Elucidating Cuproptosis-Associated Genes in the Progression from Nash to HCC Using Bulk and Single-Cell RNA Sequencing Analyses and Experimental Validation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Gene Set Enrichment Analysis (GSEA) and OS Analysis of CRGs
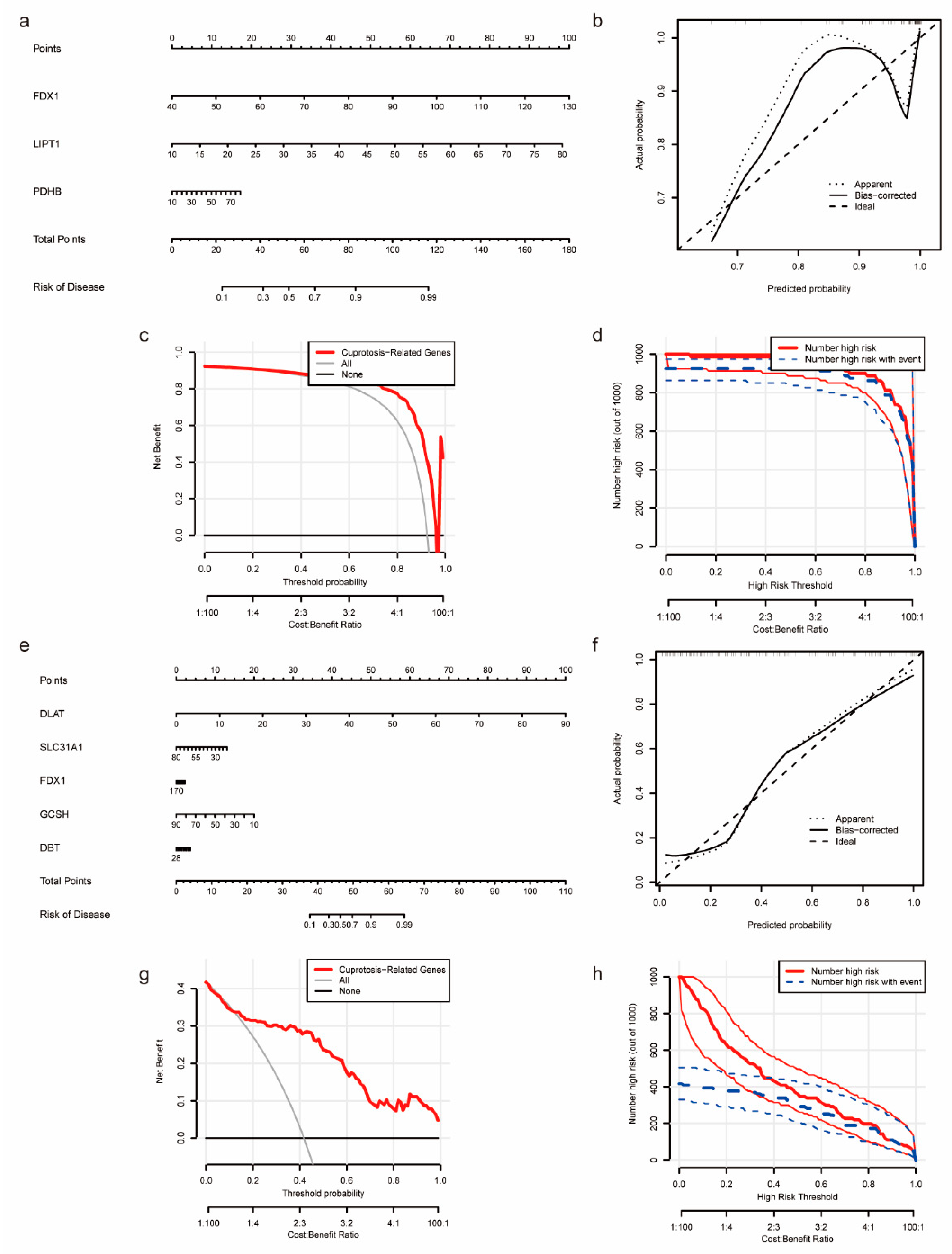
2.3. Construction of Nomogram Model for CRGs
2.4. Construction and Verification of CRC for NASH and NASH Development to HCC
2.5. Assessment of Immune Cell Infiltration CRC Using ssGSEA
2.6. Data Processing and Analysis of Single Cells
2.7. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.8. Human Tissue Specimens
2.9. Reagents, Animals, and Animal Experiments
2.10. Biochemical Analysis
2.11. Immunohistochemistry
2.12. Oil Red O Staining
2.13. Histology Examination
2.14. Statistics
3. Results
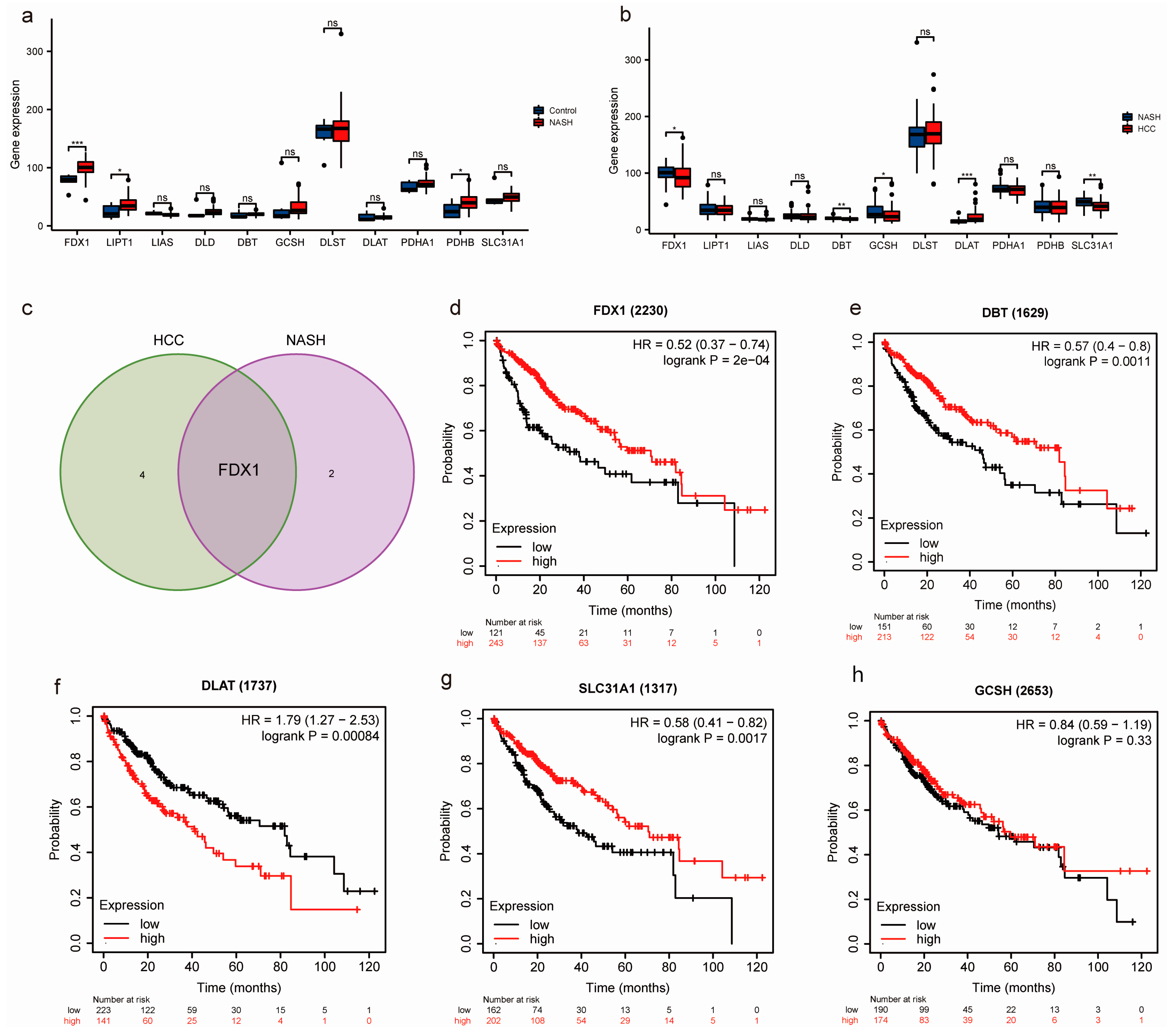
3.1. Identification of Key CRGs in the Transition of NASH to HCC
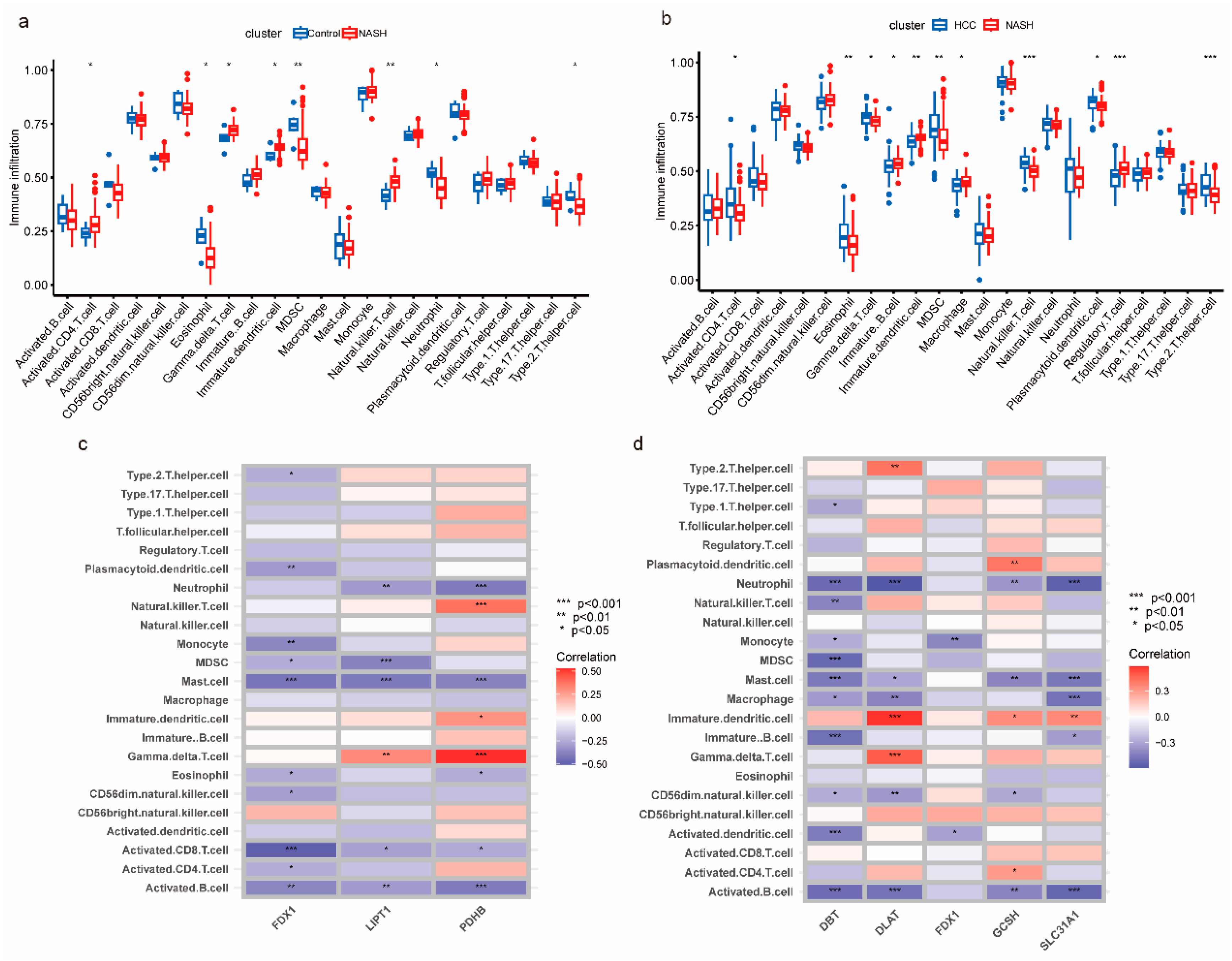
3.2. Analysis of the Immune Microenvironment of NASH to HCC
3.3. Construction of Nomogram Models Based on the Development of NASH to HCC
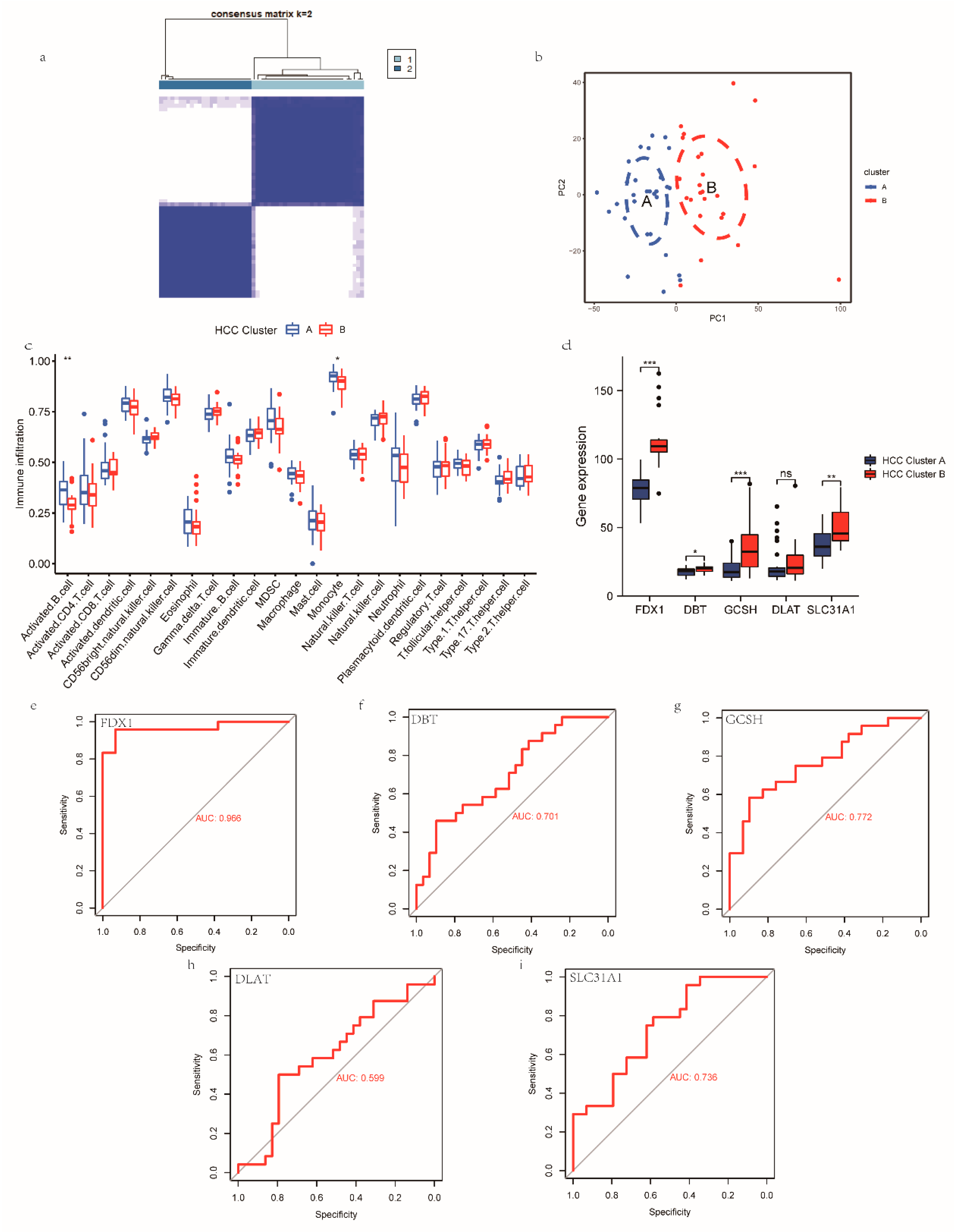
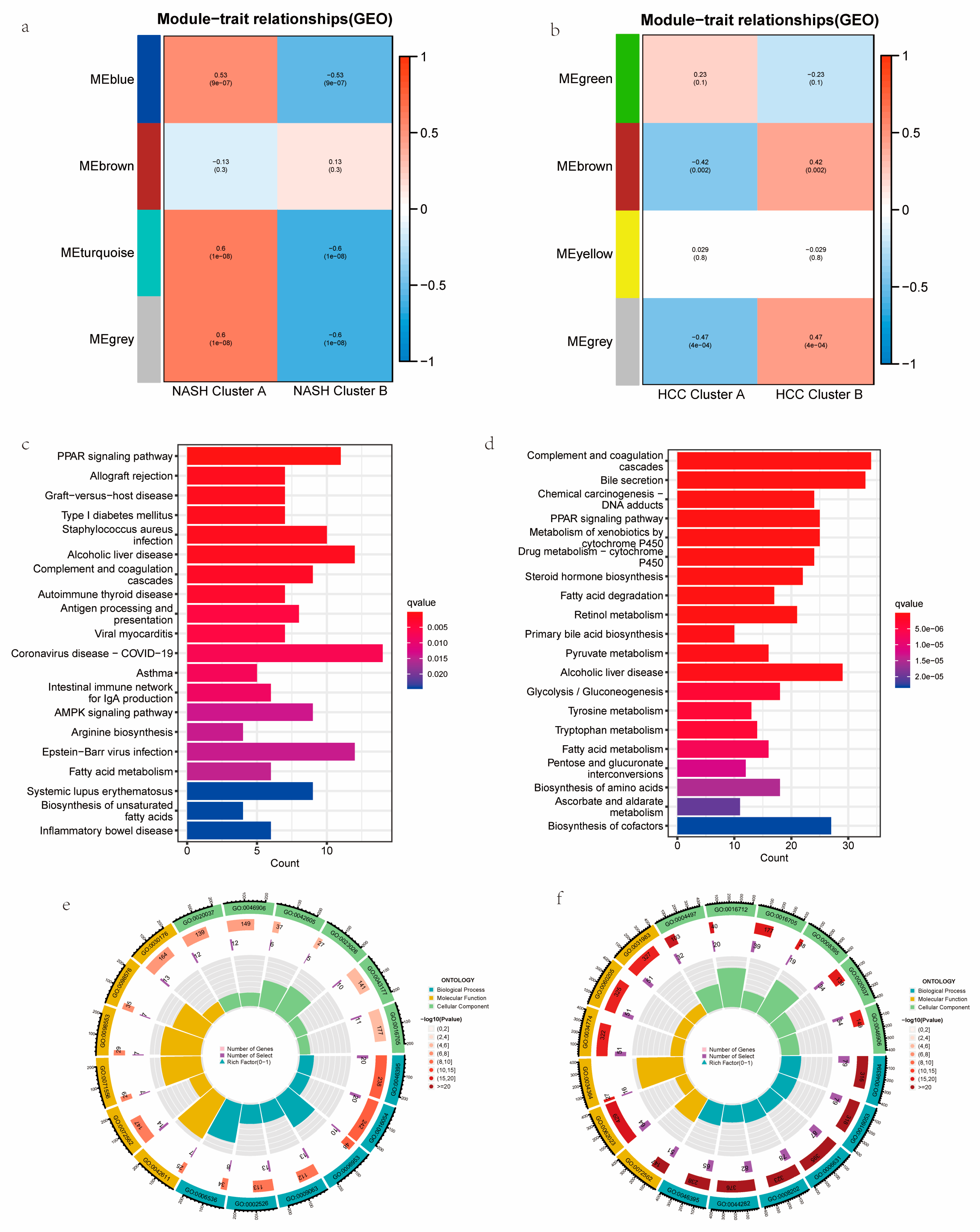
