Abstract
Diamond–Blackfan anemia (DBA) is a congenital bone marrow failure syndrome associated with malformations. DBA is related to defective ribosome biogenesis, which impairs erythropoiesis, causing hyporegenerative macrocytic anemia. The disease has an autosomal dominant inheritance and is commonly diagnosed in the first year of life, requiring continuous treatment. We present the case of a young woman who, at the age of 21, developed severe symptomatic anemia. Although, due to malformations, a congenital syndrome had been suspected since birth, a confirmation diagnosis was not made until the patient was referred to our center for an evaluation of her anemia. In her neonatal medical history, she presented with anemia that required red blood cell transfusions, but afterwards remained with a stable, mild, asymptomatic anemia throughout her childhood and adolescence. Her family history was otherwise unremarkable. To explain the symptomatic anemia, vitamin deficiencies, autoimmune diseases, bleeding causes, and myeloid and lymphoid neoplasms were investigated and ruled out. A molecular investigation showed the RPL5 gene variant c.392dup, p.(Asn131Lysfs*6), confirming the diagnosis of DBA. All family members have normal blood values and none harbored the mutation. Here, we will discuss the unusual evolution of this case and revisit the literature.
1. Introduction
Diamond–Blackfan anemia (DBA) is a rare constitutional congenital bone marrow failure syndrome mostly caused by mutations in ribosomal genes resulting in ribosomopathy, which leads to progressive macrocytic normochromic anemia and erythroblastopenia [1]. Typically, the diagnosis is made at the median age of 2 to 3 months; 95% of DBA patients will be diagnosed before the age of 2 and 99% before the age of 5 [2]. The estimated incidence of DBA is between 1:100,000 and 1:200,000 live births without differences between ethnicities [3]. Approximately 45% of patients have inherited the mutation from a parent (usually autosomal dominant inheritance) and 55% have de novo mutations [1]. In the familiar form of DBA, there is a 50% chance that the pathogenic mutation is passed onto the offspring. Autosomal dominant DBA is caused by mutations in ribosomal genes (RPL/RPS, ribosomal protein, large/small). The clinical phenotype of DBA is rarely associated with the X-linked mutations of GATA1 or TSR2, and there is only one case described with a recessive erythropoietin (EPO) mutation that presented with the typical histological findings of DBA. These entities are sometimes referred to as DBA-like [4]. A few DBA patients will present with congenital abnormalities. The most common malformations are craniofacial features (microcephaly, hypertelorism, and cleft palate), growth retardation, upper limb anomalies (classical triphalangeal thumb), genitourinary malformations, and heart defects. To characterize DBA patients, the diagnostic criteria have been defined [5]. The pathophysiology of DBA is not completely understood. RP deficiency, p53 activation, and additional mechanisms cause bone marrow failure and anemia (Figure 1). These mutations have a higher probability to lead to a specific phenotype, but this is not true for most findings [6]. Additionally, the penetrance of DBA is incomplete [7]. It is remarkable that the predominantly ribosomal mutations lead to a specific effect in hematopoiesis whilst causing relatively minor changes in the rest of the tissues. This can be explained by the high demand for ribosomal proteins due to the high proliferation of proerythroblasts and the accompanying globin synthesis. A reduction in the ribosome levels with normal ribosome composition has been shown to influence lineage commitment in hematopoiesis [8].
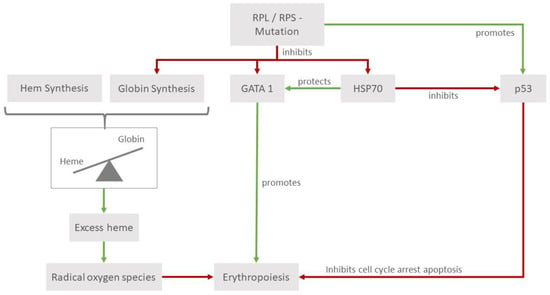
Figure 1.
