

The Prognostic and Therapeutic Role of Histone Acetylation Modification in LIHC Development and Progression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Describe the Expression Landscape of Histone Acetylation Modulator Genes
2.2. Development of a Prognostic Signature Based on Histone Acetylation Modulator Genes
2.3. Verification of the Histone Acetylation Modulator Gene-Based Signature
2.4. Functional Analyses and Genome Analyses
2.5. Analysis of Immune Infiltration and Prediction of Immunotherapy
3. Results
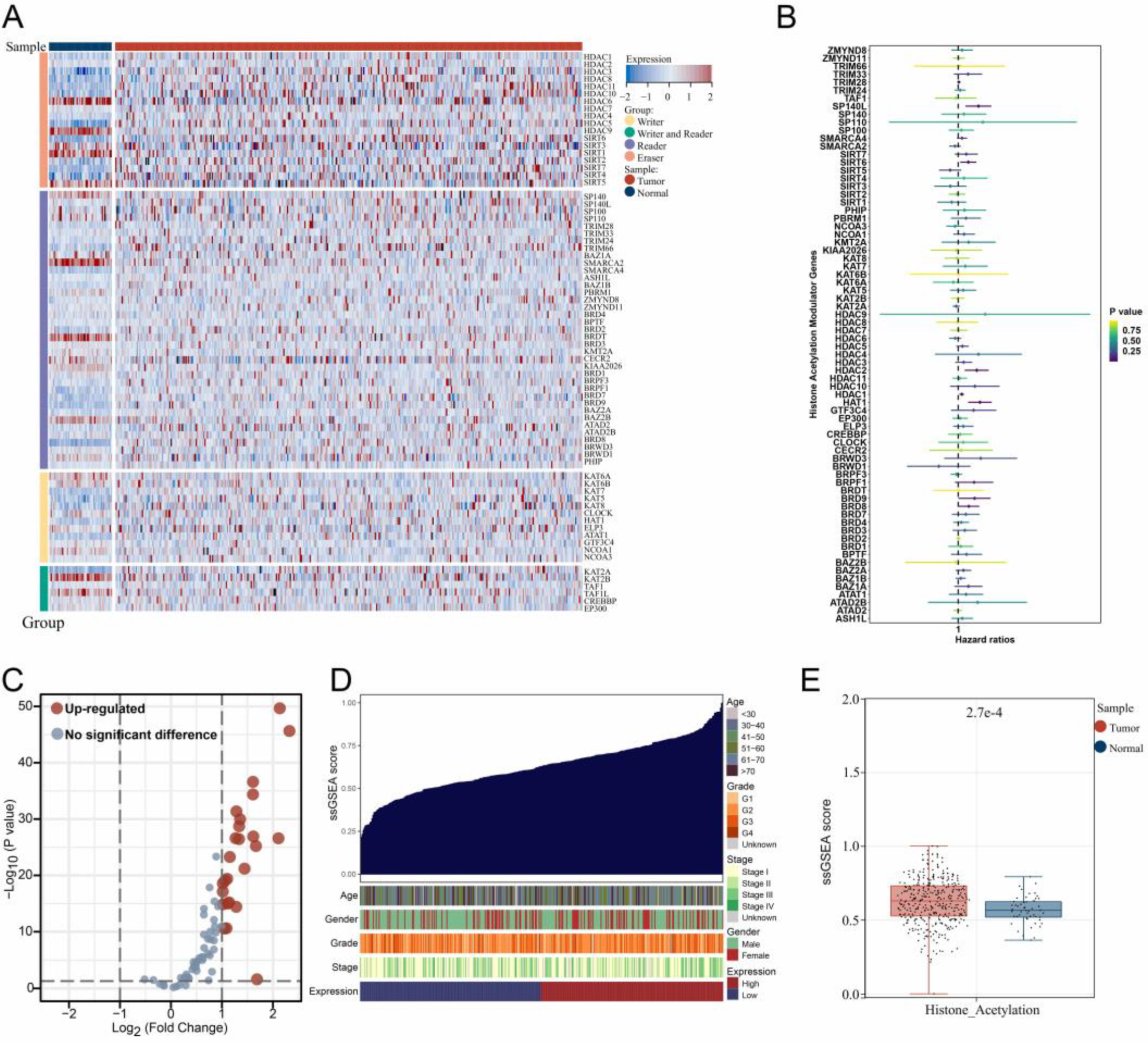
3.1. The Expression Landscape of Histone Acetylation Modulator Genes in Samples
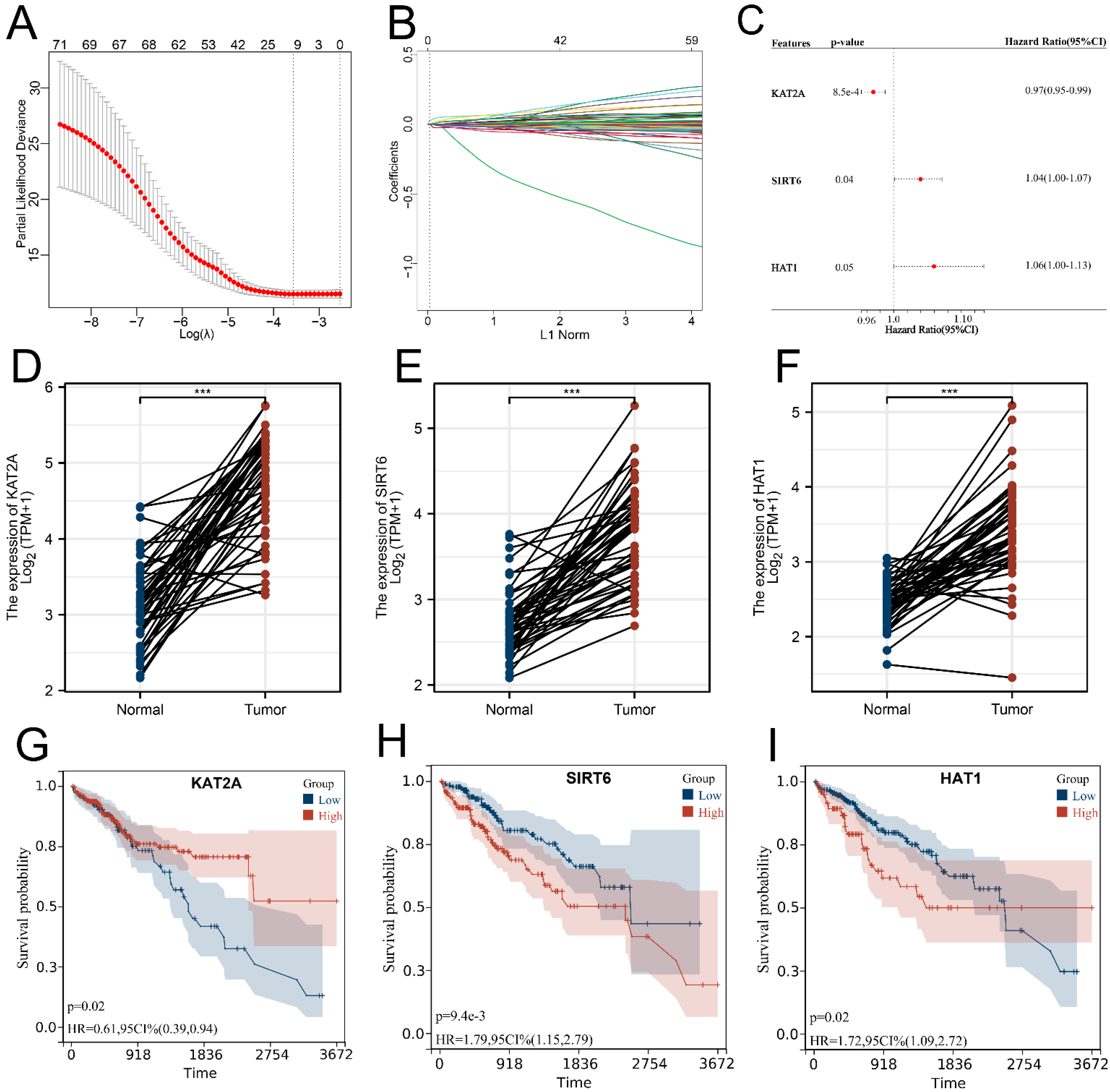
3.2. Signature Construction Based on Histone Acetylation Modulator Genes
