
Deuterium-Reinforced Polyunsaturated Fatty Acids Prevent Diet-Induced Nonalcoholic Steatohepatitis by Reducing Oxidative Stress

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model and Treatments
2.2. Blood Parameters
2.3. Liver Lipid Isolation and Determination
2.4. Histopathological Analysis
2.5. Cell Culture and Treatments
2.6. Cell Viability, Apoptosis, and ROS Assays
2.7. MitoSOX Assay
2.8. Quantitative Real-Time PCR (RT-qPCR) Analyses
2.9. Oil Red O Staining
2.10. Statistical Analyses
3. Results
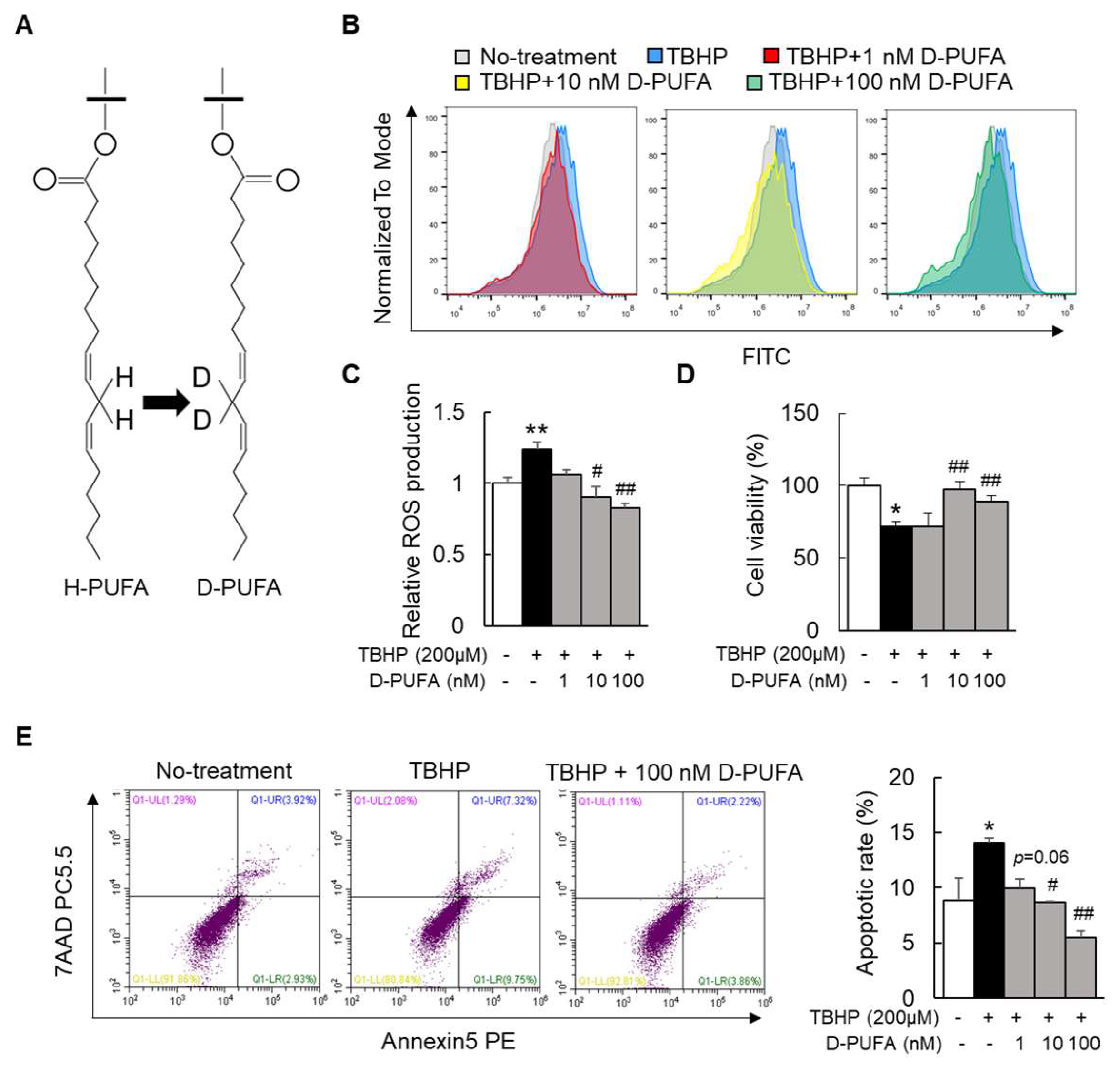
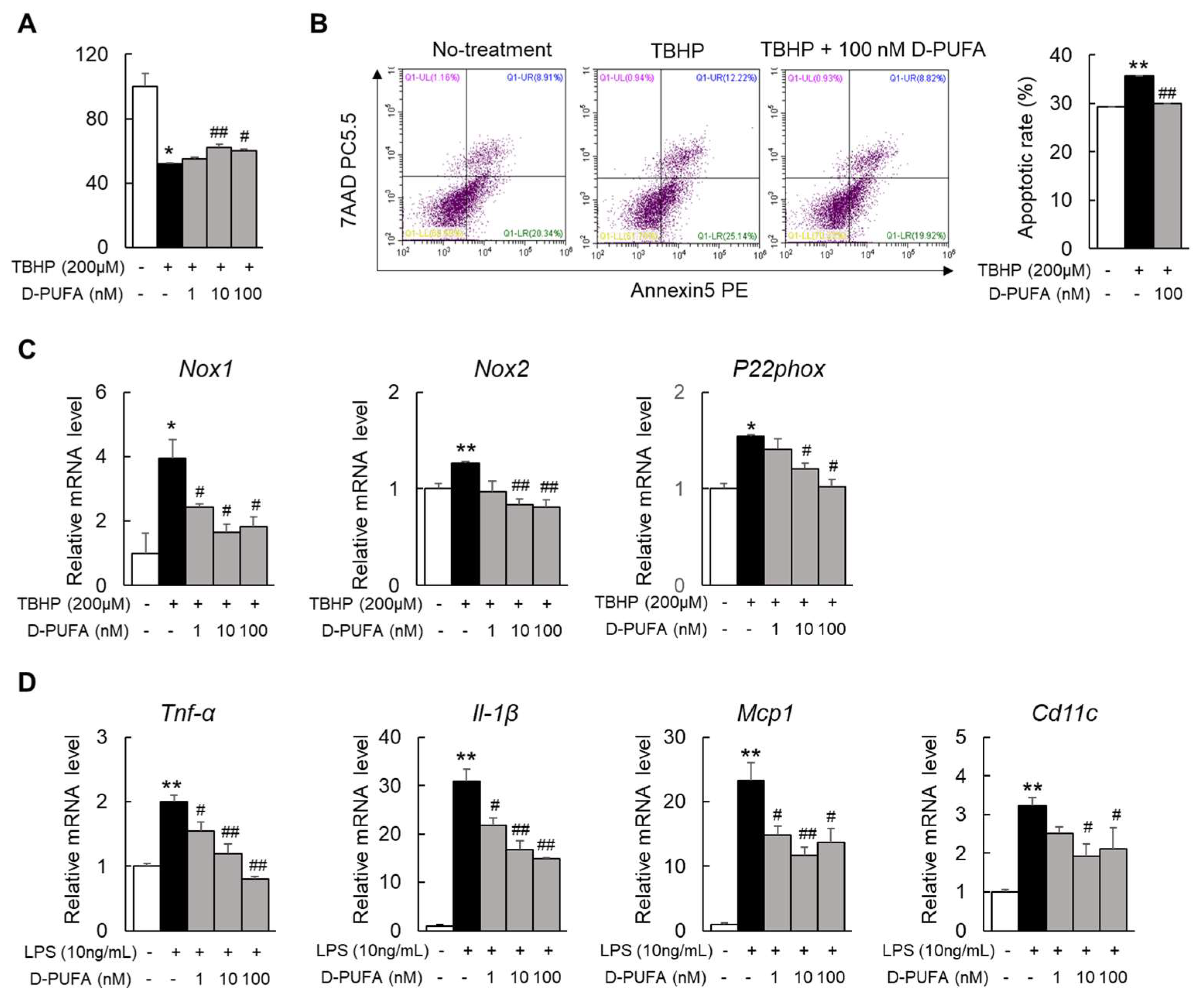
3.1. D-PUFA Attenuates Oxidative Stress and Apoptosis Induced by TBHP in Hepatocytes
3.2. D-PUFAs Reduce the Oxidative Stress and Inflammation in Macrophages
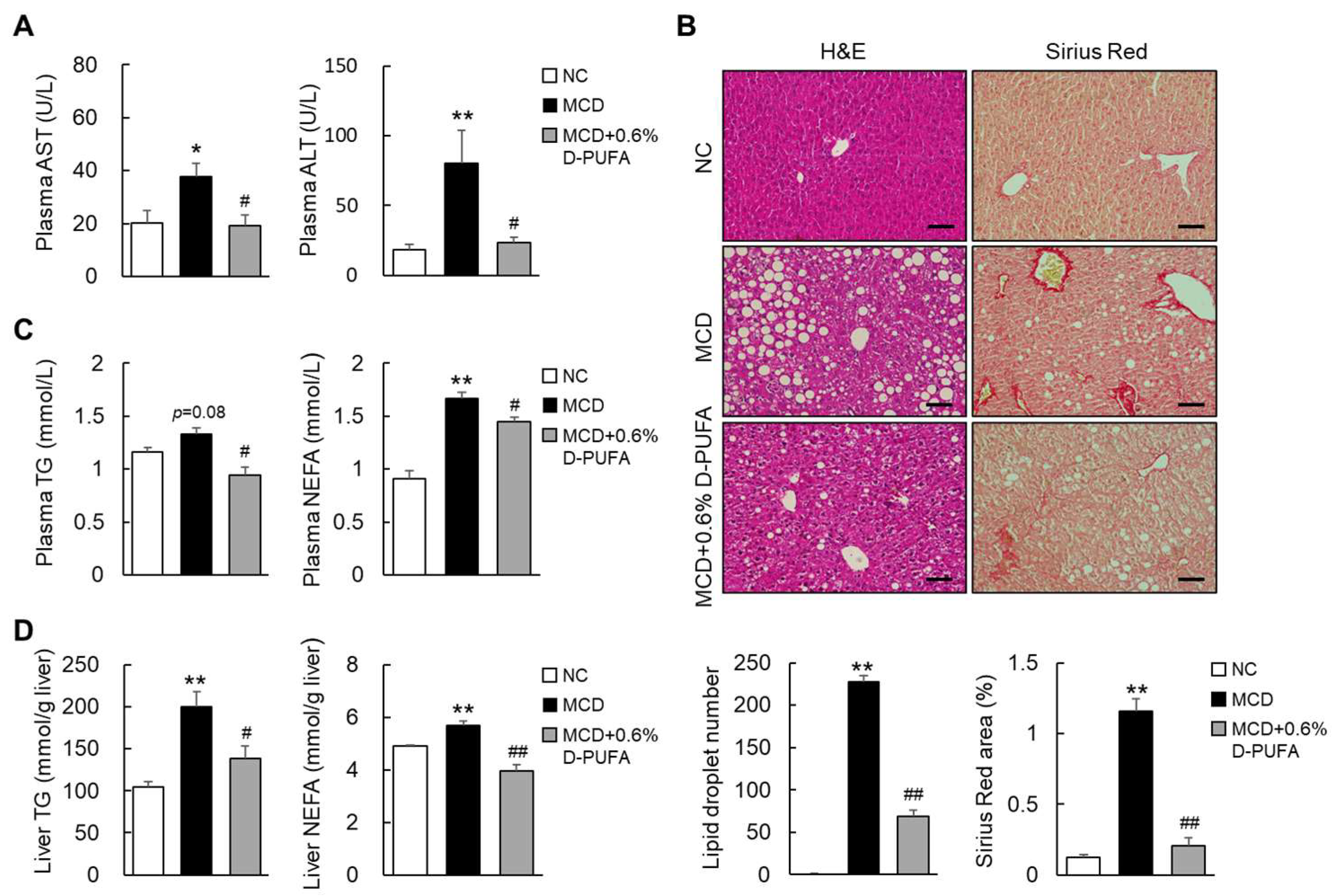
3.3. D-PUFAs Prevent MCD-Induced Steatosis in Mice
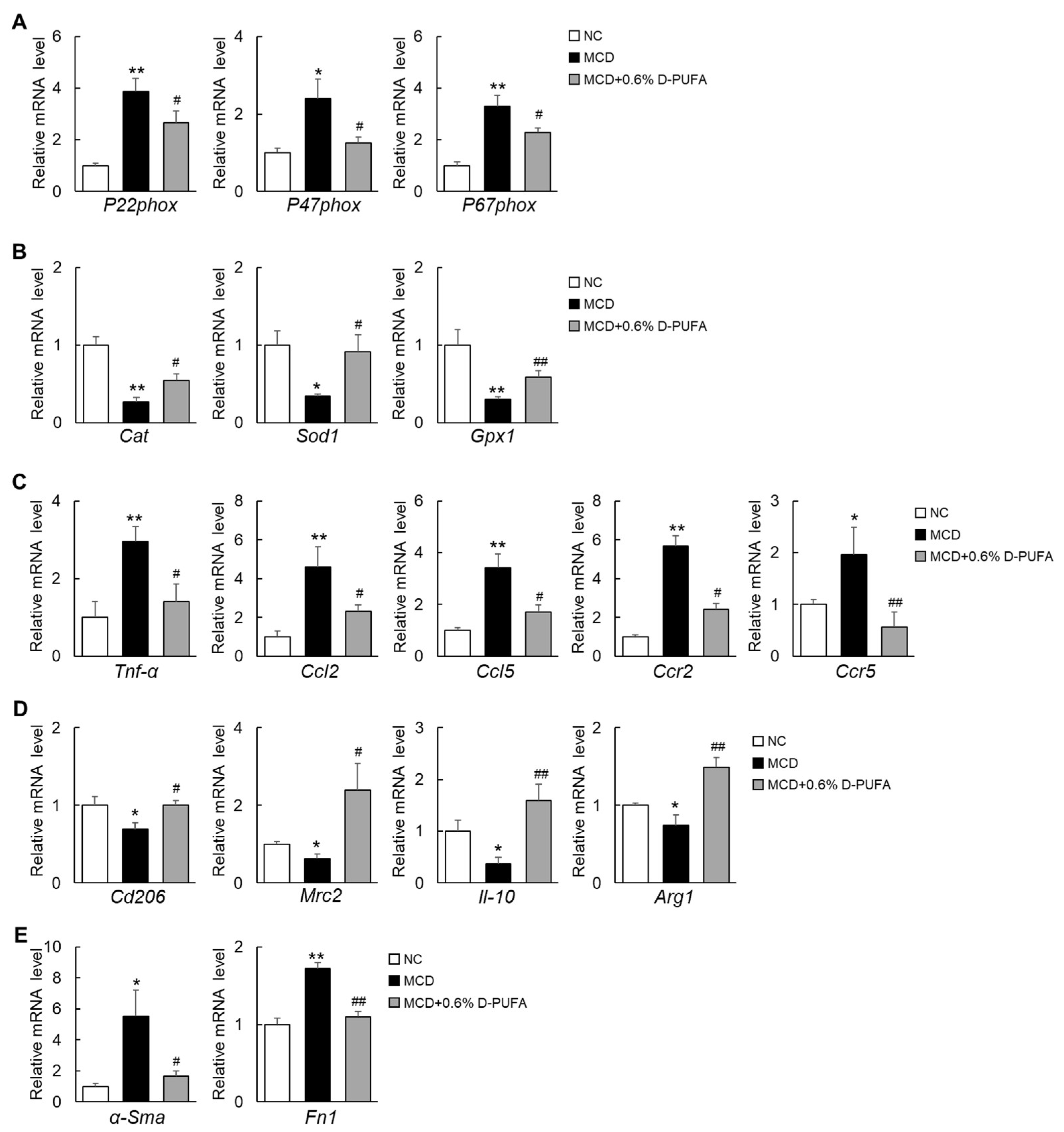
3.4. D-PUFAs Attenuate Oxidative Stress and Inflammation Induced by MCD Diet in Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cotter, T.G.; Rinella, M. Nonalcoholic fatty liver disease 2020: The state of the disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Sanyal, A.J. Management of NAFLD: A stage-based approach. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; Onali, S.; Thorburn, D.; Davidson, B.R.; Gurusamy, K.S.; Tsochatzis, E. Pharmacological interventions for non-alcohol related fatty liver disease (NAFLD): An attempted network meta-analysis. Cochrane Database Syst. Rev. 2017, 3, CD011640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Negi, C.K.; Babica, P.; Bajard, L.; Bienertova-Vasku, J.; Tarantino, G. Insights into the molecular targets and emerging pharmacotherapeutic interventions for nonalcoholic fatty liver disease. Metabolism 2022, 126, 154925. [Google Scholar] [CrossRef]