Pathophysiology of RPL and RPS mutations. RPL and RPS mutations can affect erythropoiesis in various ways that are emphasized in hematopoietic tissues. They lead to higher levels of p53, which, in its role as a tumor suppressor, leads to cell cycle arrest, thus impeding erythropoiesis. Reduced levels of heat shock protein 70 (HSP70) affect erythropoiesis through its interactions with the erythroid transcription factor GATA1 and p53. Additionally, the impaired balance in the heme/globin ratio leads to a detrimental increase in radical oxygen species.
Another influencing factor is that mutations in some RP genes lead to a reduced expression of GATA1, which is an important erythroid transcription factor regulating processes in erythroid differentiation and maturation [9]. Furthermore, a reduction or a function impairment of HSP70 proteins, which act as chaperones for GATA1, also contributes to ineffective erythroid differentiation. The tumor suppressor p53, through its relationship with GATA1, also plays a role in the pathophysiology of DBA. The models of human primary cells have shown that mutations in RPS11 and RPS19 are associated with the stabilization and increased phosphorylation of p53, leading to the activation of its targets, and ultimately promoting cell cycle arrest and apoptosis [10]. It is known that GATA1 acts as an inhibitor of p53 [11]; its impairment constitutes another modality that can interfere with the normal development of hematopoiesis. Studies in cell cultures of cord blood CD34+ cells of DBA individuals (with mutations in RPL5, RPL11, and RPS19) could show a downregulation of its chaperone HSP70, leading to the increased phosphorylation of p53. The transduction of HSP70 into RPL11-haploinsufficient CD34+ cells leads to a significant increase in the proliferation of burst forming units and colony forming units [12].
The reduced globin synthesis is another mechanism considered in the physiopathology of DBA and consequently, the relation of globin to heme diminishes, raising the amount of free intracellular heme [13]. Free intracellular heme has been proven to be toxic for cells through leading to higher levels of reactive oxygen species. The relevance of this mechanism is emphasized by the elevated levels of heme transporter FLVCR1 (feline leukemia virus subgroup C receptor-related protein 1), a membrane hem exporter in erythroid cells of DBA individuals [14].
The DBA phenotype and genotype is subject to ongoing research [15]. We present this case to highlight the existence of DBA with unusual worsening anemia in a young adult.
2. Case Presentation
A 21-year-old female patient of Tamil descent was presented in July 2021 to her family doctor due to orthostatic complaints. Persistent moderate macrocytic anemia was diagnosed. The hemoglobin was initially 82 g/L and the MCV 99.2 fl, and other values of the complete blood counts (CBC) were normal. A vitamin B12 deficiency was diagnosed and treated accordingly. After the substitution of cumulative 5 mg vitamin B12 over two months, the hemoglobin was 75 g/L with unchanged red blood cell (RBC) indices and the patient was referred to the hematology department of a tertiary hospital for further investigations. In June 2022, we saw the patient for the first time in the outpatient clinic; she was 21 years old and was in good health, but she complained of progressive fatigue. In the clinical examination, she was 145 cm tall and her weight was 40 kg (BMI 19.0). She has a series of congenital malformations (Figure 2) and no organomegaly or enlarged lymph nodes. The laboratory tests confirmed the macrocytic anemia with a hemoglobin of 74 g/L. The anemia was non-regenerative with an absolute reticulocyte count of 18 G/L, and the other CBC values were in the normal range. The ferritin, transferrin saturation, folic acid, and vitamin B12 were normal, but the holotranscobalamin (HoloTc) was reduced (37.4 pmol/L). In the evaluation of the peripheral blood film, macrocytosis was observed without other signs related to vitamin B12 deficiency. There was no evidence of hemolysis and the EPO was increased with a level of >750 mU/mL, which was interpreted in the context of anemia.