3.4. Construction, Validation, and Biological Characterization of a Cuproptosis-Related Cluster
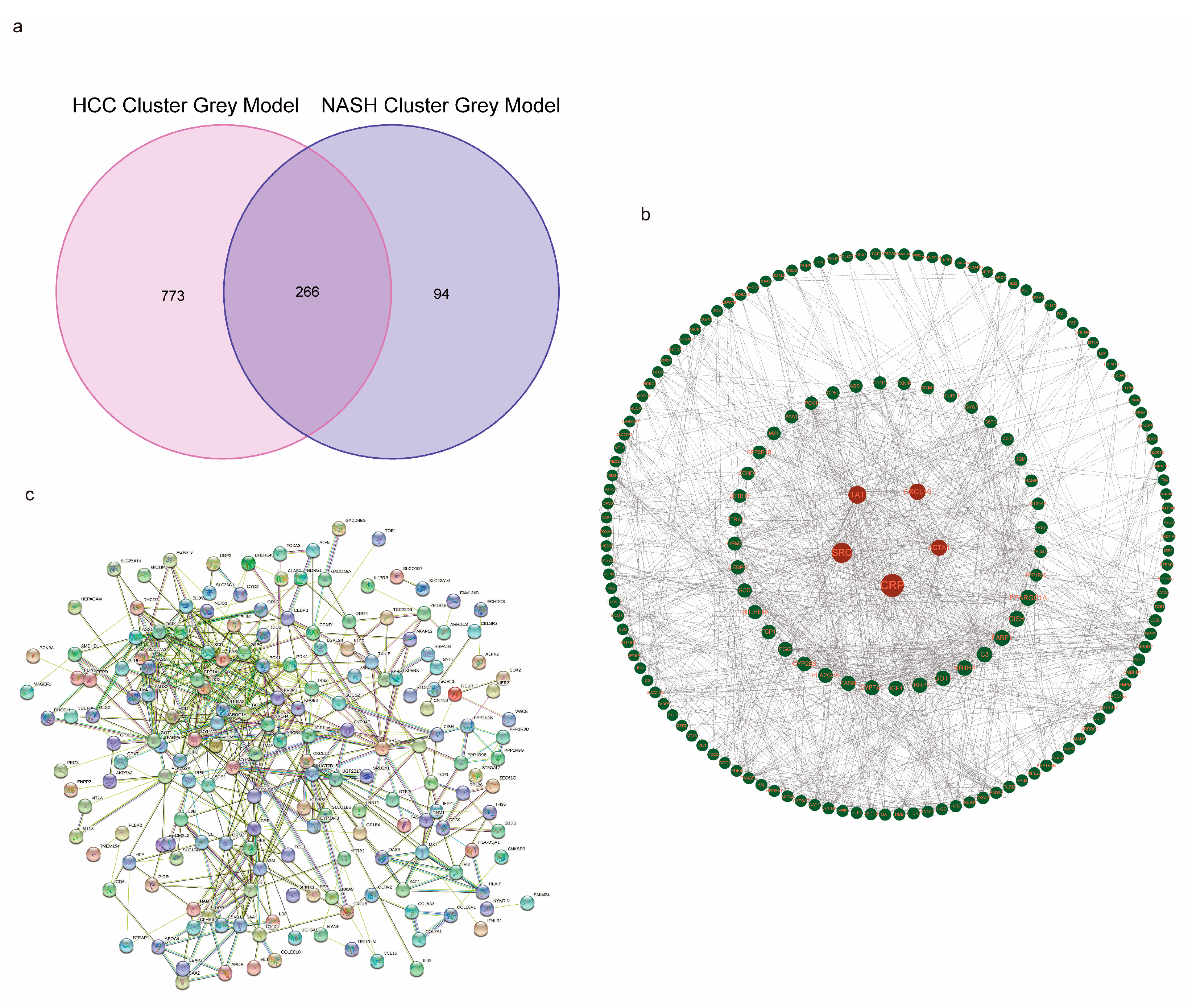
3.5. Functional Analyses Based on the NASH CRC to HCC CRC and PPI Construction
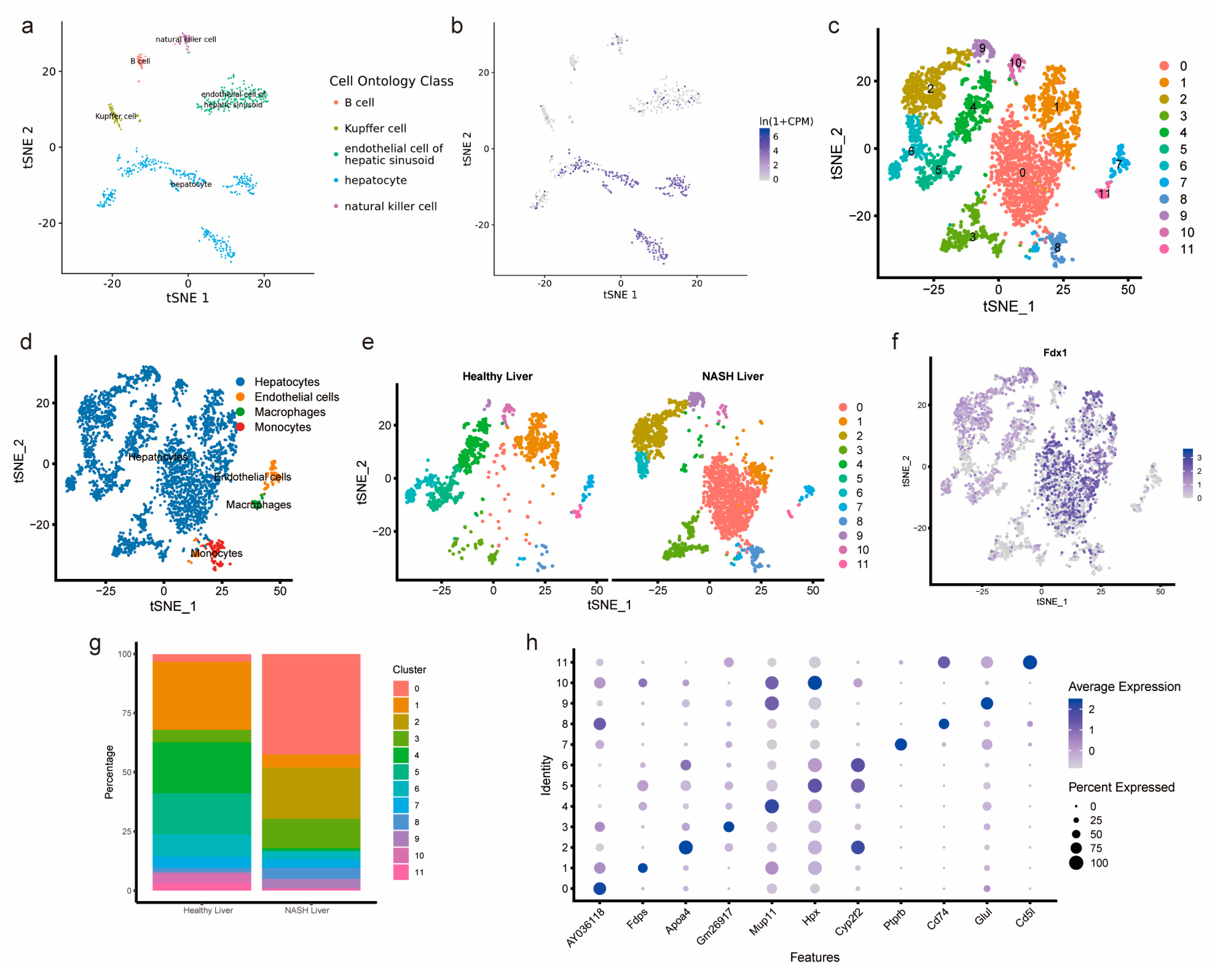
3.6. Single-Cell Analysis of Healthy Mouse Hepatocytes and Mouse NASH Hepatocytes
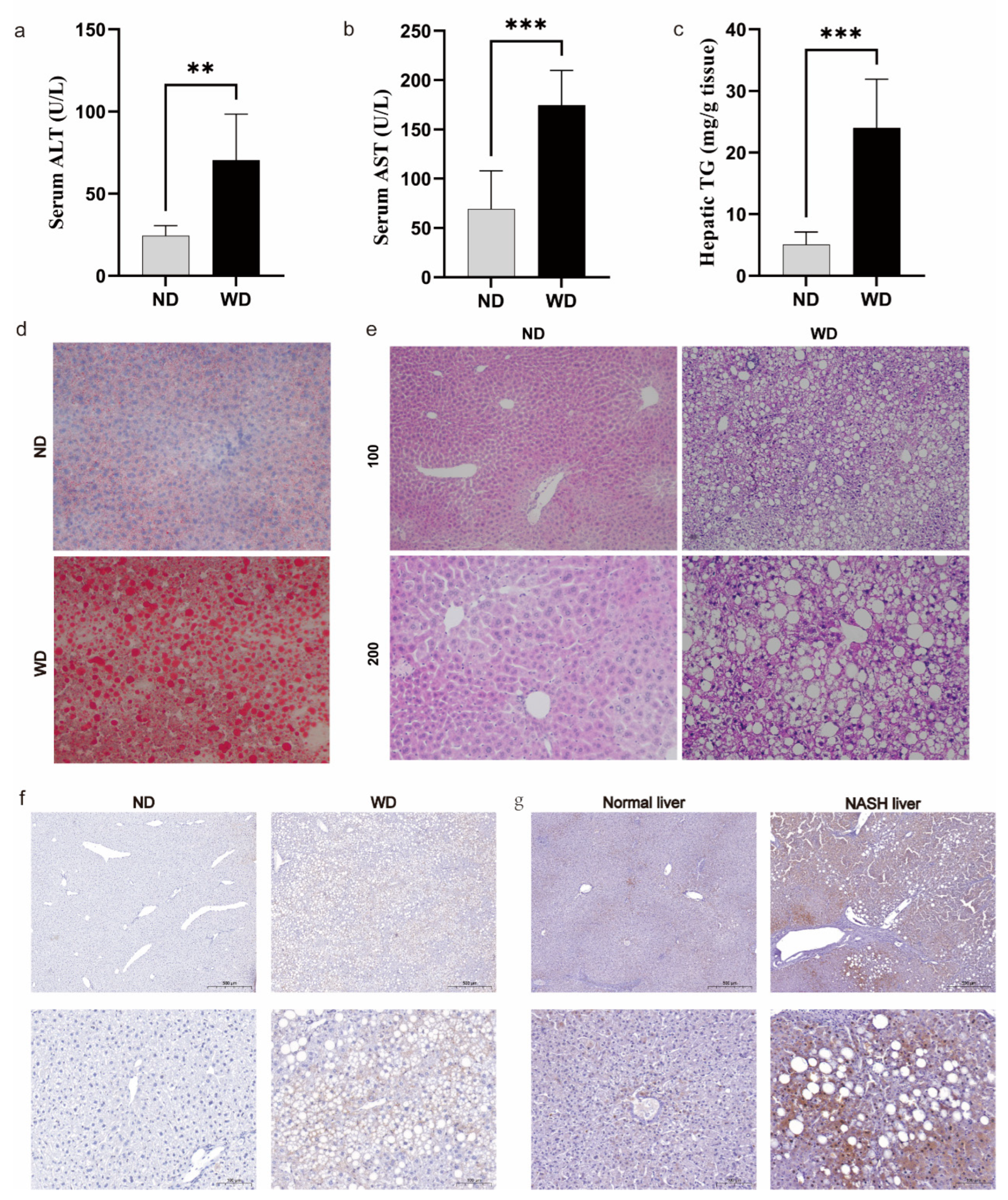
3.7. Validation of FDX1 Expression in NASH Models and Human NASH Samples
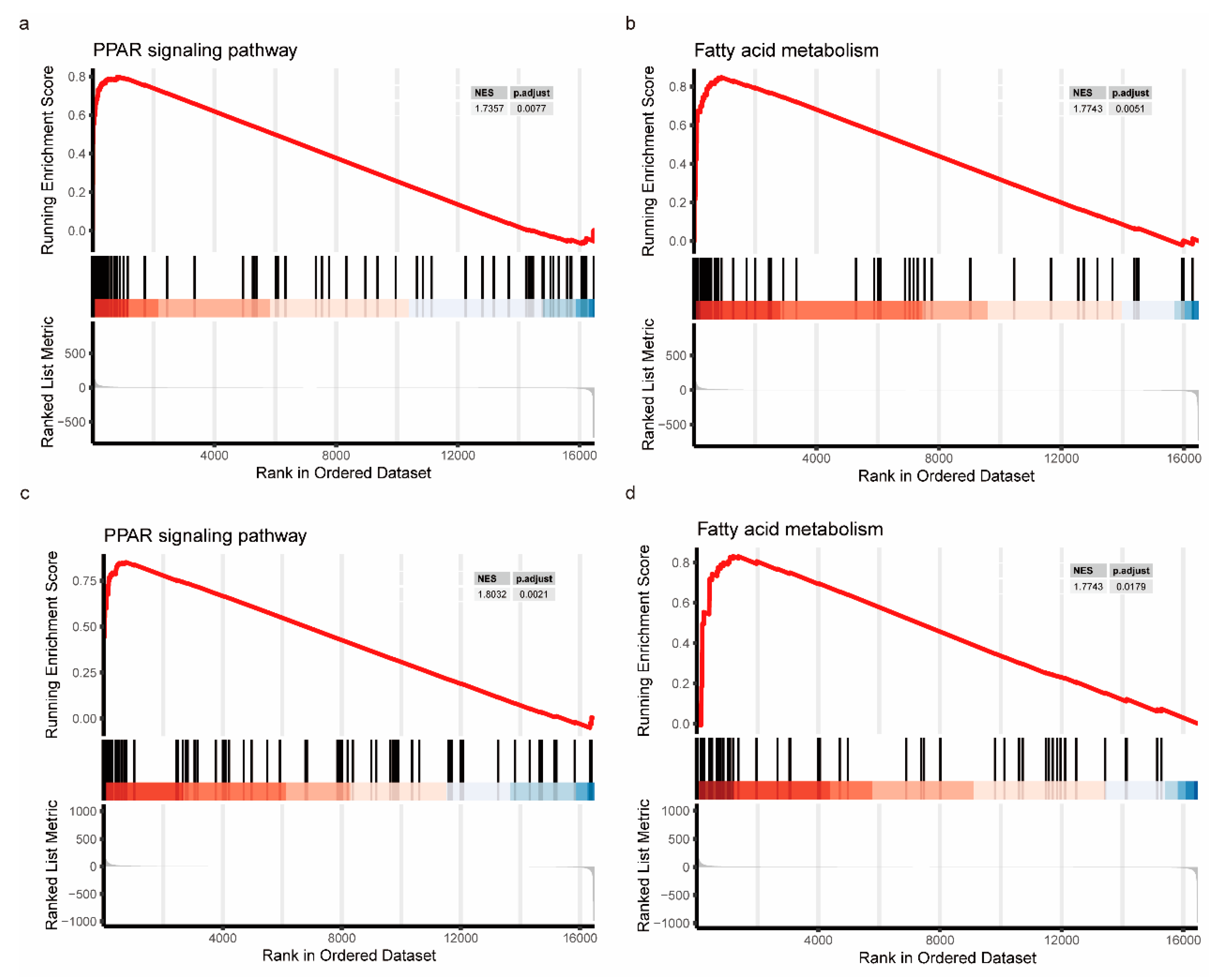
3.8. GSEA Evaluation Based on FDX1 Expression
4. Discussion
Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Foerster, F.; Gairing, S.J.; Müller, L.; Galle, P.R. NAFLD-driven HCC: Safety and efficacy of current and emerging treatment options. J. Hepatol. 2022, 76, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Chen, Z.; Kuang, H.; Liu, T.; Zhu, J.; Zhou, L.; Wang, Q.; Xiong, X.; Meng, Z.; Qiu, X.; et al. Neuregulin 4 suppresses NASH-HCC development by restraining tumor-prone liver microenvironment. Cell Metab. 2022, 34, 1359–1376.e7. [Google Scholar] [CrossRef] [PubMed]
