
Aripiprazole Attenuates Cognitive Impairments Induced by Lipopolysaccharide in Rats through the Regulation of Neuronal Inflammation, Oxidative Stress, and Apoptosis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Vehicle
2.3. Experimental Design
2.4. Novel Object Recognition (NOR)
2.5. Brain Samples Collection and Preparation of Tissue Homogenate
2.6. Analysis of Neuroinflammation Cytokines in Brain Homogenate
2.7. Measurement of Oxidative Parameters in Brain Homogenate
2.8. Determination of Apoptotic Proteins in Brain Homogenate
2.9. Statistical Analysis
3. Results
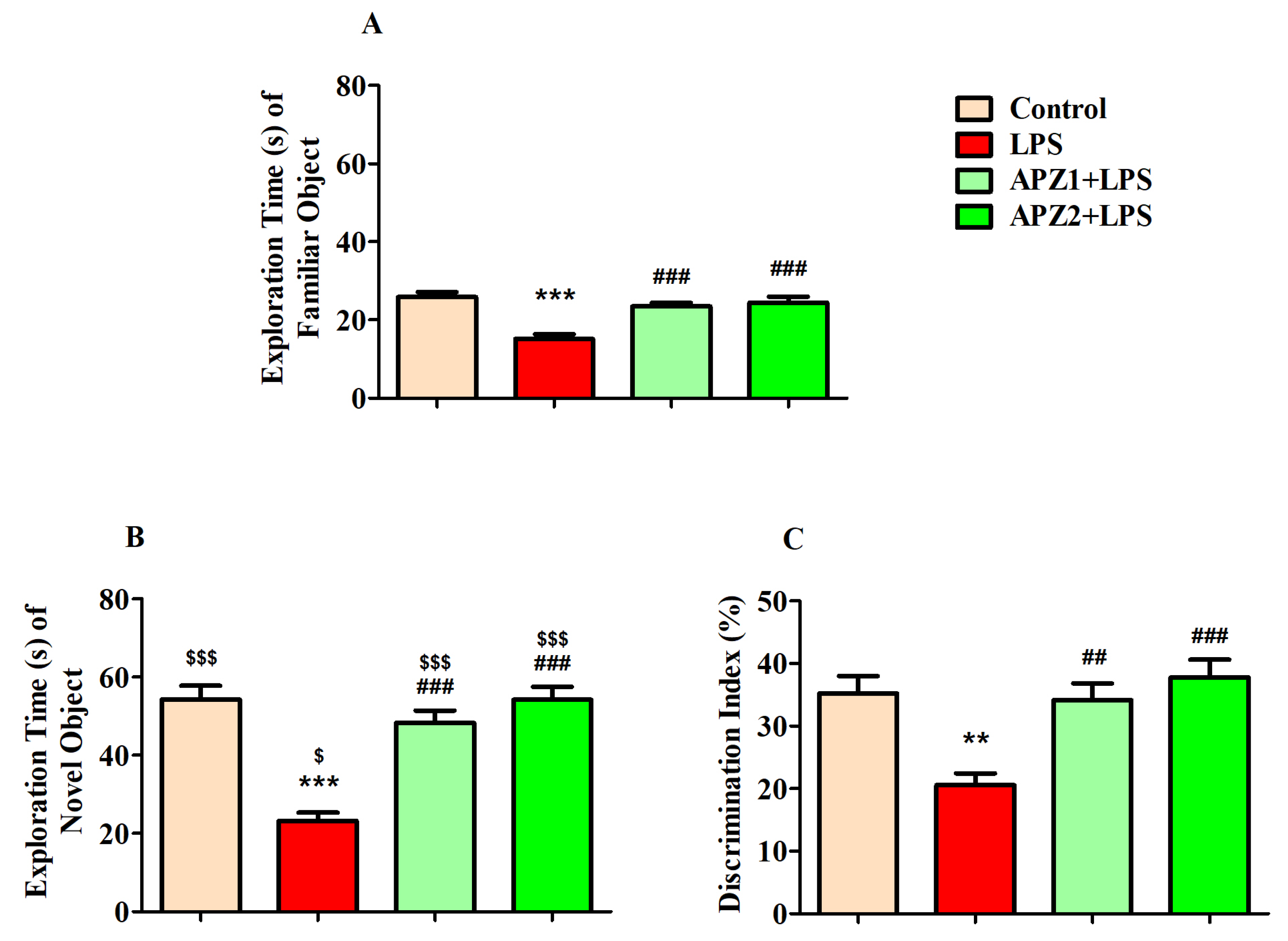
3.1. Effect of APZ on Cognitive Performance in NOR Parameters
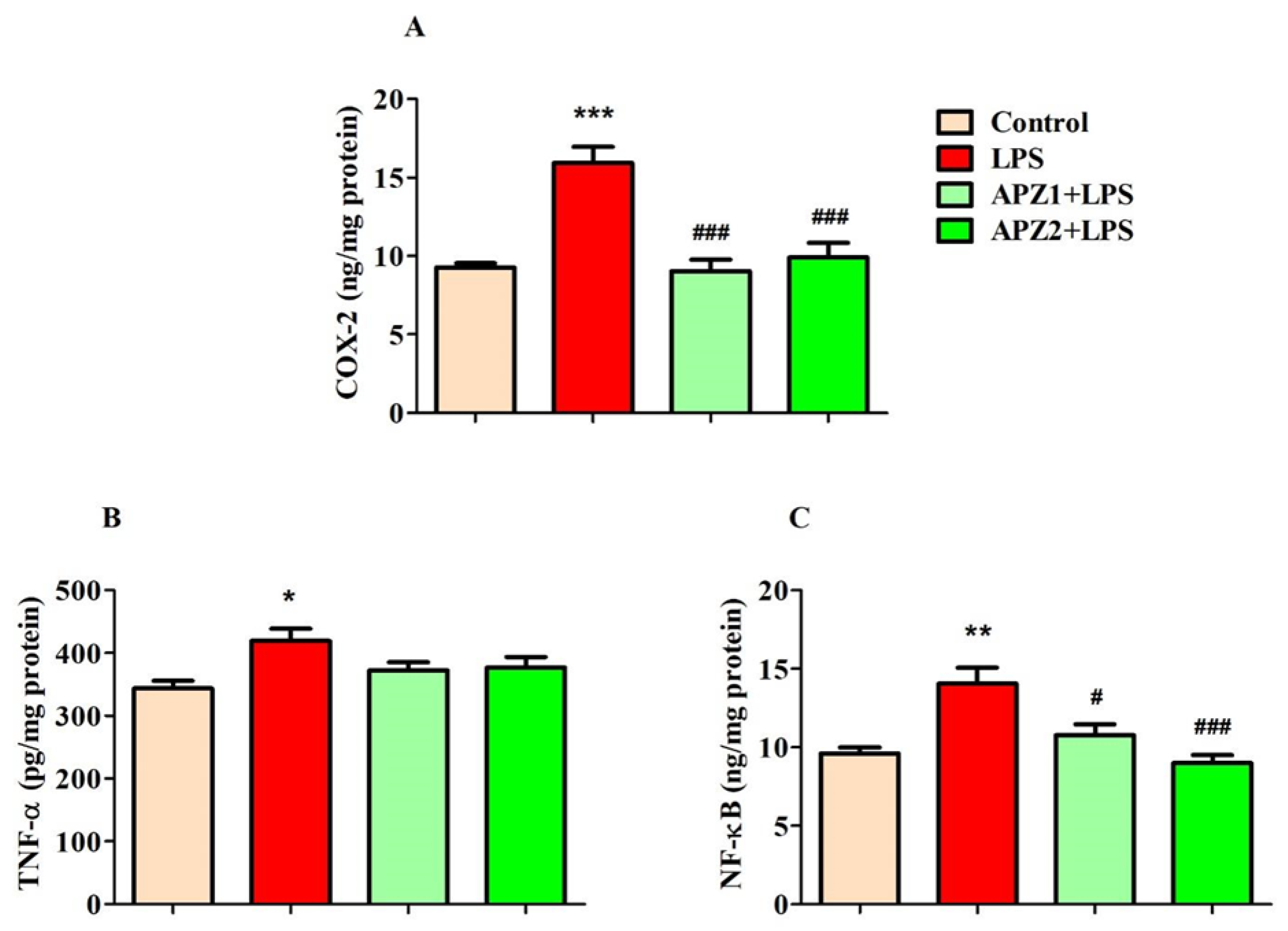
3.2. Evaluating the Influence of APZ and LPS on Neuroinflammatory Parameters
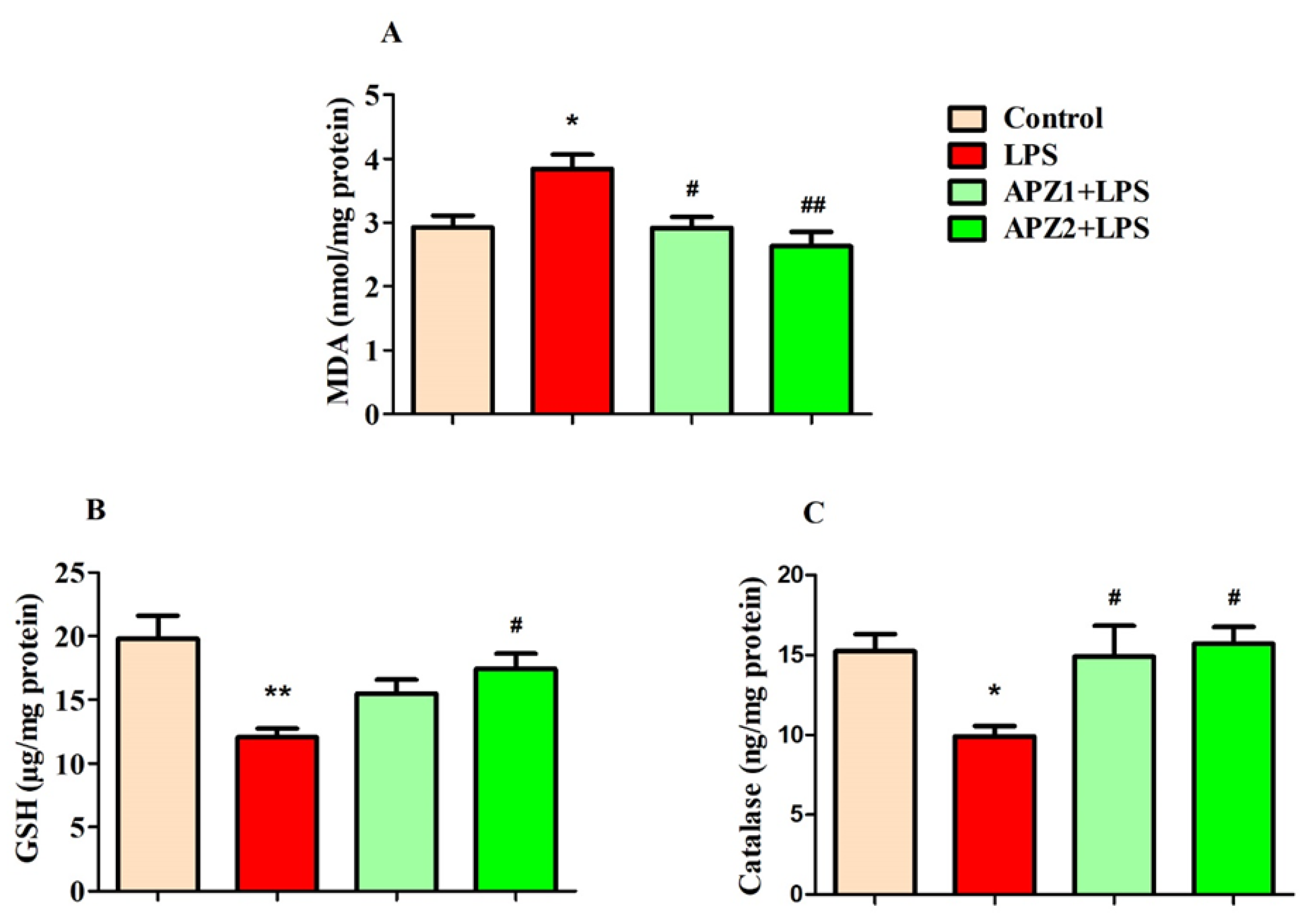
3.3. Evaluating the Influence of APZ and LPS on Oxidative Parameters
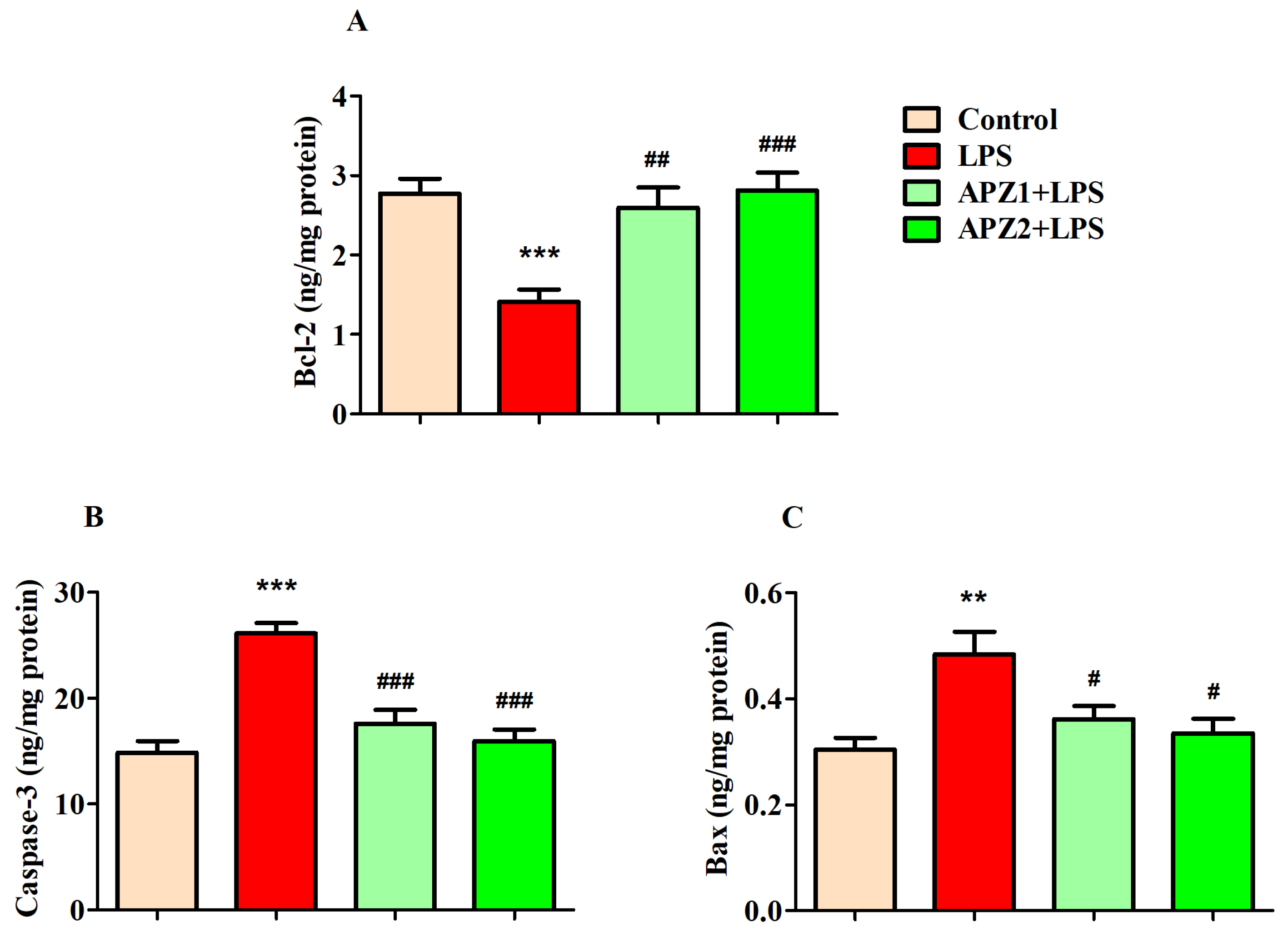
3.4. Evaluating the Influence of APZ and LPS on Apoptosis Parameters
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mohamed, W.; Kumar, J.; Alghamdi, B.S.; Soliman, A.H.; Toshihide, Y. Neurodegeneration and inflammation crosstalk: Therapeutic targets and perspectives. IBRO Neurosci. Rep. 2022, 14, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Gale, S.A.; Acar, D.; Daffner, K.R. Dementia. Am. J. Med. 2018, 131, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, L.; Zhang, C.; Guo, Y.; Li, J.; Wu, C.; Jiao, J.; Zheng, H. Ginsenoside Rg1 improves Alzheimer’s disease by regulating oxidative stress, apoptosis, and neuroinflammation through Wnt/GSK-3β/β-catenin signaling pathway. Chem. Biol. Drug Des. 2022, 99, 884–896. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, R.A.; Keefe, R.S.E.; McGuire, P.K. Cognitive impairment in schizophrenia: Aetiology, pathophysiology, and treatment. Mol. Psychiatry 2023, 28, 1902–1918. [Google Scholar] [CrossRef] [PubMed]