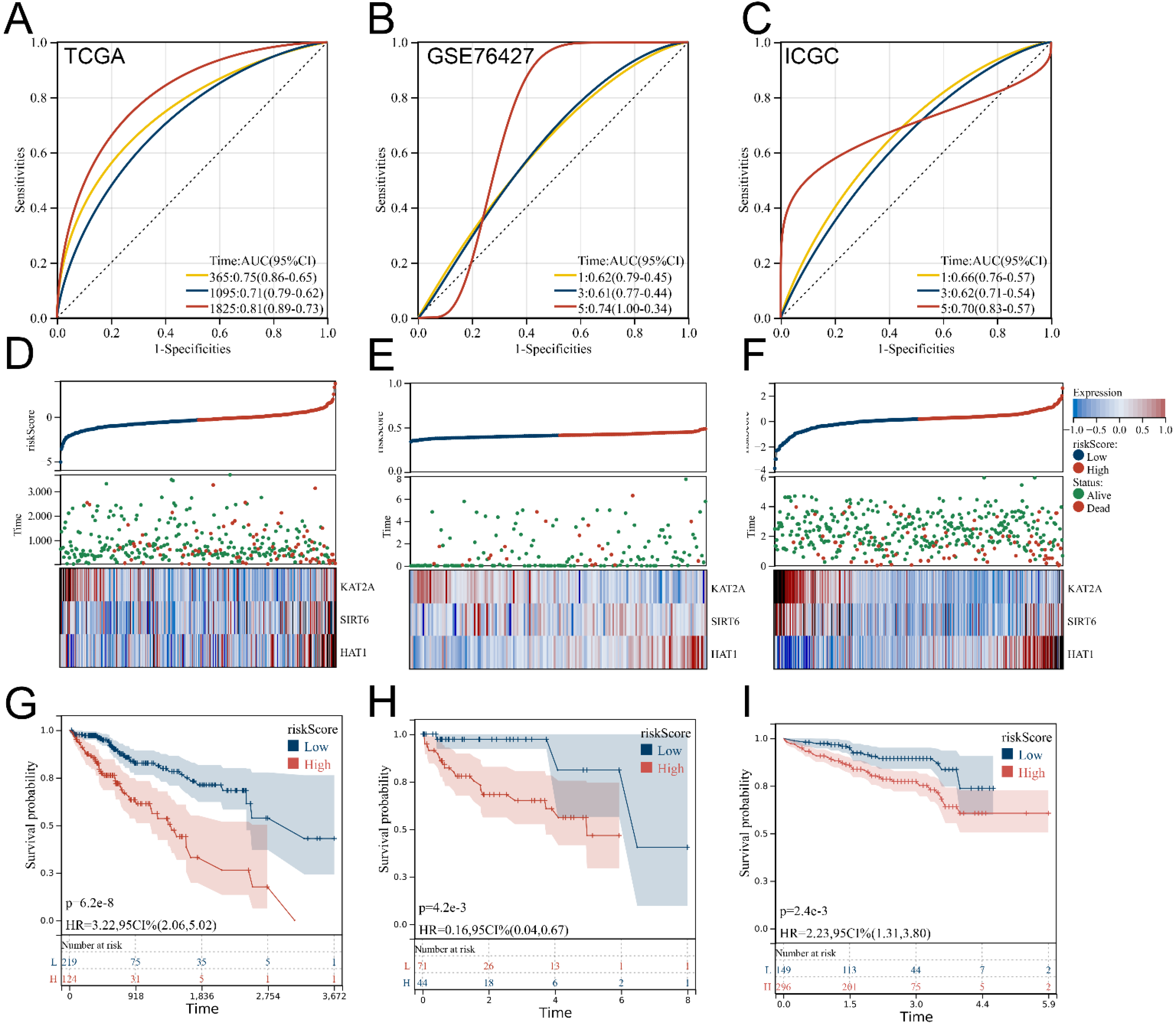
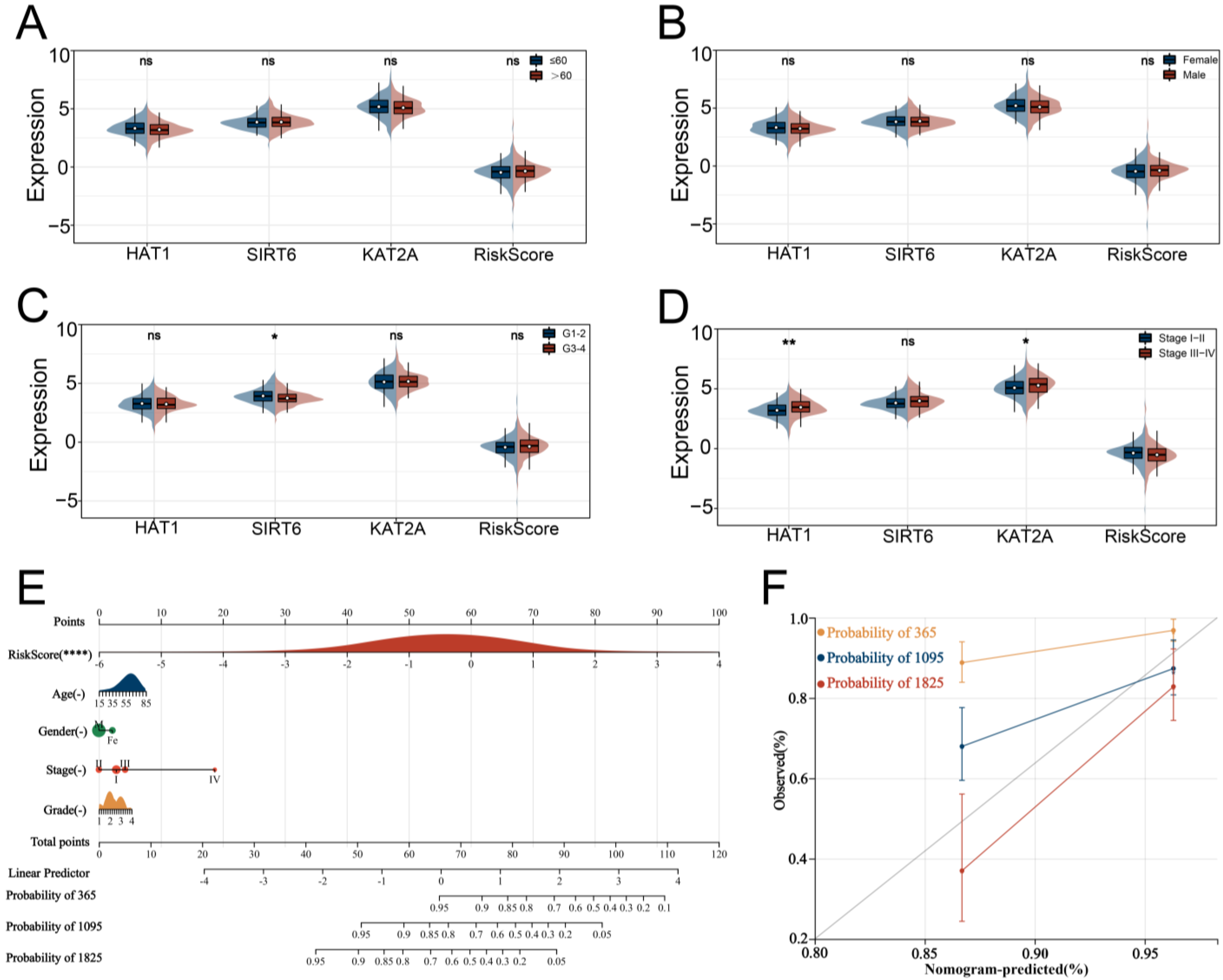
3.3. Verification and Evaluation of the Signature
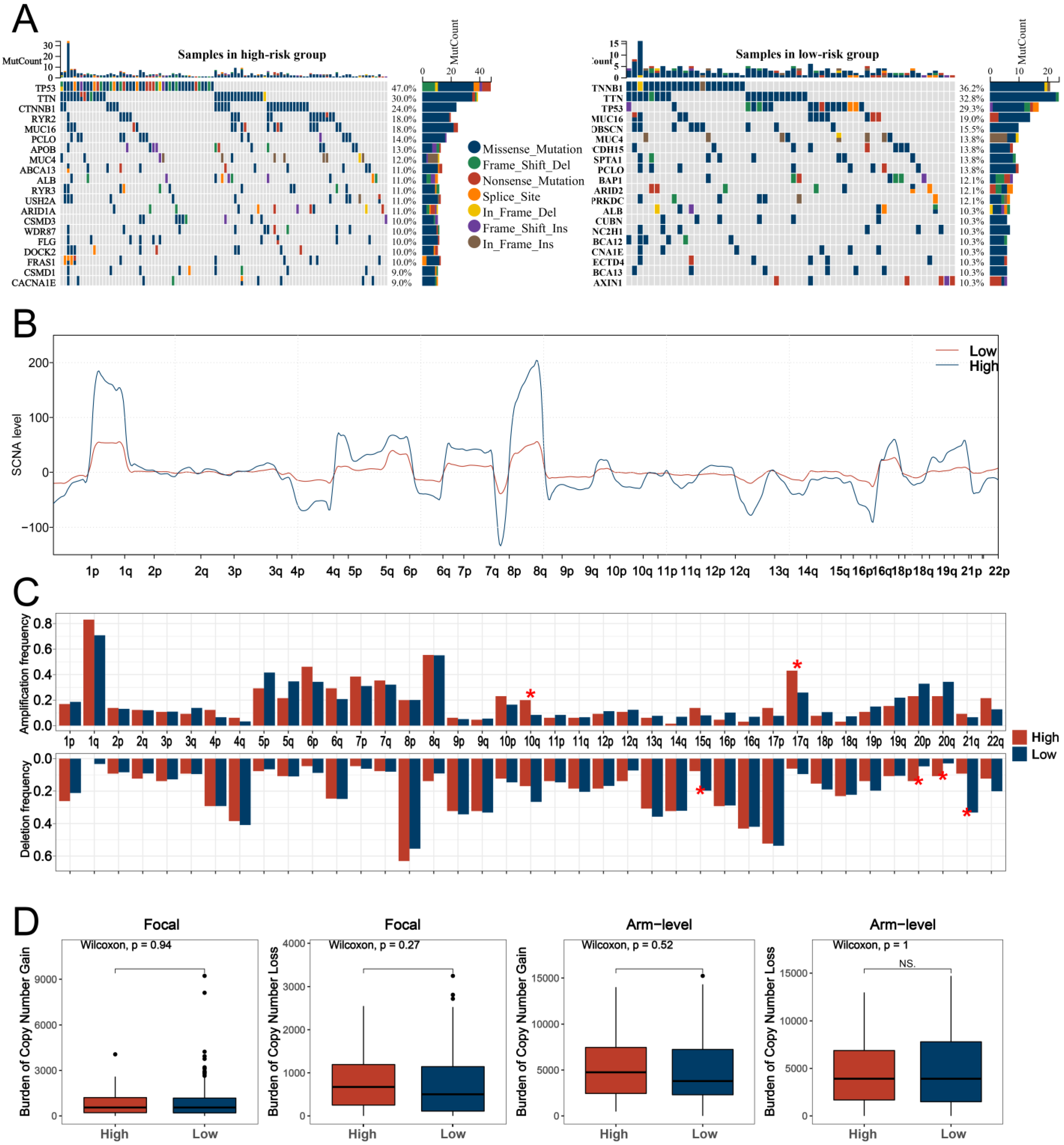
3.4. GO and KEGG Analyses and Genomic Features Analysis
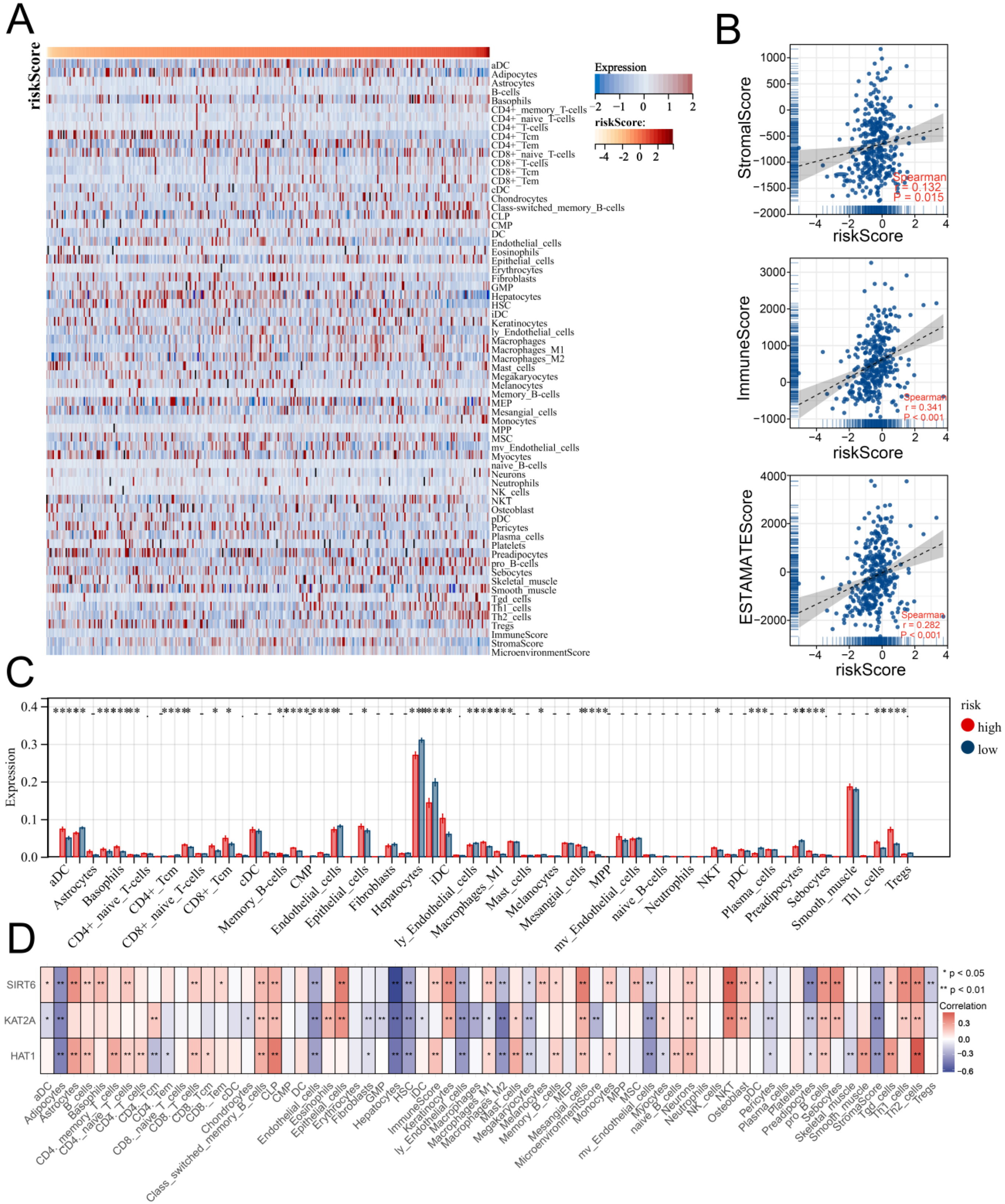
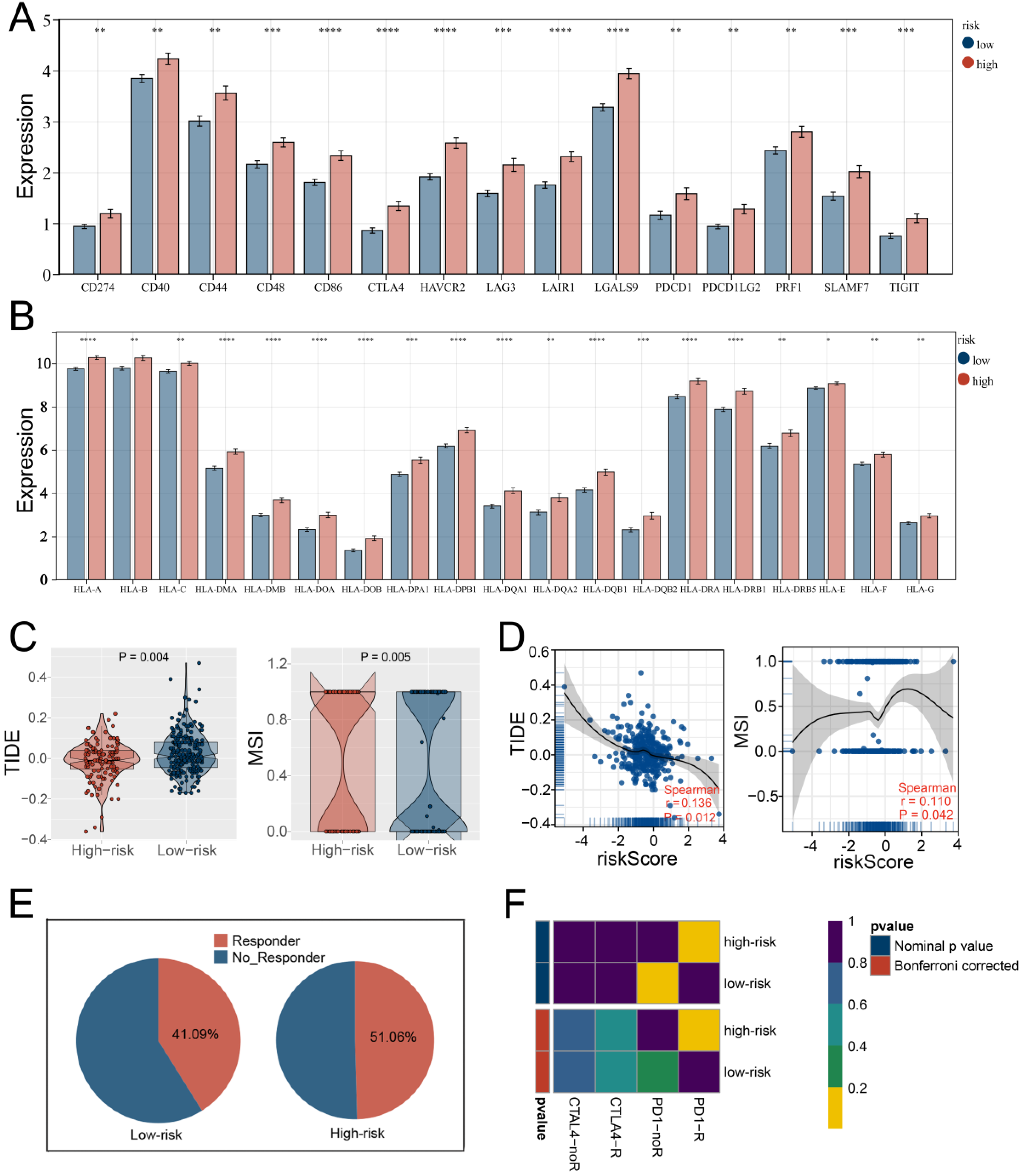
3.5. Multiple Immune Analyses Revealed High HARS Patients More Sensitive to Immunotherapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73 (Suppl. S1), 4–13. [Google Scholar] [CrossRef]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; De Baere, T.; Kulik, L.; Haber, P.K.; Greten, T.F.; Meyer, T.; Lencioni, R. Locoregional therapies in the era of molecular and immune treatments for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 293–313. