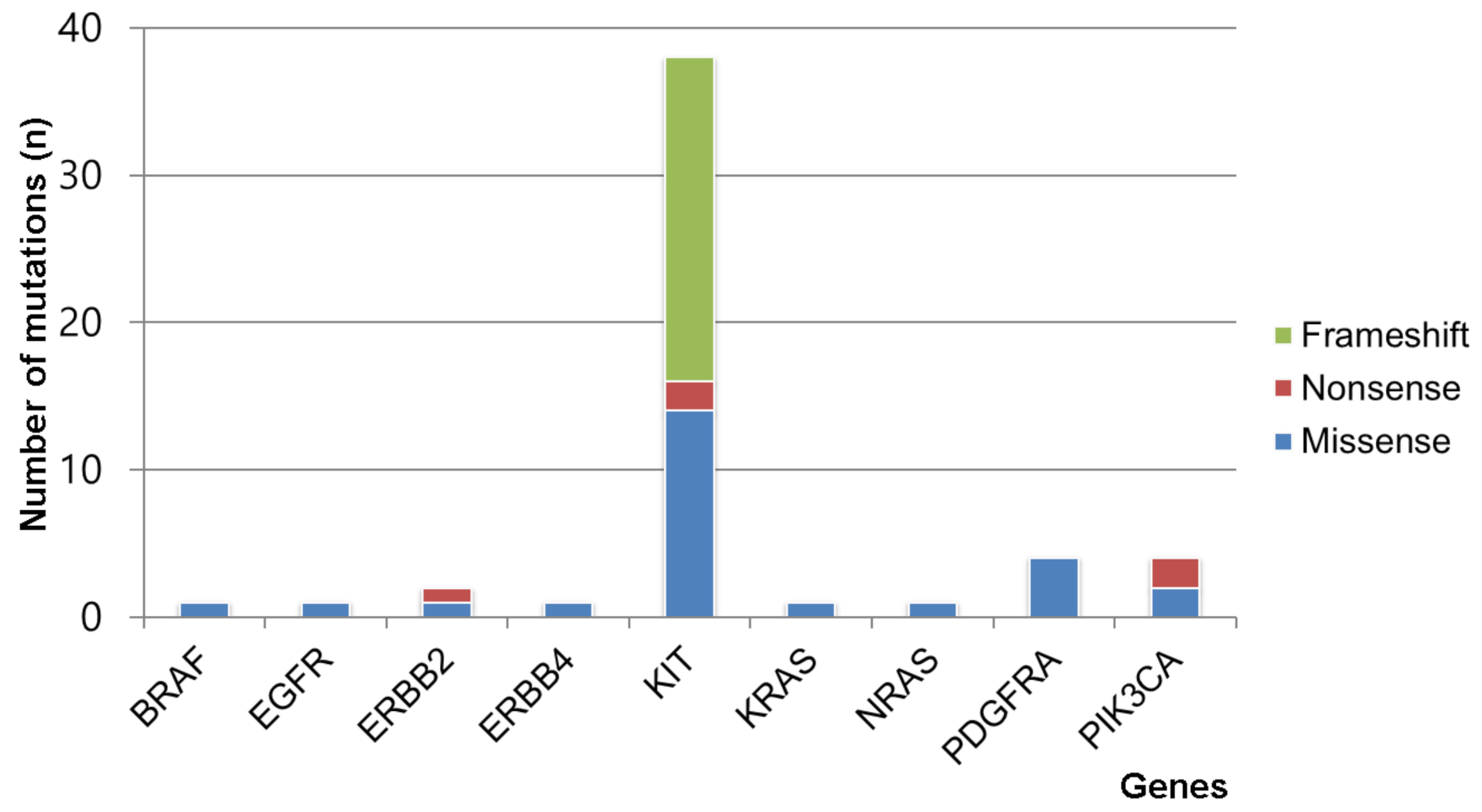
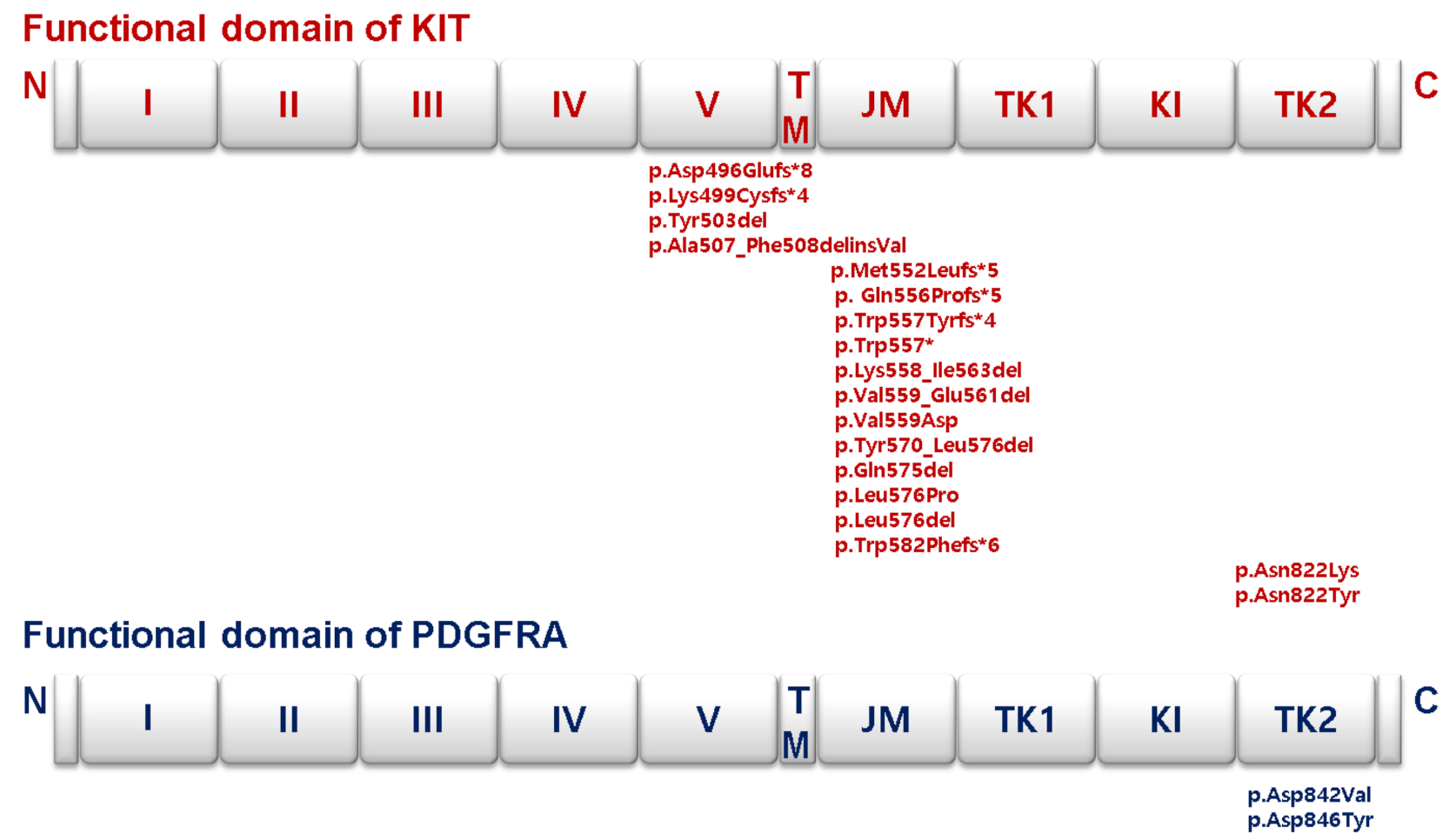
3.3. Somatic Mutation Profiles of Gastrointestinal Stromal Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Campanella, N.C.; Scapulatempo-Neto, C.; Abrahão-Machado, L.F.; De Oliveira, A.T.T.; Berardinelli, G.N.; Guimarães, D.P.; Reis, R.M. Lack of microsatellite instability in gastrointestinal stromal tumors. Oncol. Lett. 2017, 14, 5221–5228. [Google Scholar] [CrossRef] [Green Version]
- Chapusot, C.; Martin, L.; Bouvier, A.M.; Bonithon-Kopp, C.; Ecarnot-Laubriet, A.; Rageot, D.; Ponnelle, T.; Laurent-Puig, P.; Faivre, J.; Piard, F. Microsatellite instability and intratumoural heterogeneity in 100 right-sided sporadic colon carcinomas. Br. J. Cancer 2002, 87, 400–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, G.; Colombino, M.; Cossu, A.; Marchetti, A.; Botti, G.; Ascierto, P.A. Genetic instability and increased mutational load: Which diagnostic tool best direct patients with cancer to immunotherapy? J. Transl. Med. 2017, 15, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Watson, N.; Grieu, F.; Morris, M.; Harvey, J.; Stewart, C.; Schofield, L.; Goldblatt, J.; Iacopetta, B. Heterogeneous Staining for Mismatch Repair Proteins during Population-Based Prescreening for Hereditary Nonpolyposis Colorectal Cancer. J. Mol. Diagn. 2007, 9, 472–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.N.; Hile, S.E.; Eckert, K.A. Defective Mismatch Repair, Microsatellite Mutation Bias, and Variability in Clinical Cancer Phenotypes. Cancer Res. 2010, 70, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirotsu, Y.; Nagakubo, Y.; Amemiya, K.; Oyama, T.; Mochizuki, H.; Omata, M. Microsatellite Instability Status is De-termined by Targeted Sequencing with MSIcall in 25 Cancer Types. Clin. Chim. Acta 2020, 502, 207–213. [Google Scholar] [CrossRef]
- Hause, R.J.; Pritchard, C.C.; Shendure, R.J.H.J.; Salipante, C.C.P.S.J. Classification and characterization of microsatellite instability across 18 cancer types. Nat. Med. 2016, 22, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