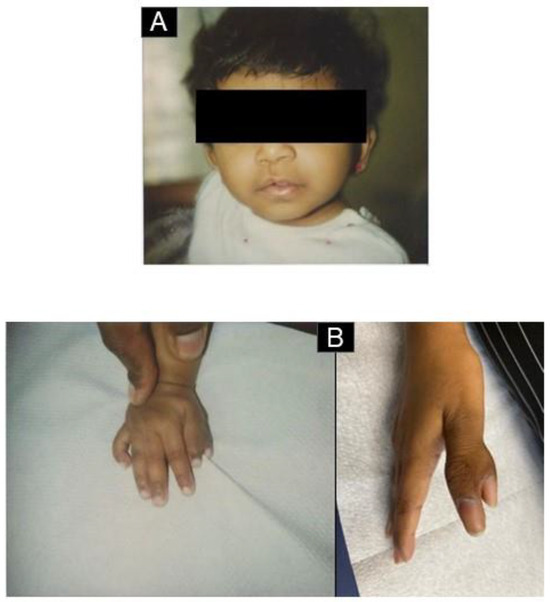
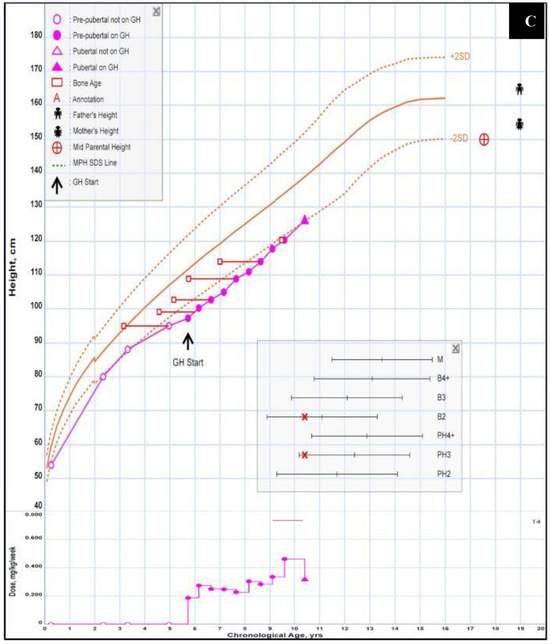
Figure 2.
Congenital anomalies. (A) Lateral cleft lip after surgical correction. (B) Polysyndactylie (“double thumb”), 2 pictures: childhood/adulthood. (C) Growth curve of the patient. As she was not treated with steroids in infancy, the accentuated growth retardation was partially corrected with the administration of somatotropin between the ages of 6 and 10 years’ treatment [15].
The review of her medical files showed that at birth, after an uncomplicated pregnancy, she had a ventricular septal defect, a double thumb on the right side, relative macrocephaly, and a lateral cleft lip. At the age of 10 weeks, she was hospitalized due to anemia-related heart failure. Her condition stabilized after transfusions. An elevated level of erythroid adenosine deaminase (eADA) activity suggested an underlying diagnosis of DBA, but at that time, genetic testing was not performed. As her hemoglobin levels were subsequently stable (above 80 g/L), therapies such as steroids were not administered. In childhood and adolescence, the hemoglobin values were always stable in the range of 85–100 g/L. There was a spontaneous closure of the ventricular septal defect around the age of 5 years with asymptomatic cardiac performance.
Due to her short stature (her height at birth was 44 cm, and weight was 2620 g, both <10th percentile, and both falling below the third percentile in childhood), she was subjected to somatropin supplementation from the age of 5 7/12 to 11 7/12. Under this regimen, she grew 28 cm (from 98 cm to 126 cm), after stopping this treatment, she reached a height of 145 cm (her father was 165 cm, and her mother was 155 cm). The cleft lip was surgically corrected with good results; the polysyndactyly does not limit the patient in her daily life.
To better characterize the worsening of the anemia observed since 2021, bone marrow (BM) investigations were performed. The aspiration showed scarce spicules and provided little information. The BM biopsy revealed normal cellularity for her age, but a highly atypical CD4-predominant T-lymphocytosis (60% of the total cellularity), an impairment of the maturation of all three cell lines, and mild myelofibrosis (maximum MF-1). The molecular pathological examinations showed no evidence of a clonal rearrangement of TCR gamma. A PET/CT scan showed no evidence of lymphoma. The karyotype analysis was unsuccessful due to insufficient metaphases, and recurrent findings typical of a myelodysplastic syndrome were not detected in fluorescence in situ hybridization (FISH). In the array-CGH, a copy neutral loss of heterozygosity of the short arm of Chromosome 3 and 8 was shown. Due to the suspicion of evolution in a myeloid disease, an NGS using the fragment analysis of 65 genes and 300 known recurrent rearrangements was evaluated. No somatic mutation was identified.