- Ballard, C.; Kales, H.C.; Lyketsos, C.; Aarsland, D.; Creese, B.; Mills, R.; Williams, H.; Sweet, R.A. Psychosis in Alzheimer’s disease. Curr. Neurol. Neurosci. Rep. 2020, 20, 57. [Google Scholar] [CrossRef] [PubMed]
- Tariot, P.N.; Cummings, J.L.; Soto-Martin, M.E.; Ballard, C.; Erten-Lyons, D.; Sultzer, D.L.; Devanand, D.P.; Weintraub, D.; McEvoy, B.; Youakim, J.M.; et al. Trial of pimavanserin in dementia-related psychosis. N. Engl. J. Med. 2021, 385, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Gettu, N.; Saadabadi, A. Aripiprazole. In StatPearls; StatPearls Publishing: St. Petersburg, FL, USA, 2023. [Google Scholar]
- Preda, A.; Shapiro, B.B. A safety evaluation of aripiprazole in the treatment of schizophrenia. Expert Opin. Drug Saf. 2020, 19, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Bervoets, C.; Morrens, M.; Vansteelandt, K.; Kok, F.; de Patoul, A.; Halkin, V.; Pitsi, D.; Constant, E.; Peuskens, J.; Sabbe, B. Effect of aripiprazole on verbal memory and fluency in schizophrenic patients: Results from the ESCAPE study. CNS Drugs 2012, 26, 975–982. [Google Scholar] [CrossRef]
- Maat, A.; Cahn, W.; Gijsman, H.J.; Hovens, J.E.; Kahn, R.S.; Aleman, A. Open, randomized trial of the effects of aripiprazole versus risperidone on social cognition in schizophrenia. Eur. Neuropsychopharmacol. 2014, 24, 575–584. [Google Scholar] [CrossRef]