- Bonneville, R.; Krook, M.A.; Kautto, E.A.; Miya, J.; Wing, M.R.; Chen, H.-Z.; Reeser, J.W.; Yu, L.; Roychowdhury, S. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precis. Oncol. 2017, 1, 1–15. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network Comprehensive molecular characterization of gastric adenocarcinoma. Nat. Cell Biol. 2014, 513, 202–209. [CrossRef] [Green Version]
- Kim, S.Y.; Choi, Y.Y.; An, J.Y.; Shin, H.B.; Jo, A.; Choi, H.; Seo, S.H.; Bang, H.-J.; Cheong, J.-H.; Hyung, W.J.; et al. The benefit of microsatellite instability is attenuated by chemotherapy in stage II and stage III gastric cancer: Results from a large cohort with subgroup analyses. Int. J. Cancer 2015, 137, 819–825. [Google Scholar] [CrossRef]
- Polom, K.; Marrelli, D.; Pascale, V.; Ferrara, F.; Voglino, C.; Marini, M.; Roviello, F. The pattern of lymph node metastases in microsatellite unstable gastric cancer. Eur. J. Surg. Oncol. (EJSO) 2017, 43, 2341–2348. [Google Scholar] [CrossRef]
- Smyth, E.C.; Wotherspoon, A.; Peckitt, C.; Gonzalez, D.; Hulkki-Wilson, S.; Eltahir, Z.; Fassan, M.; Rugge, M.; Valeri, N.; Okines, A.; et al. Mismatch Repair Deficiency, Microsatellite Instability, and Survival. JAMA Oncol. 2017, 3, 1197–1203. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.Y.; Kim, H.; Shin, S.-J.; Kim, H.Y.; Lee, J.; Yang, H.-K.; Kim, W.H.; Kim, Y.-W.; Kook, M.-C.; Park, Y.K.; et al. Microsatellite Instability and Programmed Cell Death-Ligand 1 Expression in Stage II/III Gastric Cancer. Ann. Surg. 2019, 270, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-K.; Kang, H.J.; Kim, K.-M.; Sohn, T.; Choi, N.; Ryu, M.-H.; Kim, W.H.; Yang, H.-K. Clinical Practice Guideline for Accurate Diagnosis and Effective Treatment of Gastrointestinal Stromal Tumor in Korea. Cancer Res. Treat. 2012, 44, 85–96. [Google Scholar] [CrossRef]
- Soreide, K. High-fidelity of five quasimonomorphic mononucleotide repeats to high-frequency microsatellite in-stability distribution in early-stage adenocarcinoma of the colon. Anticancer Res. 2011, 31, 967–971. [Google Scholar]