- Perumpail, B.J.; Khan, M.A.; Yoo, E.R.; Cholankeril, G.; Kim, D.; Ahmed, A. Clinical epidemiology and disease burden of nonalcoholic fatty liver disease. World J. Gastroenterol. 2017, 23, 8263–8276. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Rustichelli, A.; Dongiovanni, P. Nutrition and Genetics in NAFLD: The Perfect Binomium. Int. J. Mol. Sci. 2020, 21, 2986. [Google Scholar] [CrossRef]
- Xu, X.; Poulsen, K.L.; Wu, L.; Liu, S.; Miyata, T.; Song, Q.; Wei, Q.; Zhao, C.; Lin, C.; Yang, J. Targeted therapeutics and novel signaling pathways in non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH). Signal Transduct. Target. Ther. 2022, 7, 287. [Google Scholar] [CrossRef]
- Solimando, A.G.; Susca, N.; Argentiero, A.; Brunetti, O.; Leone, P.; De Re, V.; Fasano, R.; Krebs, M.; Petracci, E.; Azzali, I.; et al. Second-line treatments for Advanced Hepatocellular Carcinoma: A Systematic Review and Bayesian Network Meta-analysis. Clin. Exp. Med. 2022, 22, 65–74. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Golub, T.R.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 376, eabq4855, Erratum in Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Zhou, F. Cuproptosis: A new form of programmed cell death. Cell. Mol. Immunol. 2022, 19, 867–868. [Google Scholar] [CrossRef]