- Peitl, V.; Badžim, V.A.; Šiško Markoš, I.; Rendulić, A.; Matešić, K.; Karlović, D. Improvements of frontotemporal cerebral blood flow and cognitive functioning in patients with first episode of schizophrenia treated with long-acting aripiprazole. J. Clin. Psychopharmacol. 2021, 41, 638–643. [Google Scholar] [CrossRef]
- Lu, C.; Li, S.; Kang, L.; Li, Q.; Chen, H.; Lin, Y.; Zhang, H.; Tang, Z.; Bai, M.; Xiong, P. Aripiprazole combined with nerve growth factor improves cognitive function in mice with schizophrenia model. Neurosci. Lett. 2023, 812, 137410. [Google Scholar] [CrossRef] [PubMed]
- Gupta, T.; Singh, V.; Hefnawy, M.; Alanazi, M.M.; Alsuwayt, B.; Kabra, A.; Kumar, A.; Pasricha, C.; Singh, R. Ameliorating the role of aripiprazole in memory deficits induced by intracerebroventricular streptozotocin-induced dementia of Alzheimer’s type. ACS Omega 2023, 8, 25295–25302. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Kim, H.Y.; Lee, Y.S.; Heo, H.J.; Shin, H.K.; Lee, W.S.; Hong, K.W.; Kim, C.D. Augmented improvement of cognition and memory by aripiprazole add-on for cilostazol treatment in the chronic cerebral hypoperfusion mouse model. Behav. Brain Res. 2019, 365, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, P.; Kus, K.; Murawiecka, P.; Słodzińska, I.; Zaprutko, T.; Kopciuch, D.; Paczkowska, A.; Nowakowska, E. Memory deterioration based on the tobacco smoke exposure and methylazoxymethanol acetate administration vs. aripiprazole, olanzapine and enrichment environment conditions. Pharmacol. Biochem. Behav. 2020, 189, 172855. [Google Scholar] [CrossRef] [PubMed]