- He, F.; Zuo, L. Redox Roles of Reactive Oxygen Species in Cardiovascular Diseases. Int. J. Mol. Sci. 2015, 16, 27770–27780. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Fernández-Galilea, M.; Martínez-Fernández, L.; González-Muniesa, P.; Pérez-Chávez, A.; Martínez, J.A.; Moreno-Aliaga, M.J. Oxidative Stress and Non-Alcoholic Fatty Liver Disease: Effects of Omega-3 Fatty Acid Supplementation. Nutrients 2019, 11, 872. [Google Scholar] [CrossRef] [Green Version]
- Leamy, A.K.; Egnatchik, R.A.; Young, J.D. Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease. Prog. Lipid Res. 2013, 52, 165–174. [Google Scholar] [CrossRef] [Green Version]
- Novo, E.; Busletta, C.; Bonzo, L.V.d.; Povero, D.; Paternostro, C.; Mareschi, K.; Ferrero, I.; David, E.; Bertolani, C.; Caligiuri, A.; et al. Intracellular reactive oxygen species are required for directional migration of resident and bone marrow-derived hepatic pro-fibrogenic cells. J. Hepatol. 2011, 54, 964–974. [Google Scholar] [CrossRef]
- Di Domenico, F.; Tramutola, A.; Butterfield, D.A. Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic. Biol. Med. 2017, 111, 253–261. [Google Scholar] [CrossRef]
- Hill, S.; Hirano, K.; Shmanai, V.V.; Marbois, B.N.; Vidovic, D.; Bekish, A.V.; Kay, B.; Tse, V.; Fine, J.; Clarke, C.F.; et al. Isotope-reinforced polyunsaturated fatty acids protect yeast cells from oxidative stress. Free Radic. Biol. Med. 2011, 50, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Elharram, A.; Czegledy, N.M.; Golod, M.; Milne, G.L.; Pollock, E.; Bennett, B.M.; Shchepinov, M.S. Deuterium-reinforced polyunsaturated fatty acids improve cognition in a mouse model of sporadic Alzheimer’s disease. FEBS J. 2017, 284, 4083–4095. [Google Scholar] [CrossRef] [PubMed]
- Cotticelli, M.G.; Crabbe, A.M.; Wilson, R.B.; Shchepinov, M.S. Insights into the role of oxidative stress in the pathology of Friedreich ataxia using peroxidation resistant polyunsaturated fatty acids. Redox Biol. 2013, 1, 398–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, P.R.; Horrocks, M.H.; Klenerman, D.; Gandhi, S.; Abramov, A.Y.; Shchepinov, M.S. Lipid peroxidation is essential for α-synuclein-induced cell death. J. Neurochem. 2015, 133, 582–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatami, A.; Zhu, C.; Relaño-Gines, A.; Elias, C.; Galstyan, A.; Jun, M.; Milne, G.; Cantor, C.R.; Chesselet, M.-F.; Shchepinov, M.S. Deuterium-reinforced linoleic acid lowers lipid peroxidation and mitigates cognitive impairment in the Q140 knock in mouse model of Huntington’s disease. FEBS J. 2018, 285, 3002–3012. [Google Scholar] [CrossRef] [PubMed]
- Shchepinov, M.S.; Chou, V.P.; Pollock, E.; Langston, J.W.; Cantor, C.R.; Molinari, R.J.; Manning-Boğ, A.B. Isotopic reinforcement of essential polyunsaturated fatty acids diminishes nigrostriatal degeneration in a mouse model of Parkinson’s disease. Toxicol. Lett. 2011, 207, 97–103. [Google Scholar] [CrossRef]