Finally, due to the consistent suspicion of a bone marrow failure syndrome, a heterozygous mutation in the RPL5 gene, variant c.392dup, p.(Asn131Lysfs*6), was detected with the TWIST Comprehensive Exome (Twist Bioscience) NGS panel. Combined with the clinical features of the patient, the diagnosis of DBA/Aase-Smith Syndrome II could be confirmed. In the family history, all relatives have a normal height according to their ethnic background; there was no history of malformations or anemia in her parents, sister, grandparents, uncles or aunts. The genetic testing of the parents and sister did not show the mutation and their CBCs were within the normal range (Figure 3).
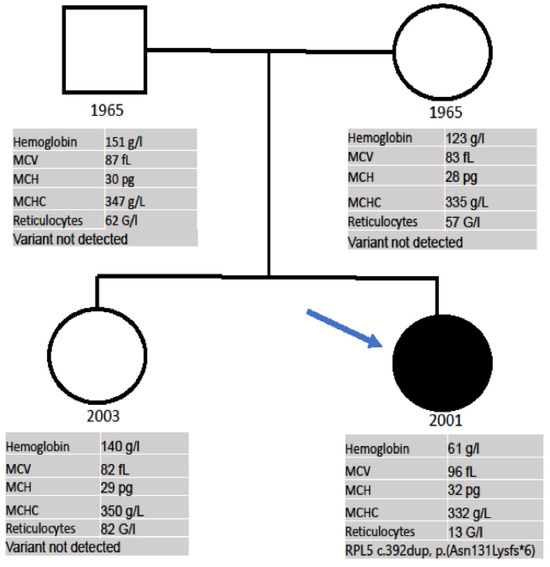
Figure 3.
Pedigree of the family. Proband is indicated by arrow. Symbols: □ = male, ○ = female; • = heterozygous mutation in the RPL5 gene, variant c.392dup, p.(Asn131Lysfs*6).
During follow-up, a worsening of the anemia was observed. At that time, the diagnosis of sarcoidosis was suspected and she received short-term steroids (prednisolone). Once the diagnosis of sarcoidosis was ruled out, the steroid treatment was quickly discontinued. This short-term administration of the steroid did not improve the anemia. The nadir values of the hemoglobin were 58 g/L and RBC transfusions were needed. The transfusion requirement was of 2 RBC units every 5 months. A 3-month trial of Eltrombopag using an aplastic anemia dose was initiated and was well-tolerated but failed to improve the anemia and was therefore withdrawn. In the last 12 months, a further worsening of the anemia (nadir Hb 40 g/L) with a greater transfusion requirement of 2 RBC units every 3 months was observed. A new attempt with steroids (0.5 mg/d) was made, and this time for longer time. A total of 28 days after the start of therapy, a reticulocyte response of 51 G/L (previously 10 G/L) with the stabilization of Hb values above 80 g/L was observed. Currently, steroid dose tapering is ongoing. The relevant aspects of this case are summarized in Table 1.

Table 1.
Summary of patient characteristics.
3. Discussion
We present here a DBA case in which anemia was diagnosed a few weeks after birth, but afterwards, the patient had an indolent course until the age of 20, when the patient began to show a clear worsening of the anemia, thus requiring transfusions and treatment. Although, due to her malformations, a congenital syndrome was suspected since birth, a confirmation diagnosis was not made until referral to our center for an evaluation of the anemia. After the exclusion of a myelodysplastic syndrome, lymphoma, and inflammatory diseases, a heterozygous gene variant in the RPL5 gene, c.392dup, p.(Asn131Lysfs*6), was detected. This frameshift mutation leads to an early stop codon, and thus, nonsense-mediated mRNA decay. Pathogenic variants in the RPL5 gene are associated with DBA [7]. The clinical, laboratory, and molecular investigation of the family did not show the mutation found in the proband in any of the other members, allowing a de novo diagnosis, which is described in 55% of DBA patients [1].