- Juncal-Ruiz, M.; Riesco-Dávila, L.; Ortiz-García de la Foz, V.; Martínez-Garcia, O.; Ramírez-Bonilla, M.; Ocejo-Viñals, J.G.; Leza, J.C.; López-Hoyos, M.; Crespo-Facorro, B. Comparison of the anti-inflammatory effect of aripiprazole and risperidone in 75 drug-naïve first episode psychosis individuals: A 3 months randomized study. Schizophr. Res. 2018, 202, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Drexhage, R.C.; Hoogenboezem, T.A.; Cohen, D.; Versnel, M.A.; Nolen, W.A.; van Beveren, N.J.; Drexhage, H.A. An activated set point of T-cell and monocyte inflammatory networks in recent-onset schizophrenia patients involves both pro- and anti-inflammatory forces. Int. J. Neuropsychopharmacol. 2011, 14, 746–755. [Google Scholar] [CrossRef]
- Stapel, B.; Sieve, I.; Falk, C.S.; Bleich, S.; Hilfiker-Kleiner, D.; Kahl, K.G. Second generation atypical antipsychotics olanzapine and aripiprazole reduce expression and secretion of inflammatory cytokines in human immune cells. J. Psychiatr. Res. 2018, 105, 95–102. [Google Scholar] [CrossRef]
- Dashti, S.; Nahavandi, A. Neuroprotective effects of aripiprazole in stress-induced depressive-like behavior: Possible role of CACNA1C. J. Chem. Neuroanat. 2022, 126, 102170. [Google Scholar] [CrossRef]
- Sun, M.; Shen, X.; Ma, Y. Rehmannioside A attenuates cognitive deficits in rats with vascular dementia (VD) through suppressing oxidative stress, inflammation and apoptosis. Biomed. Pharmacother. 2019, 120, 109492. [Google Scholar] [CrossRef]
- Nakhal, M.M.; Jayaprakash, P.; Aburuz, S.; Sadek, B.; Akour, A. Canagliflozin ameliorates oxidative stress and autistic-like features in valproic-acid-induced autism in rats: Comparison with aripiprazole action. Pharmaceuticals 2023, 16, 769. [Google Scholar] [CrossRef]