- Liu, Q.; Li, R.; Wu, H.; Liang, Z. A novel cuproptosis-related gene model predicts outcomes and treatment responses in pancreatic adenocarcinoma. BMC Cancer 2023, 23, 226. [Google Scholar] [CrossRef]
- Blockhuys, S.; Zhang, X.; Wittung-Stafshede, P. Single-cell tracking demonstrates copper chaperone Atox1 to be required for breast cancer cell migration. Proc. Natl. Acad. Sci. USA 2020, 117, 2014–2019. [Google Scholar] [CrossRef]
- Kim, Y.J.; Tsang, T.; Anderson, G.R.; Posimo, J.M.; Brady, D.C. Inhibition of BCL2 Family Members Increases the Efficacy of Copper Chelation in BRAFV600E-Driven Melanoma. Cancer Res. 2020, 80, 1387–1400. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Liu, Z.; Wang, J.; Zhang, S.; Zhang, Y. Identification of cuprotosis-mediated subtypes, the development of a prognosis model, and influence immune microenvironment in hepatocellular carcinoma. Front. Oncol. 2022, 12, 941211. [Google Scholar] [CrossRef] [PubMed]
- Lánczky, A.; Győrffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef] [PubMed]
- Powers, R.K.; Goodspeed, A.; Pielke-Lombardo, H.; Tan, A.C.; Costello, J.C. GSEA-InContext: Identifying novel and common patterns in expression experiments. Bioinformatics 2018, 34, i555–i564. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Li, C.; Luo, J.; Xu, X.; Zhou, Z.; Ying, S.; Liao, X.; Wu, K. Single cell sequencing revealed the underlying pathogenesis of the development of osteoarthritis. Gene 2020, 757, 144939. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Jiang, X.; Kuang, G.; Gong, X.; Jiang, R.; Xie, T.; Tie, H.; Wu, S.; Wang, T.; Wan, J.; Wang, B. Glycyrrhetinic acid pretreatment attenuates liver ischemia/reperfusion injury via inhibiting TLR4 signaling cascade in mice. Int. Immunopharmacol. 2019, 76, 105870. [Google Scholar] [CrossRef]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Zhang, E.; Dai, F.; Chen, T.; Liu, S.; Xiao, C.; Shen, X. Diagnostic models and predictive drugs associated with cuproptosis hub genes in Alzheimer’s disease. Front. Neurol. 2023, 13, 1064639. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ma, Y.; Guo, X.; Du, Y.; Zhu, Q.; Wang, X.; Duan, C. FDX1 can Impact the Prognosis and Mediate the Metabolism of Lung Adenocarcinoma. Front. Pharmacol. 2021, 12, 749134. [Google Scholar] [CrossRef]