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 7. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Karpinets, T.V.; Foy, B.D. Tumorigenesis: The adaptation of mammalian cells to sustained stress environment by epigenetic alterations and succeeding matched mutations. Carcinogenesis 2005, 26, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Ansari, J.; Shackelford, R.E.; El-Osta, H. Epigenetics in non-small cell lung cancer: From basics to therapeutics. Transl. Lung Cancer Res. 2016, 5, 155–171. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of RNA Synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef]
- Nagaraju, G.P.; Dariya, B.; Kasa, P.; Peela, S.; El-Rayes, B.F. Epigenetics in hepatocellular carcinoma. Semin. Cancer Biol. 2022, 86, 622–632. [Google Scholar] [CrossRef]
- Wahid, B.; Ali, A.; Rafique, S.; Idrees, M. New Insights into the Epigenetics of Hepatocellular Carcinoma. BioMed Res. Int. 2017, 2017, 1609575. [Google Scholar] [CrossRef]
- Mamdani, H.; Jalal, S.I. Histone Deacetylase Inhibition in Non-small Cell Lung Cancer: Hype or Hope? Front. Cell Dev. Biol. 2020, 8, 582370. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Zhou, J.; Jiang, J.; Yuan, J.; Zhang, Y.; Wei, X.; Loo, N.; Wang, Y.; Pan, Y.; Zhang, T.; et al. Genomic characterization of genes encoding histone acetylation modulator proteins identifies therapeutic targets for cancer treatment. Nat. Commun. 2019, 10, 733. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Brunet, J.P.; Tamayo, P.; Golub, T.R.; Mesirov, J.P. Subclass mapping: Identifying common subtypes in independent disease data sets. PLoS ONE 2007, 2, e1195. [Google Scholar] [CrossRef] [PubMed]
- Greten, T.F.; Wang, X.W.; Korangy, F. Current concepts of immune based treatments for patients with HCC: From basic science to novel treatment approaches. Gut 2015, 64, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [PubMed]
- Sprinzl, M.F.; Galle, P.R. Current progress in immunotherapy of hepatocellular carcinoma. J. Hepatol. 2017, 66, 482–484. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Minn, A.J. Combination Cancer Therapy with Immune Checkpoint Blockade: Mechanisms and Strategies. Immunity 2018, 48, 417–433. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Di Cerbo, V.; Schneider, R. Cancers with wrong HATs: The impact of acetylation. Brief. Funct. Genom. 2013, 12, 231–243. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Gui, C.Y.; Ngo, L.; Xu, W.S.; Richon, V.M.; Marks, P.A. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc. Natl. Acad. Sci. USA 2004, 101, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Gray, S.G.; Greene, C.M.; Lawless, M.W. Unmasking the pathological and therapeutic potential of histone deacetylases for liver cancer. Expert Rev. Gastroenterol. Hepatol. 