- Park, J.; Lee, S.-I.; Shin, S.; Hong, J.H.; Yoo, H.M.; Kim, J.G. Genetic profiling of somatic alterations by Oncomine Focus Assay in Korean patients with advanced gastric cancer. Oncol. Lett. 2020, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 1–14. [Google Scholar] [CrossRef]
- Fabrizio, D.A.; Jr, T.J.G.; Dunne, R.F.; Frampton, G.; Sun, J.; Gowen, K.; Kennedy, M.; Greenbowe, J.; Schrock, A.B.; Hezel, A.F.; et al. Beyond microsatellite testing: Assessment of tumor mutational burden identifies subsets of colorectal cancer who may respond to immune checkpoint inhibition. J. Gastrointest. Oncol. 2018, 9, 610–617. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Lopes, J.M.; Silva, P.; Seixas, M.; Cirnes, L.; Seruca, R. Microsatellite instability is not associated with degree of malignancy and p53 expression of gastrointestinal stromal tumours. Histopathology 1998, 33, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, T.; Chong, J.-M.; Sakurai, S.; Koshiishi, N.; Ikeno, R.; Tanaka, A.; Matsumoto, Y.; Hayashi, Y.; Koike, M.; Fukayama, M. Allelic Loss of 14q and 22q, NF2Mutation, and Genetic Instability Occur Independently of c-kitMutation in Gastrointestinal Stromal Tumor. Jpn. J. Cancer Res. 2000, 91, 1241–1249. [Google Scholar] [CrossRef] [PubMed]
- Kose, K.; Hiyama, T.; Tanaka, S.; Yoshihara, M.; Yasui, W.; Chayama, K. Nuclear and Mitochondrial DNA Microsatellite Instability in Gastrointestinal Stromal Tumors. Pathobiology 2006, 73, 93–97. [Google Scholar] [CrossRef] [PubMed]
- El-Menyar, A.; Mekkodathil, A.; Al-Thani, H. Diagnosis and management of gastrointestinal stromal tumors: An up-to-date literature review. J. Cancer Res. Ther. 2017, 13, 889–900. [Google Scholar]
- Charville, G.W.; Longacre, T.A. Surgical Pathology of Gastrointestinal Stromal Tumors: Practical Implications of Morphologic and Molecular Heterogeneity for Precision Medicine. Adv. Anat. Pathol. 2017, 24, 336–353. [Google Scholar] [CrossRef]