- Arunagiri, P.; Rajeshwaran, K.; Shanthakumar, J.; Tamilselvan, T.; Balamurugan, E. Combination of omega-3 Fatty acids, lithium, and aripiprazole reduces oxidative stress in brain of mice with mania. Biol. Trace Elem. Res. 2014, 160, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Seki, Y.; Kato, T.A.; Monji, A.; Mizoguchi, Y.; Horikawa, H.; Sato-Kasai, M.; Yoshiga, D.; Kanba, S. Pretreatment of aripiprazole and minocycline, but not haloperidol, suppresses oligodendrocyte damage from interferon-γ-stimulated microglia in co-culture model. Schizophr. Res. 2013, 151, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Mani, V.; Rashed Almutairi, S. Impact of levetiracetam on cognitive impairment, neuroinflammation, oxidative stress, and neuronal apoptosis caused by lipopolysaccharides in rats. Saudi Pharm. J. 2023, 31, 101728. [Google Scholar] [CrossRef] [PubMed]
- Mani, V.; Alshammeri, B.S. Quetiapine moderates doxorubicin-induced cognitive deficits: Influence of oxidative stress, neuroinflammation, and cellular apoptosis. Int. J. Mol. Sci. 2023, 24, 11525. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Kar, S.K.; Shukla, R. Cognitive deficits in schizophrenia: Understanding the biological correlates and remediation strategies. Clin. Psychopharmacol. Neurosci. 2018, 16, 7–17. [Google Scholar] [CrossRef]
- Feigenson, K.A.; Kusnecov, A.W.; Silverstein, S.M. Inflammation and the two-hit hypothesis of schizophrenia. Neurosci. Biobehav. Rev. 2014, 38, 72–93. [Google Scholar] [CrossRef]
- Müller, N. Inflammation in schizophrenia: Pathogenetic aspects and therapeutic considerations. Schizophr. Bull. 2018, 44, 973–982. [Google Scholar] [CrossRef]