The majority of DBA cases will present with severe anemia during the first year of life and they will require continuous treatment [15]. There are also many patients who are asymptomatic carriers, although cases of late-onset anemia during adulthood have been frequently reported [16,17,18]. Cases such as the one reported here, diagnosed in the first weeks of life and following a mild course, to then worsen again in adulthood, seem to be rare.
Furthermore, patients with inherited bone marrow failure syndromes (IBMFSs) are more likely to develop secondary malignant diseases compared to the general population. In DBA, this is not only true for MDS and AML, with an observed/expected ratio of 28.8 and 352.1, respectively [19], but also for solid tumors such as colon carcinoma, esophageal cancer, osteogenic sarcoma, squamous cell carcinoma and lung cancer. The O/R ratio for all cancers is reported to be 4.75. In other IBMFSs, like Fanconi anemia and dyskeratosis congenita, higher O/R ratios are reported but they lack the high incidence of colon cancer and osteogenic sarcoma, implying a different cancer mechanism associated with RP mutations [19]. Based on this evidence, the first hypothesis in the case presented was the progression to a myeloid disease or the appearance of other neoplasms, for which various investigations were carried out. The patient did not show either typical morphological signs or typical mutations of MDS. The PET scan ruled out an active malignant process. A gynecological examination and ultrasound were both normal.
Regarding treatment for DBA, 80% of patients initially respond to steroids. A trial of 2 mg/kg/d is frequently used; reticulocytes will typically appear after two weeks in responders. If there is no response after four weeks, the treatment should be discontinued. If there is an increase in reticulocytes and hemoglobin levels, steroids should be decreased to the minimal dosage to maintain a hemoglobin of 90 g/L. This dose should not exceed 0.3 mg/kg/d if erythrocyte transfusions are easily available (0.5 mg/kg/d if they are not) [1]. Due to the long-term effects of steroids and transfusions, the option to perform an allogeneic hematopoietic stem cell transplantation (allo-HCT) should be evaluated early. In 2020, Strahm et al. evaluated the data of 70 DBA patients that underwent allo-HCT in France and Germany from 1985 to 2017 [20]. They concluded that allo-HCT is efficient and safe in young patients (median age of 5.5 years) with steadily improving outcomes, e.g., no case of death or severe chronic GVHD after 1999 in patients who were transplanted from a matched sibling donor. The probability of survival with a median follow-up of 4.5 years was 91% with a low transplant related mortality rate. Six of 70 patients died of transplantation-related causes. The surviving patients were independent of tranfusions [20]. In this study, the difference in outcome for patients >10 years at transplantation did not reach statistical significance, while an Italian study of 30 patients (1990–2012) showed significantly better outcomes for patients younger than 10 years old with all transplantation-related deaths occurring in the older patients [21], who had severe iron overload before transplantation. Thus, allo-HCT should be considered early in young patients, preferably if a matched sibling donor is available. The patient has a HLA-identical sister, but so far, there is no indication for an allo-HCT.
Eltrombopag is an orally available mimetic of thrombopoetin, which is a regulator of platelet production and the maintenance of human stem cells. Currently, eltrombopag is used for immune thrombocytopenic purpura [22], thrombocytopenia due to hepatitis C [23], and for the treatment of severe aplastic anemia [24,25]. The hematopoietic effects are achieved through its interaction with the TPO receptor (c-mpl) and the activation of signaling cascades in its target cells [26]. Additionally, the drug is known for its capacity to chelate intracellular iron and thus reduce the labile iron pool, both decreasing reactive oxygen species and inhibiting heme synthesis. This makes it an interesting drug to address the heme/globin disbalance found in DBA progenitor cells. In ex vivo models of induced pluripotent stem cells of DBA patients, a partial rescue of the defective erythropoiesis using eltrombopag, with improved differentiation and maturation, could be shown [27]. The effect turned out to be even larger with the addition of deferasirox compared to deferoxamine, which is a less effective chelator of intracellular iron [28]. In 2020, a prospective phase 2 study of eltrombopag use in patients with moderate aplastic anemia or uni-lineage cytopenia investigated the erythropoetic response and clonal evolution under eltrombopag. One DBA patient with an RPS19 mutation who was refractory to steroids and transfusion-dependent was included and became transfusion-independent even 10 months after the discontinuation of eltrombopag before relapsing. This patient regained a response after re-establishing the treatment for the whole follow up of 5 years [29]. These findings have led to the approval of an interventional single-arm phase I/II trial that will assess the safety and efficacy of eltrombopag in DBA patients, which will be completed in 2027 (NCT04269889).
