
Donor Splice Site Variant in SLC9A6 Causes Christianson Syndrome in a Lithuanian Family: A Case Report

 ,
,  , , , ,
, , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Report
2.2. Whole Exome Sequencing (WES)
2.3. RNA Extraction and Complementary DNA Synthesis
2.4. PCR
2.5. Sanger Sequencing
2.6. In Silico Analysis
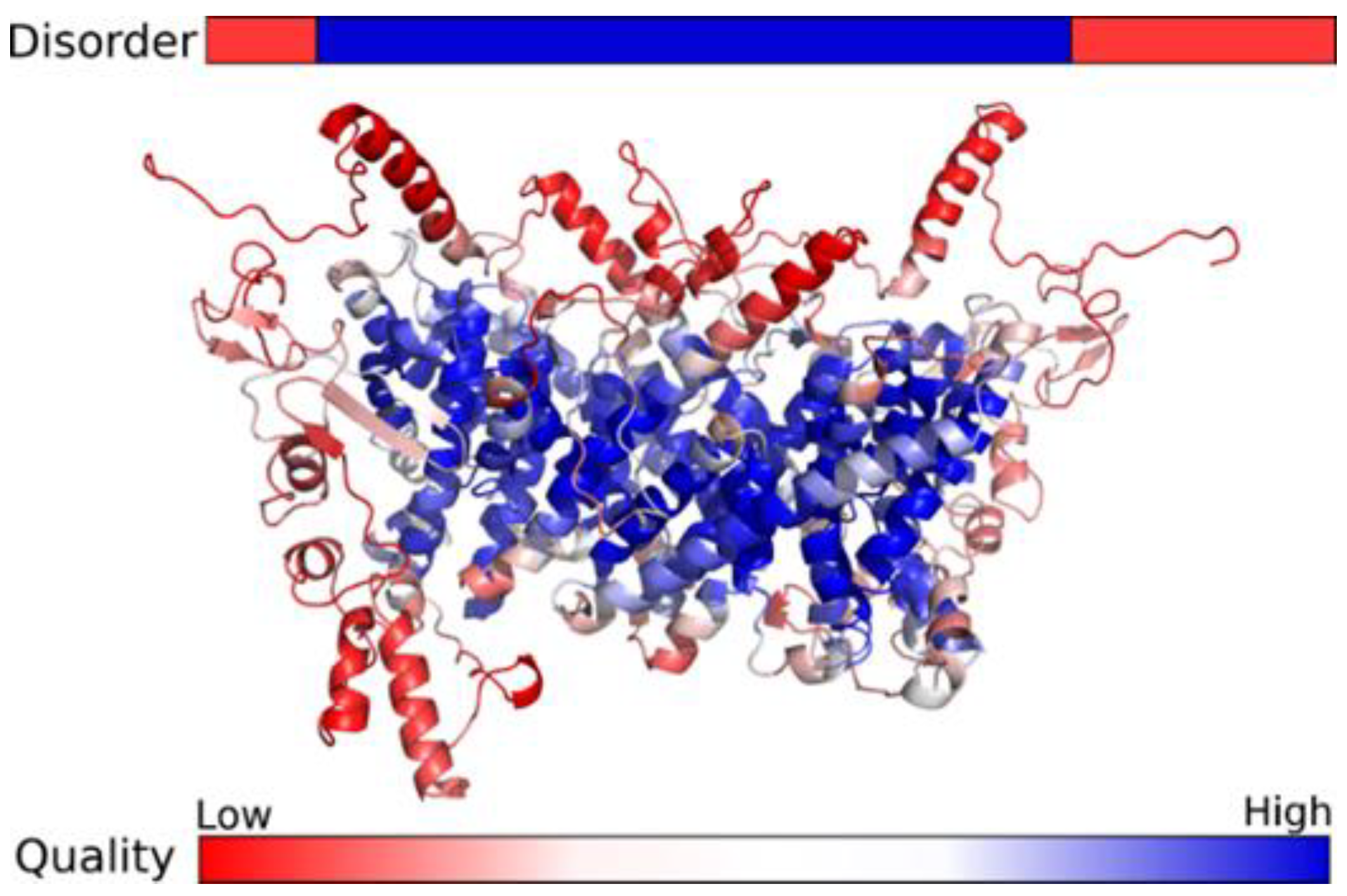
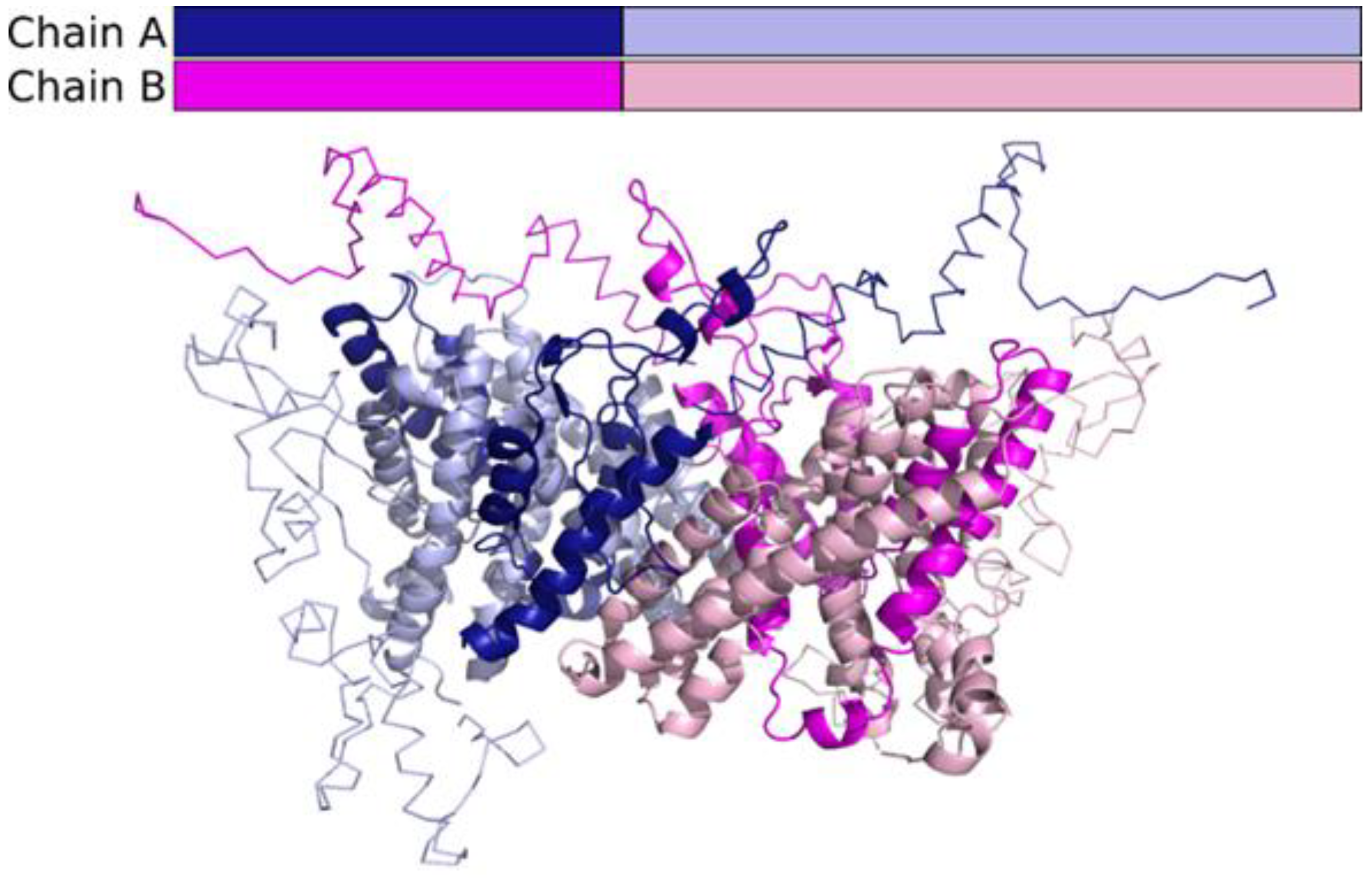
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, X.; Xie, L.; Fang, Z.; Jiang, L. Case Report: Novel SLC9A6 Splicing Variant in a Chinese Boy with Christianson Syndrome With Electrical Status Epilepticus During Sleep. Front. Neurol. 2022, 12, 796283. [Google Scholar] [CrossRef]
- Gilfillan, G.D.; Selmer, K.K.; Roxrud, I.; Smith, R.; Kyllerman, M.; Eiklid, K.; Kroken, M.; Mattingsdal, M.; Egeland, T.; Stenmark, H.; et al. SLC9A6 Mutations Cause X-Linked Mental Retardation, Microcephaly, Epilepsy, and Ataxia, a Phenotype Mimicking Angelman Syndrome. Am. J. Hum. Genet. 2008, 82, 1003–1010. [Google Scholar] [CrossRef] [Green Version]
- Pescosolido, M.F.; Stein, D.M.; Schmidt, M.; El Achkar, C.M.; Sabbagh, M.; Rogg, J.M.; Tantravahi, U.; McLean, R.L.; Liu, J.S.; Poduri, A.; et al. Genetic and phenotypic diversity of NHE6 mutations in Christianson syndrome. Ann. Neurol. 2014, 76, 581–593. [Google Scholar] [CrossRef] [Green Version]
- Schroer, R.J.; Holden, K.R.; Tarpey, P.S.; Matheus, M.G.; Griesemer, D.A.; Friez, M.J.; Fan, J.Z.; Simensen, R.J.; Strømme, P.; Stevenson, R.E.; et al. Natural History of Christianson Syndrome. Am. J. Med. Genet. Part A 2010, 152A, 2775–2783. [Google Scholar] [CrossRef] [Green Version]
- Sinajon, P.; Verbaan, D.; So, J. The expanding phenotypic spectrum of female SLC9A6 mutation carriers: A case series and review of the literature. Hum. Genet. 2016, 135, 841–850. [Google Scholar] [CrossRef]
- Pescosolido, M.F.; Kavanaugh, B.C.; Pochet, N.; Schmidt, M.; Jerskey, B.A.; Rogg, J.M.; De Jager, P.L.; Young-Pearse, T.L.; Liu, J.S.; Morrow, E.M. Complex Neurological Phenotype in Female Carriers of NHE6 Mutations. Mol. Neuropsychiatry 2019, 5, 98–108. [Google Scholar] [CrossRef]
- Schaller, L.; Lauschke, V.M. The genetic landscape of the human solute carrier (SLC) transporter superfamily. Hum. Genet. 2019, 138, 1359–1377. [Google Scholar] [CrossRef] [Green Version]