- Bian, Z.; Fan, R.; Xie, L. A Novel Cuproptosis-Related Prognostic Gene Signature and Validation of Differential Expression in Clear Cell Renal Cell Carcinoma. Genes 2022, 13, 851. [Google Scholar] [CrossRef]
- Wang, T.; Liu, Y.; Li, Q.; Luo, Y.; Liu, D.; Li, B. Cuproptosis-related gene FDX1 expression correlates with the prognosis and tumor immune microenvironment in clear cell renal cell carcinoma. Front. Immunol. 2022, 13, 999823. [Google Scholar] [CrossRef]
- Leslie, J.; Mackey, J.B.G.; Jamieson, T.; Ramon-Gil, E.; Drake, T.M.; Fercoq, F.; Clark, W.; Gilroy, K.; Hedley, A.; Nixon, C.; et al. CXCR2 inhibition enables NASH-HCC immunotherapy. Gut 2022, 71, 2093–2106. [Google Scholar] [CrossRef]
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174, Erratum in Nat. Rev. Mol. Cell Biol. 2019, 20, 508. [Google Scholar] [CrossRef] [PubMed]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Asp. Med. 2012, 33, 209–290. [Google Scholar] [CrossRef] [PubMed]
- Xing, W.; Gao, W.; Lv, X.; Zhao, Z.; Mao, G.; Dong, X.; Zhang, Z. The effects of supplementation of probiotics, prebiotics, or synbiotics on patients with non-alcoholic fatty liver disease: A meta-analysis of randomized controlled trials. Front. Nutr. 2022, 9, 1024678. [Google Scholar] [CrossRef]