4. Conclusions
More awareness of unusual DBA presentations is needed. While testing for the causative mutation in DBA is not necessary to establish the diagnosis, it is important for the follow-up of these patients, due to the increased risk of transformation into myeloid diseases and other tumors, as well as for genetic counseling. Multidisciplinary clinical follow-up is also necessary for the management of non-hematological problems. Furthermore, more data about the underlying genetic profile will allow for a better understanding of the geno- and phenotype relationship in this heterogenic disease.
Author Contributions
M.D. (Moritz Dorenkamp) and A.R. are the treating physicians. M.D. (Moritz Dorenkamp) and A.R. wrote the manuscript. N.P. performed the genetic testing and interpretation. M.D. (Miriam Diepold) advised on the pediatric aspects on the subject. All authors revised and agree with the last version of this manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Written informed consent was obtained from the patient.
Data Availability Statement
All data generated for this manuscript is included in the published article.
Acknowledgments
We thank our patient and her family for consenting to this publication.
Conflicts of Interest
M.D. has no competing interests. N.P. has no competing interests. M.Di. has no competing interests. A.R. has received research funding from Novartis, CSL Behring, and AG Alexion, has received honoraria from AG Alexion and Novartis, has received honoraria for advisory board meetings from AG Alexion, BMS, Novartis, OrPhaSwiss GmbH, and Swedish Orphan Biovitrum AG.
References
- Costa, L.M.D.; Marie, I.; Leblanc, T.M. Diamond—Blackfan anemia. Hematol. Am. Soc. Hematol. Educ. Program 2021, 2021, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Willig, T.-N.; Niemeyer, C.M.; Leblanc, T.; Tiemann, C.; Robert, A.; Budde, J.; Lambiliotte, A.; Kohne, E.; Souillet, G.; Eber, S.; et al. Identification of new prognosis factors from the clinical and epidemiologic analysis of a registry of 229 Diamond-Blackfan anemia patients. Pediatr. Res. 1999, 46, 553. [Google Scholar] [CrossRef]
- Costa, L.D.; Leblanc, T.; Mohandas, N. Diamond-Blackfan anemia. Blood 2020, 136, 1262–1273. [Google Scholar] [CrossRef]
- Sieff, C. Diamond-Blackfan Anemia. GeneReviews®. Published 25 June 2009. Available online: https://www.ncbi.nlm.nih.gov/books/NBK7047/ (accessed on 30 August 2023).
- Vlachos, A.; Ball, S.; Dahl, N.; Alter, B.P.; Sheth, S.; Ramenghi, U.; Meerpohl, J.; Karlsson, S.; Liu, J.M.; Leblanc, T.; et al. Diagnosing and treating Diamond Blackfan anaemia: Results of an international clinical consensus conference. Br. J. Haematol. 2008, 142, 859–876. [Google Scholar] [CrossRef] [PubMed]
- Gazda, H.T.; Sheen, M.R.; Vlachos, A.; Choesmel, V.; O’Donohue, M.-F.; Schneider, H.; Darras, N.; Hasman, C.; Sieff, C.A.; Newburger, P.E.; et al. Ribosomal Protein L5 and L11 Mutations Are Associated with Cleft Palate and Abnormal Thumbs in Diamond-Blackfan Anemia Patients. Am. J. Hum. Genet. 2008, 83, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Ulirsch, J.C.; Verboon, J.M.; Kazerounian, S.; Guo, M.H.; Yuan, D.; Ludwig, L.S.; Handsaker, R.E.; Abdulhay, N.J.; Fiorini, C.; Genovese, G.; et al. The Genetic Landscape of Diamond-Blackfan Anemia. Am. J. Hum. Genet. 2018, 103, 930–947. [Google Scholar] [CrossRef] [PubMed]