- Fuster, D.G.; Alexander, R.T. Traditional and emerging roles for the SLC9 Na+/H+ exchangers. Pflugers Arch. Eur. J. Physiol. 2014, 466, 61–76. [Google Scholar] [CrossRef] [Green Version]
- Donowitz, M.; Ming Tse, C.; Fuster, D. SLC9/NHE gene family, a plasma membrane and organellar family of Na+/H+ exchangers. Mol. Asp. Med. 2013, 34, 236–251. [Google Scholar] [CrossRef] [Green Version]
- Kondapalli, K.C.; Prasad, H.; Rao, R. An inside job: How endosomal Na+ /H+ exchangers link to autism and neurological disease. Front. Cell. Neurosci. 2014, 8, 172. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Schlesinger, P.H.; Slack, N.M.; Friedman, P.A.; Blair, H.C. High Capacity Na+/H+ Exchange Activity in Mineralizing Osteoblasts. J. Cell Physiol. 2011, 226, 1702–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Numata, M.; Orlowski, J. Molecular Cloning and Characterization of a Novel (Na+,K +)/H+ Exchanger Localized to the transGolgi Network. J. Biol. Chem. 2001, 276, 17387–17394. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. Mutationtaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Bëroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [Green Version]
- Divina, P.; Kvitkovicova, A.; Buratti, E.; Vorechovsky, I. Ab initio prediction of mutation-induced cryptic splice-site activation and exon skipping. Eur. J. Hum. Genet. 2009, 17, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, 222–230. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Dapkunas, J.; Timinskas, A.; Olechnovic, K.; Margelevicius, M.; Diciunas, R.; Venclovas, C. The PPI3D web server for searching, analyzing and modeling protein-protein interactions in the context of 3D structures. Bioinformatics 2017, 33, 935–937. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; DiMaio, F.; Wang, R.Y.; Kim, D.; Miles, C.; Brunette, T.; Thompson, J.; Baker, D. High-resolution comparative modeling with RosettaCM. Structure 2013, 21, 1735–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef]
- Raman, S.; Vernon, R.; Thompson, J.; Tyka, M.; Sadreyev, R.; Pei, J.; Kim, D.; Kellogg, E.; DiMaio, F.; Lange, O.; et al. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins 2009, 77, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Olechnovič, K.; Venclovas, Č. VoroMQA: Assessment of protein structure quality using interatomic contact areas. Proteins 2017, 85, 1131–1145. [Google Scholar] [CrossRef]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise disordered region predictions with annotated protein-binding activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef]
