
Ocular Toxoplasmosis Associated with Unilateral Pigmentary Retinopathy That May Mimic Retinitis Pigmentosa: Diagnostic Dilemmas


{kind=link}
{kind=link}
Abstract
:1. Introduction
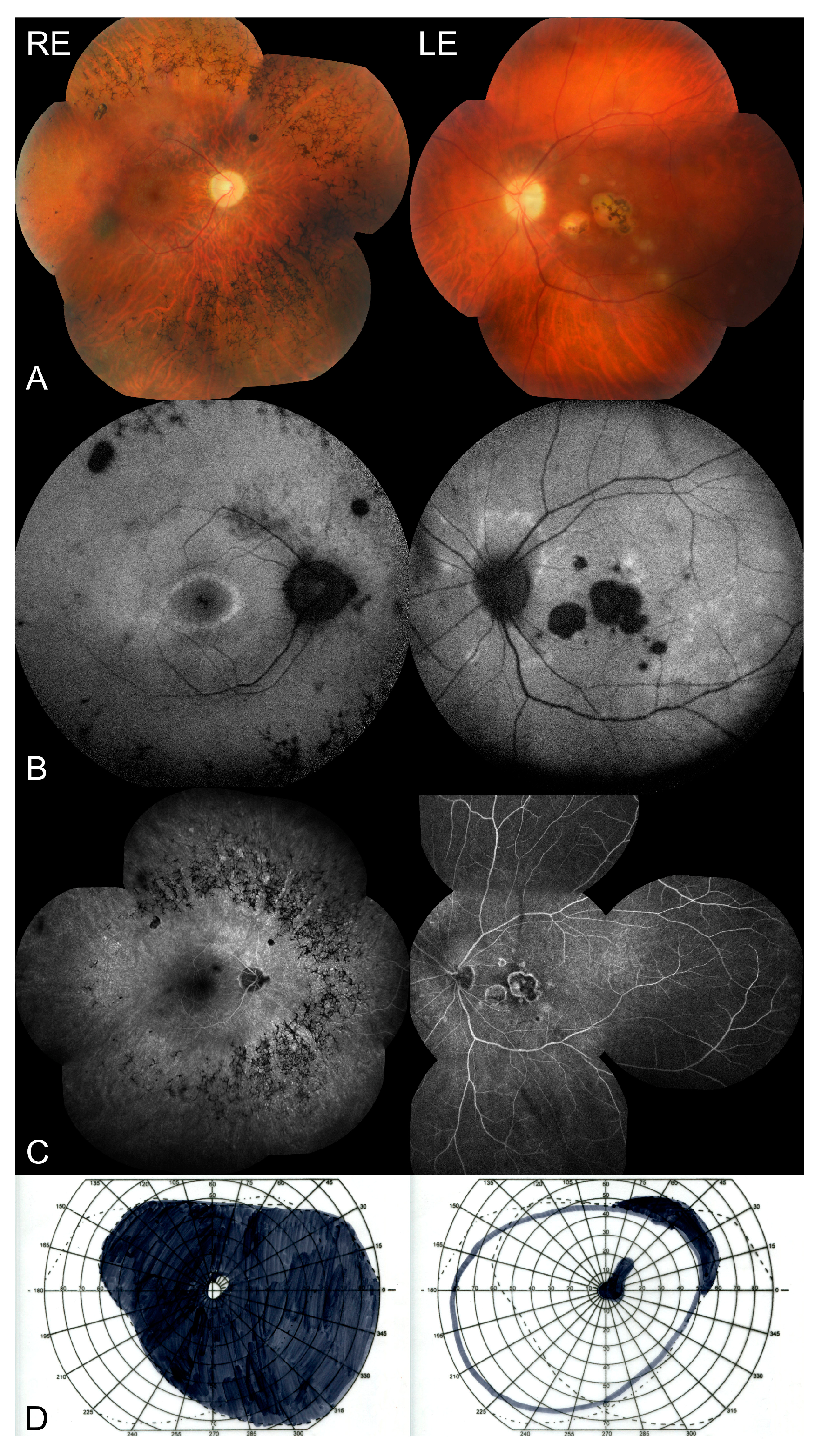
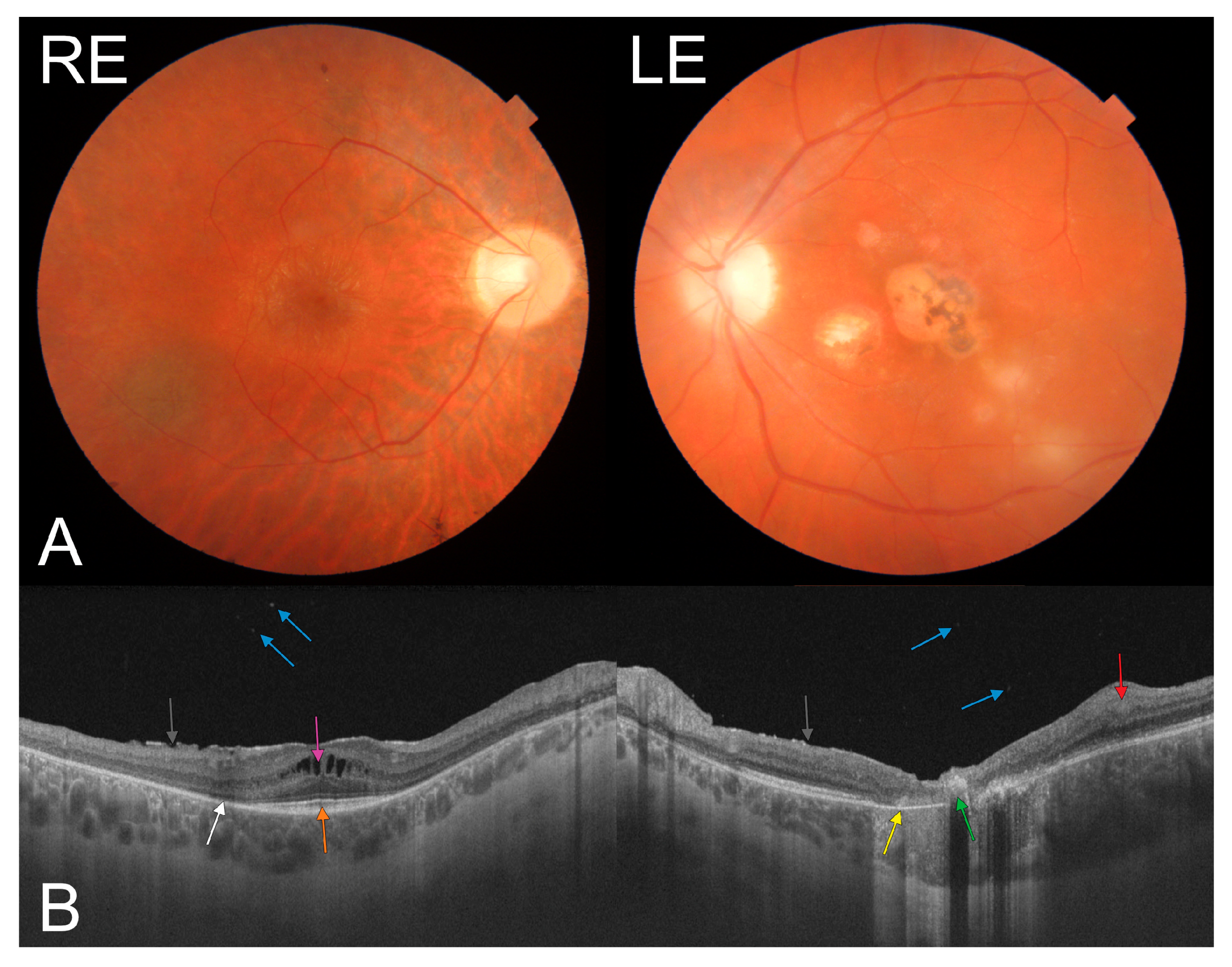
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rassi, A.; Todorich, B.; Faia, L.J.; Trese, M.; Drenser, K.; Capone, A., Jr. Congenital toxoplasmosis associated with tractional retinal detachment. Retin. Cases Brief Rep. 2021, 15, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Toutée, A.; Orès, R.; Mrejen, S.; Bodaghi, B.; Kobal, A.; Merabet, L.; Brignole-Baudouin, F.; Sahel, J.A.; Nghiem-Buffet, S.; Errera, M.H. New optical coherence tomography angiography findings on an intraretinal vascular process secondary to toxoplasma retinochoroiditis: 2 case reports. Retin. Cases Brief Rep. 2019. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.R.; Cunningham, E.T., Jr. Atypical presentations of ocular toxoplasmosis. Curr. Opin. Ophthalmol. 2002, 13, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Lewis, R.A.; Traboulsi, E.I. Pigmentary retinopathy in systemic inherited disease. In Genetic Diseases of the Eye, 2nd ed.; Traboulsi, E., Ed.; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; Sorrentino, F.S.; Parmeggiani, F. Retinitis pigmentosa: Genes and disease mechanisms. Curr. Genom. 2011, 12, 238–249. [Google Scholar] [CrossRef]
- Weller, J.M.; Michelson, G.; Juenemann, A.G. Unilateral retinitis pigmentosa: 30 years follow-up. BMJ Case Rep. 2014, 2014, bcr2013202236. [Google Scholar] [CrossRef] [PubMed]