- Ota, T.; Gayet, C.; Ginsberg, H.N. Inhibition of apolipoprotein B100 secretion by lipid-induced hepatic endoplasmic reticulum stress in rodents. J. Clin. Investig. 2008, 118, 316–332. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cai, B.; Yang, X.; Sonubi, O.O.; Zheng, Z.; Ramakrishnan, R.; Shi, H.; Valenti, L.; Pajvani, U.B.; Sandhu, J.; et al. Cholesterol Stabilizes TAZ in Hepatocytes to Promote Experimental Non-alcoholic Steatohepatitis. Cell Metab. 2020, 31, 969–986. [Google Scholar] [CrossRef]
- Xu, L.; Nagata, N.; Nagashimada, M.; Zhuge, F.; Ni, Y.; Chen, G.; Kamei, J.; Ishikawa, H.; Komatsu, Y.; Kaneko, S.; et al. A porcine placental extract prevents steatohepatitis by suppressing activation of macrophages and stellate cells in mice. Oncotarget 2018, 9, 15047–15060. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.; Ma, J.; Liu, Y.; He, P.; Dong, W. Combining α-Hederin with cisplatin increases the apoptosis of gastric cancer in vivo and in vitro via mitochondrial related apoptosis pathway. Biomed. Pharm. 2019, 120, 109477. [Google Scholar] [CrossRef]
- Xie, C.; Ge, M.; Jin, J.; Xu, H.; Mao, L.; Geng, S.; Wu, J.; Zhu, J.; Li, X.; Zhong, C. Mechanism investigation on Bisphenol S-induced oxidative stress and inflammation in murine RAW264.7 cells: The role of NLRP3 inflammasome, TLR4, Nrf2 and MAPK. J. Hazard. Mater. 2020, 394, 122549. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.; Banga, J.; Silveyra, P. Oxidative stress and cellular pathways of asthma and inflammation: Therapeutic strategies and pharmacological targets. Pharmacol. Ther. 2018, 181, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhao, E.; Ilyas, G.; Lalazar, G.; Lin, Y.; Haseeb, M.; Tanaka, K.E.; Czaja, M.J. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015, 11, 271–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutti, S.; Jindal, A.; Locatelli, I.; Vacchiano, M.; Gigliotti, L.; Bozzola, C.; Albano, E. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology 2014, 59, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Massart, J.; Robin, M.-A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; Nie, J.; Zhang, J.; Kimball, S.R.; Zhang, H.; Zhang, W.J.; Jefferson, L.S.; Cheng, Z.; Ji, Q.; et al. ALCAT1 controls mitochondrial etiology of fatty liver diseases, linking defective mitophagy to steatosis. Hepatology 2015, 61, 486–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdecantos, M.P.; Pérez-Matute, P.; González-Muniesa, P.; Prieto-Hontoria, P.L.; Moreno-Aliaga, M.J.; Martínez, J.A. Lipoic acid improves mitochondrial function in nonalcoholic steatosis through the stimulation of sirtuin 1 and sirtuin 3. Obesity 2012, 20, 1974–1983. [Google Scholar] [CrossRef]
- Borrelli, A.; Bonelli, P.; Tuccillo, F.M.; Goldfine, I.D.; Evans, J.L.; Buonaguro, F.M.; Mancini, A. Role of gut microbiota and oxidative stress in the progression of non-alcoholic fatty liver disease to hepatocarcinoma: Current and innovative therapeutic approaches. Redox Biol. 2018, 15, 467–479. [Google Scholar] [CrossRef]
- Seki, S.; Kitada, T.; Yamada, T.; Sakaguchi, H.; Nakatani, K.; Wakasa, K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J. Hepatol. 2002, 37, 56–62. [Google Scholar] [CrossRef]
