
Prader–Willi Syndrome with Angelman Syndrome in the Offspring



 ,
, 
Abstract
1. Introduction
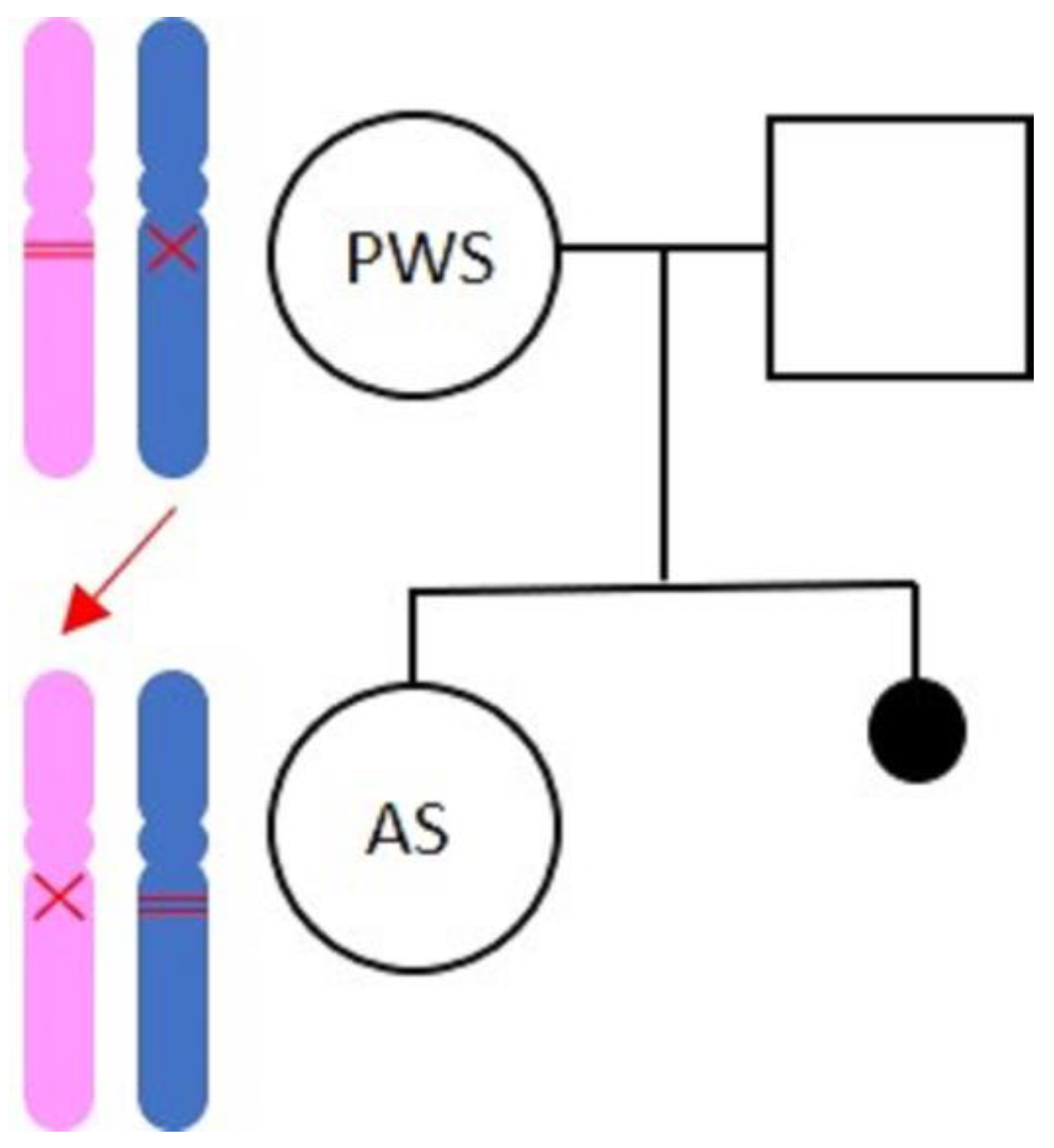
2. Case Report
2.1. Materials and Methods
2.2. The Mother with Prader–Willi Syndrome
2.3. The Daughter with AS
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buiting, K.; Williams, C.; Horsthemke, B. Angelman syndrome—Insights into a rare neurogenetic disorder. Nat. Rev. Neurol. 2016, 12, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Angulo, M.A.; Butler, M.G.; Cataletto, M.E. Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J. Endocrinol. Investig. 2015, 38, 1249–1263. [Google Scholar] [CrossRef] [PubMed]
- Holm, V.A.; Cassidy, S.B.; Butler, M.G.; Hanchett, J.M.; Greenswag, L.R.; Whitman, B.Y.; Greenberg, F. Prader-Willi syndrome: Consensus diagnostic criteria. Pediatrics 1993, 91, 398. [Google Scholar] [PubMed]
- Clayton-Smith, J.; Pembrey, M.E. Angelman syndrome. J. Med. Genet. 1992, 29, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Buiting, K. Prader-Willi syndrome and Angelman syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2010, 154, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Kalsner, L.; Chamberlain, S.J. Prader-Willi, Angelman, and 15q11–q13 duplication syndromes. Pediatr. Clin. 2015, 62, 587–606. [Google Scholar] [CrossRef] [PubMed]
- Verrotti, A.; Cusmai, R.; Laino, D.; Carotenuto, M.; Esposito, M.; Falsaperla, R.; Margari, L.; Rizzo, R.; Savasta, S.; Grosso, S.; et al. Long-term outcome of epilepsy in patients with Prader-Willi syndrome. J. Neurol. 2015, 262, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Crino, A.; Schiaffini, R.; Ciampalini, P.; Spera, S.; Beccaria, L.; Benzi, F.; Bosio, L.; Corrias, A.; Gargantini, L.; Salvatoni, A.; et al. Hypogonadism and pubertal development in Prader-Willi syndrome. Eur. J. Pediatr. 2003, 162, 327–333. [Google Scholar] [CrossRef]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef]
- Radicioni, A.; Di Giorgio, G.; Grugni, G.; Cuttini, M.; Losacco, V.; Anzuini, A.; Spera, S.; Marzano, C.; Lenzi, A.; Cappa, M. Multiple forms of hypogonadism of central, peripheral or combined origin in males with Prader–Willi syndrome. Clin. Endocrinol. 2012, 76, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Eiholzer, U.; l’Allemand, D.; Rousson, V.; Schlumpf, M.; Gasser, T.; Girard, J.; Gruters, A.; Simoni, M. Hypothalamic and gonadal components of hypogonadism in boys with Prader-Labhart- Willi syndrome. J. Clin. Endocrinol. Metab. 2006, 91, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Siemensma, E.P.; van Alfen-van der Velden, A.A.; Otten, B.J.; Laven, J.S.; Hokken-Koelega, A.C. Ovarian function and reproductive hormone levels in girls with Prader-Willi syndrome: A longitudinal study. J. Clin. Endocrinol. Metab. 2012, 97, E1766–E1773. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.; Eldar-Geva, T.; Bennaroch, F.; Pollak, Y.; Gross-Tsur, V. Sexual dichotomy of gonadal function in Prader–Willi syndrome from early infancy through the fourth decade. Hum. Reprod. 2015, 30, 2587–2596. [Google Scholar] [CrossRef]