- Hood, D.C.; Bach, M.; Brigell, M.; Keating, D.; Kondo, M.; Lyons, J.S.; Marmor, M.F.; McCulloch, D.L.; Plamowski-Wolfe, A.M. ISCEV standard for clinical multifocal electroretinography (mfERG) (2011 edition). Doc. Ophthalmol. 2012, 124, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Silveira, C.; Belfort, R., Jr.; Nussenblatt, R.; Farah, M.; Takahashi, W.; Imamura, P.; Burnier, M., Jr. Unilateral pigmentary retinopathy associated with ocular toxoplasmosis. Am. J. Ophthalmol. 1989, 107, 682–684. [Google Scholar] [CrossRef]
- Chen, H.; Wu, D.; Huang, S.; Jiang, F. Unilateral retinitis pigmentosa with amblyopia in the fellow eye. Graefes Arch. Clin. Exp. Ophthalmol. 2006, 244, 1701–1704. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, D.; Cunningham, E.T., Jr.; Pavesio, C.; Garweg, J.G.; Zierhut, M. Controversies in ocular toxoplasmosis. Ocul. Immunol. Inflamm. 2011, 19, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Ozgonul, C.; Besirli, C.G. Recent developments in the diagnosis and treatment of ocular toxoplasmosis. Ophthalmic Res. 2017, 57, 112. [Google Scholar] [CrossRef] [PubMed]
- Pociej-Marciak, W.; Karska-Basta, I.; Kuźniewski, M.; Kubicka-Trząska, A.; Romanowska-Dixon, B. Sudden Visual Deterioration as the First Symptom of Chronic Kidney Failure. Case Rep. Ophthalmol. 2015, 28, 394–400. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, R.; Holder, G.E.; Moore, A.T.; Webster, A.R. Unilateral retinitis pigmentosa occurring in an individual with a germline mutation in the RP1 gene. Arch. Ophthalmol. 