- Kozlowski, L.P.; Bujnicki, J.M. MetaDisorder: A meta-server for the prediction of intrinsic disorder in proteins. BMC Bioinform. 2012, 13, 111. [Google Scholar] [CrossRef] [Green Version]
- Dufner-Almeida, L.G.; do Carmo, R.T.; Masotti, C.; Haddad, L.A. Understanding Human DNA Variants Affecting Pre-MRNA Splicing in the NGS Era. Adv. Genet. 2019, 103, 39–90. [Google Scholar] [CrossRef]
- Krawczak, M.; Thomas, N.S.; Hundrieser, B.; Mort, M.; Wittig, M.; Hampe, J.; Cooper, D.N. Single Base-Pair Substitutions in Exon–Intron Junctions of Human Genes: Nature, Distribution, and Consequences for mRNA Splicing. Hum. Mutat. 2007, 28, 150–158. [Google Scholar] [CrossRef]
- Abramowicz, A.; Gos, M. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keravnou, A.; Bashiardes, E.; Barberis, V.; Michailidou, K.; Soteriou, M.; Tanteles, G.A.; Cariolou, M.A. Identification of novel splice mutation in SMAD3 in two Cypriot families with nonsyndromic thoracic aortic aneurysm. Two case reports. Mol. Genet. Genom. Med. 2020, 8, e1378. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.B.; Mansour, R.B.; Driss, F.; Baklouti-Gargouria, S.; Siala, O.; Mkaouar-Rebai, E.; Fakhfakh, F. Evaluation of the effect of c.2946 + 1G > T mutation on splicing in the SCN1A gene. Comput. Biol. Chem. 2015, 54, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Balteanu, V.A.; Carsai, T.C.; Vlaic, A. Identification of an intronic regulatory mutation at the buffalo αs1-casein gene that triggers the skipping of exon 6. Mol. Biol. Rep. 2013, 40, 4311–4316. [Google Scholar] [CrossRef]
- Ieda, D.; Hori, I.; Nakamura, Y.; Ohashi, K.; Negishi, Y.; Hattori, A.; Arisaka, A.; Hasegawa, S.; Saitoh, S. A novel splicing mutation in SLC9A6 in a boy with Christianson syndrome. Hum. Genome Var. 2019, 6, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Yan, R.; Wen, S.; Li, X.; Li, T.; Cai, Z.; Li, X.; Du, H.; Chen, H. A splice mutation and mRNA decay of EXT2 provoke hereditary multiple exostoses. PLoS ONE. 2014, 9, e94848. [Google Scholar] [CrossRef] [Green Version]