- Yang, L.; Zhou, R.; Tong, Y.; Chen, P.; Shen, Y.; Miao, S.; Liu, X. Neuroprotection by dihydrotestosterone in LPS-induced neuroinflammation. Neurobiol. Dis. 2020, 140, 104814. [Google Scholar] [CrossRef]
- Yoo, S.; Kim, M.Y.; Cho, J.Y. Syk and Src-targeted anti-inflammatory activity of aripiprazole, an atypical antipsychotic. Biochem. Pharmacol. 2018, 148, 1–12. [Google Scholar] [CrossRef]
- Sobiś, J.; Kunert, Ł.; Rykaczewska-Czerwińska, M.; Świętochowska, E.; Gorczyca, P. The effect of aripiprazole on leptin levels of patients with chronic schizophrenia and a comparison of leptin, acute phase protein, and cytokine levels with regard to body mass and body composition indexes. Endokrynol. Polska 2022, 73, 35–42. [Google Scholar] [CrossRef]
- Salim, S. Oxidative stress and the central nervous system. J. Pharmacol. Exp. Ther. 2017, 360, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Flatow, J.; Buckley, P.; Miller, B.J. Meta-analysis of oxidative stress in schizophrenia. Biol. Psychiatry 2013, 74, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Do, K.Q. Linking early-life NMDAR hypofunction and oxidative stress in schizophrenia pathogenesis. Nat. Rev. Neurosci. 2016, 17, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Koga, M.; Serritella, A.V.; Sawa, A.; Sedlak, T.W. Implications for reactive oxygen species in schizophrenia pathogenesis. Schizophr. Res. 2016, 176, 52–71. [Google Scholar] [CrossRef] [PubMed]
- Radi, E.; Formichi, P.; Battisti, C.; Federico, A. Apoptosis and oxidative stress in neurodegenerative diseases. J. Alzheimers Dis. 2014, 42, S125–S152. [Google Scholar] [CrossRef] [PubMed]