2011, 129, 954–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chhabra, M.S.; Prakash, G.; Vashisht, N.; Garg, S.P. Retinitis pigmentosa and congenital toxoplasmoisis: A rare coexistence. Indian J. Ophthalmol. 2007, 55, 303–304. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karska-Basta, I.; Romanowska-Dixon, B.; Pojda-Wilczek, D.; Mackiewicz, N. Ocular Toxoplasmosis Associated with Unilateral Pigmentary Retinopathy That May Mimic Retinitis Pigmentosa: Diagnostic Dilemmas. Medicina 2021, 57, 892. https://doi.org/10.3390/medicina57090892
Karska-Basta I, Romanowska-Dixon B, Pojda-Wilczek D, Mackiewicz N. Ocular Toxoplasmosis Associated with Unilateral Pigmentary Retinopathy That May Mimic Retinitis Pigmentosa: Diagnostic Dilemmas. Medicina. 2021; 57(9):892. https://doi.org/10.3390/medicina57090892
Chicago/Turabian StyleKarska-Basta, Izabella, Bożena Romanowska-Dixon, Dorota Pojda-Wilczek, and Natalia Mackiewicz. 2021. "Ocular Toxoplasmosis Associated with Unilateral Pigmentary Retinopathy That May Mimic Retinitis Pigmentosa: Diagnostic Dilemmas" Medicina 57, no. 9: 892. https://doi.org/10.3390/medicina57090892
APA StyleKarska-Basta, I., Romanowska-Dixon, B., Pojda-Wilczek, D., & Mackiewicz, N. (2021). Ocular Toxoplasmosis Associated with Unilateral Pigmentary Retinopathy That May Mimic Retinitis Pigmentosa: Diagnostic Dilemmas. Medicina, 57(9), 892. https://doi.org/10.3390/medicina57090892