- Attanasio, C.; David, A.; Neerman-Arbez, M. Outcome of donor splice site mutations accounting for congenital afibrinogenemia reflects order of intron removal in the fibrinogen alpha gene (FGA). Blood 2003, 101, 1851–1856. [Google Scholar] [CrossRef] [Green Version]
- Lv, X.; Wu, W.Q.; Zhang, J.X.; Miao, L.F.; Yu, B.Z.; Chen, F.F.; Cui, Y.X.; Xia, Z.K.; Liu, Z.H.; Li, X.J. Comparative Functional Analysis in vitro of 2 COL4A5 Splicing Mutations at the Same Site in 2 Unrelated Alport Syndrome Chinese Families. Cytogenet. Genome Res. 2020, 160, 238–244. [Google Scholar] [CrossRef]
- Wang, Z.; Burge, C.B. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 2008, 14, 802–813. [Google Scholar] [CrossRef] [Green Version]
- Saha, K.; England, W.; Fernandez, M.M.; Biswas, T.; Spitale, R.C.; Ghosh, G. Structural disruption of exonic stem–loops immediately upstream of the intron regulates mammalian splicing. Nucleic Acids Res. 2020, 48, 6294–6309. [Google Scholar] [CrossRef]
- Takahara, K.; Schwarze, U.; Imamura, Y.; Hoffman, G.G.; Toriello, H.; Smith, L.T.; Byers, P.H.; Greenspan, D.S. Order of intron removal influences multiple splice outcomes, including a two-exon skip, in a COL5A1 acceptor-site mutation that results in abnormal pro-α1(V) N-propeptides and Ehlers-Danlos syndrome type I. Am. J. Hum. Genet. 2002, 71, 451–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ymoens, S.; Malfait, F.; Vlummens, P.; Hermanns-Lê, T.; Syx, D.; De Paepe, A. A novel splice variant in the N-propeptide of COL5A1 causes an EDS phenotype with severe kyphoscoliosis and eye involvement. PLoS ONE. 2011, 6, e20121. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Qiu, W.; Liu, H.; Ye, X.; Sun, Y.; Fan, Y.; Yu, Y. RT-PCR analysis of mRNA revealed the splice-altering effect of rare intronic variants in monogenic disorders. Ann. Hum. Genet. 2020, 84, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Hosoki, K.; Matsushita, M.; Funatsuka, M.; Saito, K.; Kanazawa, H.; Goto, Y.; Saitoh, S. A loss-of-function mutation in the SLC9A6 gene causes X-linked mental retardation resembling Angelman syndrome. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2011, 156, 799–807. [Google Scholar] [CrossRef]
- Kurosaki, T.; Maquat, L.E. Nonsense-mediated mRNA decay in humans at a glance. J. Cell Sci. 2016, 129, 461–467. [Google Scholar] [CrossRef] [Green Version]