- Khajuria, R.K.; Munschauer, M.; Ulirsch, J.C.; Fiorini, C.; Ludwig, L.S.; McFarland, S.K.; Abdulhay, N.J.; Specht, H.; Keshishian, H.; Mani, D.; et al. Ribosome Levels Selectively Regulate Translation and Lineage Commitment in Human Hematopoiesis. Cell 2018, 173, 90–103.e19. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Ghazvinian, R.; Do, R.; Thiru, P.; Vergilio, J.-A.; Beggs, A.H.; Sieff, C.A.; Orkin, S.H.; Nathan, D.G.; Lander, E.S.; et al. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J. Clin. Investig. 2012, 122, 2439–2443. [Google Scholar] [CrossRef]
- Moniz, H.; Gastou, M.; Leblanc, T.; Hurtaud, C.; Crétien, A.A.; Lécluse, Y.; Raslova, H.; Larghero, J.; Croisille, L.; Faubladier, M.; et al. Primary hematopoietic cells from DBA patients with mutations in RPL11 and RPS19 genes exhibit distinct erythroid phenotype in vitro. Cell Death Dis. 2012, 3, e356. [Google Scholar] [CrossRef] [PubMed]
- Trainor, C.D.; Mas, C.; Archambault, P.; Di Lello, P.; Omichinski, J.G. GATA-1 associates with and inhibits p53. Blood 2009, 114, 165–173. [Google Scholar] [CrossRef]
- Gastou, M.; Rio, S.; Dussiot, M.; Karboul, N.; Moniz, H.; Leblanc, T.; Sevin, M.; Gonin, P.; Larghéro, J.; Garrido, C.; et al. The severe phenotype of Diamond-Blackfan anemia is modulated by heat shock protein 70. Blood Adv. 2017, 1, 1959–1976. [Google Scholar] [CrossRef] [PubMed]
- Rio, S.; Gastou, M.; Karboul, N.; Derman, R.; Suriyun, T.; Manceau, H.; Leblanc, T.; El Benna, J.; Schmitt, C.; Azouzi, S.; et al. Regulation of globin-heme balance in Diamond-Blackfan anemia by HSP70/GATA1. Blood 2019, 133, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Keel, S.B.; Shimamura, A.; Liu, L.; Gerds, A.T.; Li, H.Y.; Wood, B.L.; Scott, B.L.; Abkowitz, J.L. Delayed globin synthesis leads to excess heme and the macrocytic anemia of Diamond Blackfan anemia and del(5q) myelodysplastic syndrome. Sci. Transl. Med. 2016, 8, 338ra67. [Google Scholar] [CrossRef] [PubMed]
- Iskander, D.; Roy, N.B.; Payne, E.; Drasar, E.; Hennessy, K.; Harrington, Y.; Christodoulidou, C.; Karadimitris, A.; Batkin, L.; de la Fuente, J. Diamond-Blackfan anemia in adults: In pursuit of a common approach for a rare disease. Blood Rev. 2023, 61, 101097. [Google Scholar] [CrossRef] [PubMed]
- Farruggia, P.; Quarello, P.; Garelli, E.; Paolicchi, O.; Ruffo, G.B.; Cuccia, L.; Cannella, S.; Bruno, G.; D’Angelo, P. The spectrum of non-classical Diamond-Blackfan anemia: A case of late beginning transfusion dependency associated to a new RPL5 mutation. Pediatr. Rep. 2012, 4, 91–93. [Google Scholar] [CrossRef]
- Ballester, E.F.; Gil-Fernández, J.J.; Blanco, M.V.; Mesa, J.M.; García, J.D.; Tamayo, A.T.; Burgaleta, C. Adult-onset Diamond-Blackfan anemia with a novel mutation in the exon 5 of RPL 11: Too late and too rare. Clin. Case Rep. 2015, 3, 392–395. [Google Scholar] [CrossRef]
- Tamefusa, K.; Muraoka, M.; Washio, K.; Wakamatsu, M.; Shimada, A. Late-onset familial Diamond–Blackfan anemia with neutropenia caused by RPL35A variant. Pediatr. Int. 2022, 64, e15275. [Google Scholar] [CrossRef]