2019, 13, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Arede, L.; Pina, C. Buffering noise: KAT2A modular contributions to stabilization of transcription and cell identity in cancer and development. Exp. Hematol. 2021, 93, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, F.; Carafa, V.; Favale, G.; Altucci, L.; Mai, A.; Rotili, D. The Two-Faced Role of SIRT6 in Cancer. Cancers 2021, 13, 1156. [Google Scholar] [CrossRef] [PubMed]
- Han, N.; Shi, L.; Guo, Q.; Sun, W.; Yu, Y.; Yang, L.; Zhang, X.; Zhang, M. HAT1 induces lung cancer cell apoptosis via up regulating Fas. Oncotarget 2017, 8, 89970–89977. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, J.; Han, S.; Gu, J.; Wu, C.; Mu, X. The Prognostic and Therapeutic Role of Histone Acetylation Modification in LIHC Development and Progression. Medicina 2023, 59, 1682. https://doi.org/10.3390/medicina59091682
Gao J, Han S, Gu J, Wu C, Mu X. The Prognostic and Therapeutic Role of Histone Acetylation Modification in LIHC Development and Progression. Medicina. 2023; 59(9):1682. https://doi.org/10.3390/medicina59091682
Chicago/Turabian StyleGao, Ji, Sheng Han, Jian Gu, Chen Wu, and Xiaoxin Mu. 2023. "The Prognostic and Therapeutic Role of Histone Acetylation Modification in LIHC Development and Progression" Medicina 59, no. 9: 1682. https://doi.org/10.3390/medicina59091682
APA StyleGao, J., Han, S., Gu, J., Wu, C., & Mu, X. (2023). The Prognostic and Therapeutic Role of Histone Acetylation Modification in LIHC Development and Progression. Medicina, 59(9), 1682. https://doi.org/10.3390/medicina59091682