- Roxrud, I.; Raiborg, C.; Gilfillan, G.D.; Strømme, P.; Stenmark, H. Dual degradation mechanisms ensure disposal of NHE6 mutant protein associated with neurological disease. Exp. Cell Res. 2009, 315, 3014–3027. [Google Scholar] [CrossRef]
- Ouyang, Q.; Joesch-Cohen, L.; Mishra, S.; Riaz, H.A.; Schmidt, M.; Morrow, E.M. Functional assessment in vivo of the mouse homolog of the human ala-9-ser NHE6 variant. eNeuro 2019, 6, ENEURO.0046-192019. [Google Scholar] [CrossRef]
- Brett, C.L.; Wei, Y.; Donowitz, M.; Rao, R. Human Na+/H+ exchanger isoform 6 is found in recycling endosomes of cells, not in mitochondria. Am. J. Physiol.-Cell Physiol. 2002, 282, 1031–1041. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Kallay, L.; Brett, C.L.; Rao, R. Mutational analysis of the intramembranous H10 loop of yeast Nhx1 reveals a critical role in ion homoeostasis and vesicle trafficking. Biochem. J. 2006, 398, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Ohgaki, R.; Matsushita, M.; Kanazawa, H.; Ogihara, S.; Hoekstra, D.; van Ijzendoorn, S.C. The Na_/H_ Exchanger NHE6 in the Endosomal Recycling System Is Involved in the Development of Apical Bile Canalicular Surface Domains in HepG2 Cells. Mol. Biol. Cell 2010, 21, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Orlowski, J.; Grinstein, S. Na+/H+ exchangers. Compr. Physiol. 2011, 1, 2083–2100. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, E.; Sakaguchi, M.; Wakabayashi, S.; Shigekawa, M.; Mihara, K. NHE6 Protein Possesses a Signal Peptide Destined for Endoplasmic Reticulum Membrane and Localizes in Secretory Organelles of the Cell. J. Biol. Chem. 2001, 276, 49221–49227. [Google Scholar] [CrossRef] [Green Version]
- Ilie, A.; Gao, A.Y.L.; Boucher, A.; Park, J.; Berghuis, A.M.; Hoffer, M.J.V.; Hilhorst-Hofstee, Y.; McKinney, R.A.; Orlowski, J. A potential gain-of-function variant of SLC9A6 leads to endosomal alkalinization and neuronal atrophy associated with Christianson Syndrome. Neurobiol. Dis. 2019, 121, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Q.; Lizarraga, S.B.; Schmidt, M.; Yang, U.; Gong, J.; Ellisor, D.; Kauer, J.A.; Morrow, E.M. Christianson syndrome protein NHE6 modulates TrkB endosomal signaling required for neuronal circuit development. Neuron 2013, 80, 97–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathje, M.; Fang, H.; Bachman, J.L.; Anggono, V.; Gether, U.; Huganir, R.L.; Madsen, K.L. AMPA receptor pHluorin-GluA2 reports NMDA receptor-induced intracellular acidification in hippocampal neurons. Proc. Natl. Acad. Sci. USA 2013, 110, 14426–14431. [Google Scholar] [CrossRef] [Green Version]