- Vlachos, A.; Rosenberg, P.S.; Atsidaftos, E.; Kang, J.; Onel, K.; Sharaf, R.N.; Alter, B.P.; Lipton, J.M. Increased risk of colon cancer and osteogenic sarcoma in diamond-Blackfan anemia. Blood 2018, 132, 2205–2208. [Google Scholar] [CrossRef]
- Strahm, B.; Loewecke, F.; Niemeyer, C.M.; Albert, M.; Ansari, M.; Bader, P.; Bertrand, Y.; Burkhardt, B.; Da Costa, L.M.; Ferster, A.; et al. Favorable outcomes of hematopoietic stem cell transplantation in children and adolescents with Diamond-Blackfan anemia. Blood Adv. 2020, 4, 1760–1769. [Google Scholar] [CrossRef]
- Fagioli, F.; Quarello, P.; Zecca, M.; Lanino, E.; Corti, P.; Favre, C.; Ripaldi, M.; Ramenghi, U.; Locatelli, F.; Prete, A. Haematopoietic stem cell transplantation for Diamond Blackfan anaemia: A report from the Italian Association of Paediatric Haematology and Oncology Registry. Br. J. Haematol. 2014, 165, 673–681. [Google Scholar] [CrossRef]
- Cheng, G.; Saleh, M.N.; Marcher, C.; Vasey, S.; Mayer, B.; Aivado, M.; Arning, M.; Stone, N.L.; Bussel, J.B. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): A 6-month, randomised, phase 3 study. Lancet 2011, 377, 393–402. [Google Scholar] [CrossRef]
- Afdhal, N.H.; Dusheiko, G.M.; Giannini, E.G.; Chen, P.; Han, K.; Mohsin, A.; Rodriguez–Torres, M.; Rugina, S.; Bakulin, I.; Lawitz, E.; et al. Eltrombopag increases platelet numbers in thrombocytopenic patients with hcv infection and cirrhosis, allowing for effective antiviral therapy. Gastroenterology 2014, 146, 442–452.e1. [Google Scholar] [CrossRef]
- Townsley, D.M.; Scheinberg, P.; Winkler, T.; Desmond, R.; Dumitriu, B.; Rios, O.; Weinstein, B.; Valdez, J.; Lotter, J.; Feng, X.; et al. Eltrombopag Added to Standard Immunosuppression for Aplastic Anemia. N. Engl. J. Med. 2017, 376, 1540–1550. [Google Scholar] [CrossRef]
- McHutchison, J.G.; Dusheiko, G.; Shiffman, M.L.; Rodriguez-Torres, M.; Sigal, S.; Bourliere, M.; Berg, T.; Gordon, S.C.; Campbell, F.M.; Theodore, D.; et al. Eltrombopag for Thrombocytopenia in Patients with Cirrhosis Associated with Hepatitis C. N. Engl. J. Med. 2007, 357, 2227–2236. [Google Scholar] [CrossRef]
- Bussel, J.; Kulasekararaj, A.; Cooper, N.; Verma, A.; Steidl, U.; Semple, J.W.; Will, B. Mechanisms and therapeutic prospects of thrombopoietin receptor agonists. Semin. Hematol. 2019, 56, 262–278. [Google Scholar] [CrossRef]
- Qanash, H.; Li, Y.; Smith, R.H.; Linask, K.; Young-Baird, S.; Hakami, W.; Keyvanfar, K.; Choy, J.S.; Zou, J.; Larochelle, A. Eltrombopag improves erythroid differentiation in a human induced pluripotent stem cell model of diamond blackfan anemia. Cells 2021, 10, 734. [Google Scholar] [CrossRef] [PubMed]
- Hoffbrand, A.V.; Taher, A.; Cappellini, M.D. How I treat transfusional iron overload. Blood 2012, 120, 3657–3669. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fan, X.; Desmond, R.; Winkler, T.; Young, D.J.; Dumitriu, B.; Townsley, D.M.; Gutierrez-Rodrigues, F.; Lotter, J.; Valdez, J.; Sellers, S.E.; et al. Eltrombopag for patients with moderate aplastic anemia or uni-lineage cytopenias. Blood Adv. 2020, 4, 1700–1710. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).