- Jarskog, L.F.; Gilmore, J.H.; Selinger, E.S.; Lieberman, J.A. Cortical Bcl-2 protein expression and apoptotic regulation in schizophrenia. Biol. Psychiatry 2000, 48, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Dirican, E.; Özcan, H.; Karabulut Uzunçakmak, S.; Takım, U. Evaluation expression of the Caspase-3 and Caspase-9 apoptotic genes in schizophrenia patients. Clin. Psychopharmacol. Neurosci. 2023, 21, 171–178. [Google Scholar] [CrossRef]
- Abekawa, T.; Ito, K.; Nakagawa, S.; Nakato, Y.; Koyama, T. Effects of aripiprazole and haloperidol on progression to schizophrenia-like behavioural abnormalities and apoptosis in rodents. Schizophr. Res. 2011, 125, 77–87. [Google Scholar] [CrossRef]
- Park, S.W.; Lee, J.G.; Ha, E.K.; Choi, S.M.; Cho, H.Y.; Seo, M.K.; Kim, Y.H. Differential effects of aripiprazole and haloperidol on BDNF-mediated signal changes in SH-SY5Y cells. Eur. Neuropsychopharmacol. 2009, 19, 356–362. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mani, V.; Alshammeri, B.S. Aripiprazole Attenuates Cognitive Impairments Induced by Lipopolysaccharide in Rats through the Regulation of Neuronal Inflammation, Oxidative Stress, and Apoptosis. Medicina 2024, 60, 46. https://doi.org/10.3390/medicina60010046
Mani V, Alshammeri BS. Aripiprazole Attenuates Cognitive Impairments Induced by Lipopolysaccharide in Rats through the Regulation of Neuronal Inflammation, Oxidative Stress, and Apoptosis. Medicina. 2024; 60(1):46. https://doi.org/10.3390/medicina60010046
Chicago/Turabian StyleMani, Vasudevan, and Bander Shehail Alshammeri. 2024. "Aripiprazole Attenuates Cognitive Impairments Induced by Lipopolysaccharide in Rats through the Regulation of Neuronal Inflammation, Oxidative Stress, and Apoptosis" Medicina 60, no. 1: 46. https://doi.org/10.3390/medicina60010046