- Kerner-Rossi, M.; Gulinello, M.; Walkley, S.; Dobrenis, K. Pathobiology of Christianson syndrome: Linking disrupted endosomal-lysosomal function with intellectual disability and sensory impairments. Neurobiol. Learn. Mem. 2019, 165, 106867. [Google Scholar] [CrossRef] [PubMed]
- Prasad, H.; Rao, R. The Na+/H+ exchanger NHE6 modulates endosomal pH to control processing of amyloid precursor protein in a cell culture model of Alzheimer disease. J. Biol. Chem. 2015, 290, 5311–5327. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petraitytė, G.; Mikštienė, V.; Siavrienė, E.; Cimbalistienė, L.; Maldžienė, Ž.; Rančelis, T.; Vaitėnienė, E.M.; Ambrozaitytė, L.; Dapkūnas, J.; Dzindzalieta, R.; et al. Donor Splice Site Variant in SLC9A6 Causes Christianson Syndrome in a Lithuanian Family: A Case Report. Medicina 2022, 58, 351. https://doi.org/10.3390/medicina58030351
Petraitytė G, Mikštienė V, Siavrienė E, Cimbalistienė L, Maldžienė Ž, Rančelis T, Vaitėnienė EM, Ambrozaitytė L, Dapkūnas J, Dzindzalieta R, et al. Donor Splice Site Variant in SLC9A6 Causes Christianson Syndrome in a Lithuanian Family: A Case Report. Medicina. 2022; 58(3):351. https://doi.org/10.3390/medicina58030351
Chicago/Turabian StylePetraitytė, Gunda, Violeta Mikštienė, Evelina Siavrienė, Loreta Cimbalistienė, Živilė Maldžienė, Tautvydas Rančelis, Evelina Marija Vaitėnienė, Laima Ambrozaitytė, Justas Dapkūnas, Ramūnas Dzindzalieta, and et al. 2022. "Donor Splice Site Variant in SLC9A6 Causes Christianson Syndrome in a Lithuanian Family: A Case Report" Medicina 58, no. 3: 351. https://doi.org/10.3390/medicina58030351
APA StylePetraitytė, G., Mikštienė, V., Siavrienė, E., Cimbalistienė, L., Maldžienė, Ž., Rančelis, T., Vaitėnienė, E. M., Ambrozaitytė, L., Dapkūnas, J., Dzindzalieta, R., Pranckevičienė, E., Kučinskas, V., Utkus, A., & Preikšaitienė, E. (2022). Donor Splice Site Variant in SLC9A6 Causes Christianson Syndrome in a Lithuanian Family: A Case Report. Medicina, 58(3), 351. https://doi.org/10.3390/medicina58030351