- Gong, W.J.; Liu, J.Y.; Yin, J.Y.; Cui, J.J.; Xiao, D.; Zhuo, W.; Luo, C.; Liu, R.J.; Li, X.; Zhang, W.; et al. Resistin facilitates metastasis of lung adenocarcinoma through the TLR4/Src/EGFR/PI3K/NF-κB pathway. Cancer Sci. 2018, 109, 2391–2400. [Google Scholar] [CrossRef]
- Ibrahim, S.; Zhu, X.; Luo, X.; Feng, Y.; Wang, J. PIK3R3 regulates ZO-1 expression through the NF-kB pathway in inflammatory bowel disease. Int. Immunopharmacol. 2020, 85, 106610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fan, L.; Wu, J.; Xu, H.; Leung, W.Y.; Fu, K.; Wu, J.; Liu, K.; Man, K.; Yang, X.; et al. Macrophage p38α promotes nutritional steatohepatitis through M1 polarization. J. Hepatol. 2019, 71, 163–174, Erratum in J. Hepatol. 2020, 73, 742–743. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Zhang, S.; Zhang, D.; Tao, Q.; Zhang, T.; Liu, G.; Liu, X.; Zhao, T. Identification of SERPINE1, PLAU and ACTA1 as biomarkers of head and neck squamous cell carcinoma based on integrated bioinformatics analysis. Int. J. Clin. Oncol. 2019, 24, 1030–1041. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Z.; Luan, T.; Wan, J.; Du, H.; Hu, J.; Liu, H.; Gong, X.; Kuang, G.; Wang, B. Elucidating Cuproptosis-Associated Genes in the Progression from Nash to HCC Using Bulk and Single-Cell RNA Sequencing Analyses and Experimental Validation. Medicina 2023, 59, 1639. https://doi.org/10.3390/medicina59091639
Zhao Z, Luan T, Wan J, Du H, Hu J, Liu H, Gong X, Kuang G, Wang B. Elucidating Cuproptosis-Associated Genes in the Progression from Nash to HCC Using Bulk and Single-Cell RNA Sequencing Analyses and Experimental Validation. Medicina. 2023; 59(9):1639. https://doi.org/10.3390/medicina59091639
Chicago/Turabian StyleZhao, Zizuo, Tiankuo Luan, Jingyuan Wan, Hui Du, Jun Hu, Hao Liu, Xia Gong, Ge Kuang, and Bin Wang. 2023. "Elucidating Cuproptosis-Associated Genes in the Progression from Nash to HCC Using Bulk and Single-Cell RNA Sequencing Analyses and Experimental Validation" Medicina 59, no. 9: 1639. https://doi.org/10.3390/medicina59091639
APA StyleZhao, Z., Luan, T., Wan, J., Du, H., Hu, J., Liu, H., Gong, X., Kuang, G., & Wang, B. (2023). Elucidating Cuproptosis-Associated Genes in the Progression from Nash to HCC Using Bulk and Single-Cell RNA Sequencing Analyses and Experimental Validation. Medicina, 59(9), 1639. https://doi.org/10.3390/medicina59091639