- Mansouri, A.; Gattolliat, C.-H.; Asselah, T. Mitochondrial Dysfunction and Signaling in Chronic Liver Diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [Green Version]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Mofrad, P.S.; Contos, M.J.; Sargeant, C.; Luketic, V.A.; Sterling, R.K.; Stravitz, R.T.; Shiffman, M.L.; Clore, J.; Mills, A.S. A pilot study of vitamin E versus vitamin E and pioglitazone for the treatment of nonalcoholic steatohepatitis. Clin. Gastroenterol. Hepatol. 2004, 2, 1107–1115. [Google Scholar] [CrossRef]
- Chen, X.; Xue, H.; Fang, W.; Chen, K.; Chen, S.; Yang, W.; Shen, T.; Chen, X.; Zhang, P.; Ling, W. Adropin protects against liver injury in nonalcoholic steatohepatitis via the Nrf2 mediated antioxidant capacity. Redox Biol. 2019, 21, 101068. [Google Scholar] [CrossRef] [PubMed]
- Bellenger, J.; Bellenger, S.; Bataille, A.; Massey, K.A.; Nicolaou, A.; Rialland, M.; Tessier, C.; Kang, J.X.; Narce, M. High pancreatic n-3 fatty acids prevent STZ-induced diabetes in fat-1 mice: Inflammatory pathway inhibition. Diabetes 2011, 60, 1090–1099. [Google Scholar] [CrossRef] [Green Version]
- Okada, L.S.D.R.R.; Oliveira, C.P.; Stefano, J.T.; Nogueira, M.A.; Silva, I.D.C.G.d.; Cordeiro, F.B.; Alves, V.A.F.; Torrinhas, R.S.; Carrilho, F.J.; Puri, P.; et al. Omega-3 PUFA modulate lipogenesis, ER stress, and mitochondrial dysfunction markers in NASH—Proteomic and lipidomic insight. Clin. Nutr. 2018, 37, 1474–1484. [Google Scholar] [CrossRef]
- Arendt, B.M.; Comelli, E.M.; Ma, D.W.L.; Lou, W.; Teterina, A.; Kim, T.; Fung, S.K.; Wong, D.K.H.; McGilvray, I.; Fischer, S.E.; et al. Altered hepatic gene expression in nonalcoholic fatty liver disease is associated with lower hepatic n-3 and n-6 polyunsaturated fatty acids. Hepatology 2015, 61, 1565–1578. [Google Scholar] [CrossRef] [Green Version]