- Szucs, Z.; Thway, K.; Fisher, C.; Bulusu, R.; Constantinidou, A.; Benson, C.; Van Der Graaf, W.T.A.; Jones, R.L. Molecular subtypes of gastrointestinal stromal tumors and their prognostic and therapeutic implications. Future Oncol. 2017, 13, 93–107. [Google Scholar] [CrossRef] [Green Version]
- Mei, L.; Du, W.; Idowu, M.; Von Mehren, M.; Boikos, S.A. Advances and Challenges on Management of Gastrointestinal Stromal Tumors. Front. Oncol. 2018, 8, 135. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, H.; Wardelmann, E.; Sihto, H.; Eriksson, M.; Hall, K.S.; Reichardt, A.; Hartmann, J.T.; Pink, D.; Cameron, S.; Hohenberger, P.; et al. Effect of KIT and PDGFRA Mutations on Survival in Patients With Gastrointestinal Stromal Tumors Treated With Adjuvant Imatinib: An Exploratory Analysis of a Randomized Clinical Trial. JAMA Oncol. 2017, 3, 602–609. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Oda, Y. Gastrointestinal stromal tumor: Recent advances in pathology and genetics. Pathol. Int. 2014, 65, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Maki, R.G.; Corless, C.L.; Antonescu, C.R.; Harlow, A.; Griffith, D.; Town, A.; McKinley, A.; Ou, W.-B.; Fletcher, J.A.; et al. Primary and Secondary Kinase Genotypes Correlate With the Biological and Clinical Activity of Sunitinib in Imatinib-Resistant Gastrointestinal Stromal Tumor. J. Clin. Oncol. 2008, 26, 5352–5359. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.; Sul, H.J.; Kim, J.G. Rare Occurrence of Microsatellite Instability in Gastrointestinal Stromal Tumors. Medicina 2021, 57, 174. https://doi.org/10.3390/medicina57020174
Park J, Sul HJ, Kim JG. Rare Occurrence of Microsatellite Instability in Gastrointestinal Stromal Tumors. Medicina. 2021; 57(2):174. https://doi.org/10.3390/medicina57020174
Chicago/Turabian StylePark, Joonhong, Hae Jung Sul, and Jeong Goo Kim. 2021. "Rare Occurrence of Microsatellite Instability in Gastrointestinal Stromal Tumors" Medicina 57, no. 2: 174. https://doi.org/10.3390/medicina57020174
APA StylePark, J., Sul, H. J., & Kim, J. G. (2021). Rare Occurrence of Microsatellite Instability in Gastrointestinal Stromal Tumors. Medicina, 57(2), 174. https://doi.org/10.3390/medicina57020174