- Eldar-Geva, T.; Hirsch, H.J.; Rabinowitz, R.; Benarroch, F.; Rubinstein, O.; Gross-Tsur, V. Primary ovarian dysfunction contributes to the hypogonadism in women with Prader-Willi Syndrome. Horm. Res. 2009, 72, 153–159. [Google Scholar] [CrossRef]
- Eldar-Geva, T.; Hirsch, H.J.; Benarroch, F.; Rubinstein, O.; Gross-Tsur, V. Hypogonadism in females with Prader-Willi syndrome from infancy to adulthood: Variable combinations of a primary gonadal defect and hypothalamic dysfunction. Eur. J. Endocrinol. 2010, 162, 377–384. [Google Scholar] [CrossRef]
- Irizarry, K.A.; Miller, M.; Freemark, M.; Haqq, A.M. Prader Willi syndrome: Genetics, metabolomics, hormonal function, and new approaches to therapy. Adv. Pediatr. 2016, 63, 47. [Google Scholar] [CrossRef] [PubMed]
- Young, J. Approach to the male patient with congenital hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 2012, 97, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Bakker, N.E.; Wolffenbuttel, K.P.; Looijenga, L.H.; Hokken-Koelega, A.C. Testes in infants with Prader-Willi syndrome: Human chorionic gonadotropin treatment, surgery and histology. J. Urol. 2015, 193, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Zitzmann, M.; Nieschlag, E. Hormone substitution in male hypogonadism. Mol. Cell. Endocrinol. 2000, 161, 73–88. [Google Scholar] [CrossRef]
- Noordam, C.; Hoybye, C.; Eiholzer, U. Prader-Willi Syndrome and Hypogonadism: A Review Article. Int. J. Mol. Sci. 2021, 22, 2705. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.B.; Driscoll, D.J. Prader-Willi syndrome. Eur. J. Hum. Genet. 2009, 17, 3–13. [Google Scholar] [CrossRef]
- Laxova, R.; Gilderdale, S.; Ridler, M.A. An aetiological study of fifty-three female patients from a subnormality hospital and of their offspring. J. Ment. Defic. Res. 1973, 17, 193–225. [Google Scholar] [CrossRef] [PubMed]
- Hockey, A.; Opitz, J.M.; Reynolds, J.F. X-linked intellectual handicap and precocious puberty with obesity in carrier females. Am. J. Med. Genet. 1986, 23, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Hockey, A.; Byrne, G.; Cohen, A. Precocious puberty in the male offspring of a mother and daughter with the Prader-Willi syndrome. Am. J. Med. Genet. 1987, 26. [Google Scholar] [CrossRef] [PubMed]
- Zellweger, H. Can women with the Prader-Labhart-Willi syndrome (PLWS) reproduce? Does the deletion (15)(q11–13) occur in individuals not affected with PLWS? Am. J. Med. Genet. 1988, 29, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Akefeldt, A.; Tornhage, C.J.; Gillberg, C. A woman with Prader-Willi syndrome gives birth to a healthy baby girl. Dev. Med. Child. Neurol. 1999, 41, 789–790. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Mogensen, H.; Hamborg-Petersen, B.; Graem, N.; Østergaard, J.; Brøndum-Nielsen, K. Fertility in Prader-Willi syndrome: A case report with Angelman syndrome in the offspring. Acta Paediatr. 2001, 90, 455–459. [Google Scholar] [CrossRef]