- Shefer-Weinberg, D.; Sasson, S.; Schwartz, B.; Argov-Argaman, N.; Tirosh, O. Deleterious effect of n-3 polyunsaturated fatty acids in non-alcoholic steatohepatitis in the fat-1 mouse model. Clin. Nutr. Exp. 2017, 12, 37–49. [Google Scholar] [CrossRef]
- Berbée, J.F.P.; Mol, I.M.; Milne, G.L.; Pollock, E.; Hoeke, G.; Lütjohann, D.; Monaco, C.; Rensen, P.C.N.; van der Ploeg, L.H.T.; Shchepinov, M.S. Deuterium-reinforced polyunsaturated fatty acids protect against atherosclerosis by lowering lipid peroxidation and hypercholesterolemia. Atherosclerosis 2017, 264, 100–107. [Google Scholar] [CrossRef]
- Lamberson, C.R.; Xu, L.; Muchalski, H.; Montenegro-Burke, J.R.; Shmanai, V.V.; Bekish, A.V.; McLean, J.A.; Clarke, C.F.; Shchepinov, M.S.; Porter, N.A. Unusual kinetic isotope effects of deuterium reinforced polyunsaturated fatty acids in tocopherol-mediated free radical chain oxidations. J. Am. Chem. Soc. 2014, 136, 838–841. [Google Scholar] [CrossRef] [Green Version]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Hussain, T.; Tan, B.; Yin, Y.; Blachier, F.; Tossou, M.C.B.; Rahu, N. Oxidative Stress and Inflammation: What Polyphenols Can Do for Us? Oxid. Med. Cell. Longev. 2016, 2016, 7432797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamidzadeh, K.; Christensen, S.M.; Dalby, E.; Chandrasekaran, P.; Mosser, D.M. Macrophages and the Recovery from Acute and Chronic Inflammation. Annu. Rev. Physiol. 2017, 79, 567–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyati, K.K.; Masuda, K.; Zaman, M.M.