- Ostergaard, J.R. Phenotype of a child with Angelman syndrome born to a woman with Prader-Willi syndrome. Am. J. Med. Genet. A 2015, 167, 2138–2144. [Google Scholar] [CrossRef]
- Watson, S.L.; Richards, D.A.; Miodrag, N.; Fedoroff, J.P. Sex and genes, part 1: Sexuality and Down, Prader-Willi, and Williams syndromes. Intellect. Dev. Disabil. 2012, 50, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Coppola, G.; Operto, F.F.; Auricchio, G.; D’Amico, A.; Fortunato, D.; Pascotto, A. Temporal lobe dual pathology in malignant migrating partial seizures in infancy. Epileptic Disord. 2007, 9, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Fedoroff, J.; Richards, D. Sexual disorders and intellectual disabilities. Handb. Clin. Sex. Ment. Health Prof. 2010, 451–468. [Google Scholar]
- Kramers-Olen, A. Sexuality, intellectual disability, and human rights legislation. S. Afr. J. Psychol. 2016, 46, 504–516. [Google Scholar] [CrossRef]


{kind=link}
| Case | Diagnosis | Deliveries | Age at Delivery | Healthy Children | Affected Children | Reference |
|---|---|---|---|---|---|---|
| 1 | Clinical | 4 | 29 y | 1 | 1: clinical PWS 2: congenital heart defects | Laxova et al., 1973 [22] |
| 2 | Clinical | 1 | 28 y | 1 | Laxova et al., 1973 [22] | |
| 3 | Cytogenetical (deletion) | 2 | Unknown | 1 | 1: precocious puberty | Hockey et al., 1986 [23] Hockey et al., 1987 [24] |
| 4 | Molecular (UDP) | 1 | 33 y | 1 | Åkefeldt et al., 1999 [26] | |
| 5 | Molecular (deletion) | 1 | 32 y | 1: Angelman syndrome | Schulze et al., 2001 [27] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greco, D.; Vetri, L.; Ragusa, L.; Vinci, M.; Gloria, A.; Occhipinti, P.; Costanzo, A.A.; Quatrosi, G.; Roccella, M.; Buono, S.; et al. Prader–Willi Syndrome with Angelman Syndrome in the Offspring. Medicina 2021, 57, 460. https://doi.org/10.3390/medicina57050460
Greco D, Vetri L, Ragusa L, Vinci M, Gloria A, Occhipinti P, Costanzo AA, Quatrosi G, Roccella M, Buono S, et al. Prader–Willi Syndrome with Angelman Syndrome in the Offspring. Medicina. 2021; 57(5):460. https://doi.org/10.3390/medicina57050460
Chicago/Turabian StyleGreco, Donatella, Luigi Vetri, Letizia Ragusa, Mirella Vinci, Angelo Gloria, Paola Occhipinti, Angela Antonia Costanzo, Giuseppe Quatrosi, Michele Roccella, Serafino Buono, and et al. 2021. "Prader–Willi Syndrome with Angelman Syndrome in the Offspring" Medicina 57, no. 5: 460. https://doi.org/10.3390/medicina57050460
APA StyleGreco, D., Vetri, L., Ragusa, L., Vinci, M., Gloria, A., Occhipinti, P., Costanzo, A. A., Quatrosi, G., Roccella, M., Buono, S., & Romano, C. (2021). Prader–Willi Syndrome with Angelman Syndrome in the Offspring. Medicina, 57(5), 460. https://doi.org/10.3390/medicina57050460