-U.; Dubey, P.K.; Millrine, D.; Chalise, J.P.; Higa, M.; Li, S.; Standley, D.M.; Saito, K.; et al. TLR4-induced NF-κB and MAPK signaling regulate the IL-6 mRNA stabilizing protein Arid5a. Nucleic Acids Res. 2017, 45, 2687–2703. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Tabas, I.; Pajvani, U.B. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology 2020, 158, 1913–1928. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Zhang, O.; Hui, C.; Huang, Y.; Shao, H.; Song, M.; Gao, L.; Jin, S.; Ding, C.; Xu, L. Deuterium-Reinforced Polyunsaturated Fatty Acids Prevent Diet-Induced Nonalcoholic Steatohepatitis by Reducing Oxidative Stress. Medicina 2022, 58, 790. https://doi.org/10.3390/medicina58060790
Li H, Zhang O, Hui C, Huang Y, Shao H, Song M, Gao L, Jin S, Ding C, Xu L. Deuterium-Reinforced Polyunsaturated Fatty Acids Prevent Diet-Induced Nonalcoholic Steatohepatitis by Reducing Oxidative Stress. Medicina. 2022; 58(6):790. https://doi.org/10.3390/medicina58060790
Chicago/Turabian StyleLi, Haoran, Ouyang Zhang, Chenmin Hui, Yaxin Huang, Hengrong Shao, Menghui Song, Lingjia Gao, Shengnan Jin, Chunming Ding, and Liang Xu. 2022. "Deuterium-Reinforced Polyunsaturated Fatty Acids Prevent Diet-Induced Nonalcoholic Steatohepatitis by Reducing Oxidative Stress" Medicina 58, no. 6: 790. https://doi.org/10.3390/medicina58060790
APA StyleLi, H., Zhang, O., Hui, C., Huang, Y., Shao, H., Song, M., Gao, L., Jin, S., Ding, C., & Xu, L. (2022). Deuterium-Reinforced Polyunsaturated Fatty Acids Prevent Diet-Induced Nonalcoholic Steatohepatitis by Reducing Oxidative Stress. Medicina, 58(6), 790. https://doi.org/10.3390/medicina58060790