
Design of an Epitope-Based Vaccine Against MERS-CoV

Abstract
1. Introduction
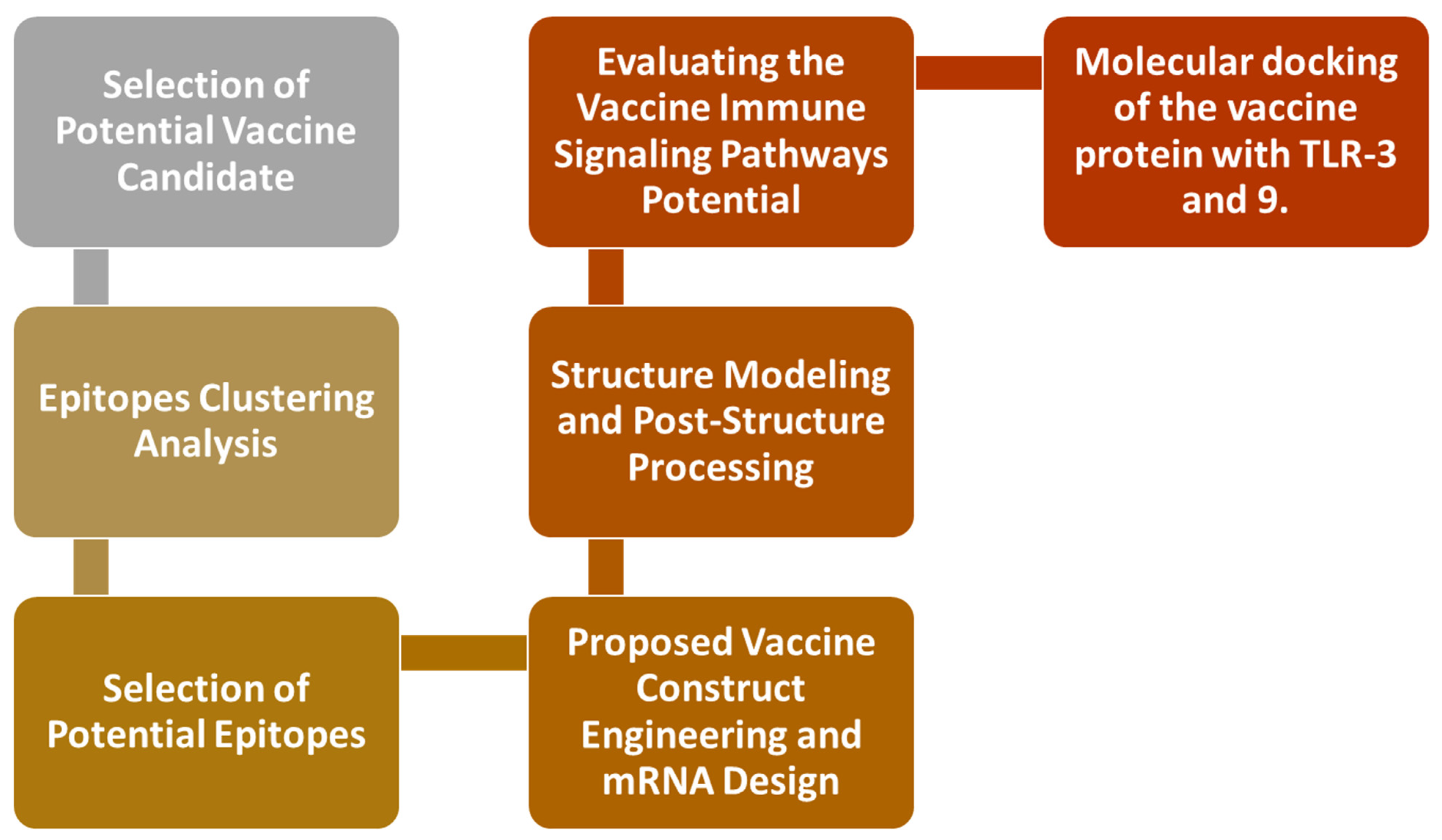
2. Research Methods
2.1. Epitopes Clustering Analysis
2.2. Selection of Potential Epitopes
2.3. Proposed Vaccine Construct Engineering and mRNA Design
2.4. Structure Modeling and Post-Structure Processing
2.5. Evaluating the Vaccine’s Immune Signaling Pathway Potential
2.6. The Molecular Docking of the Vaccine Protein with TLR-3 and 9
3. Results and Discussion
3.1. Multi-Epitope Vaccine Construction and Refinement
3.2. Disulfide Engineering
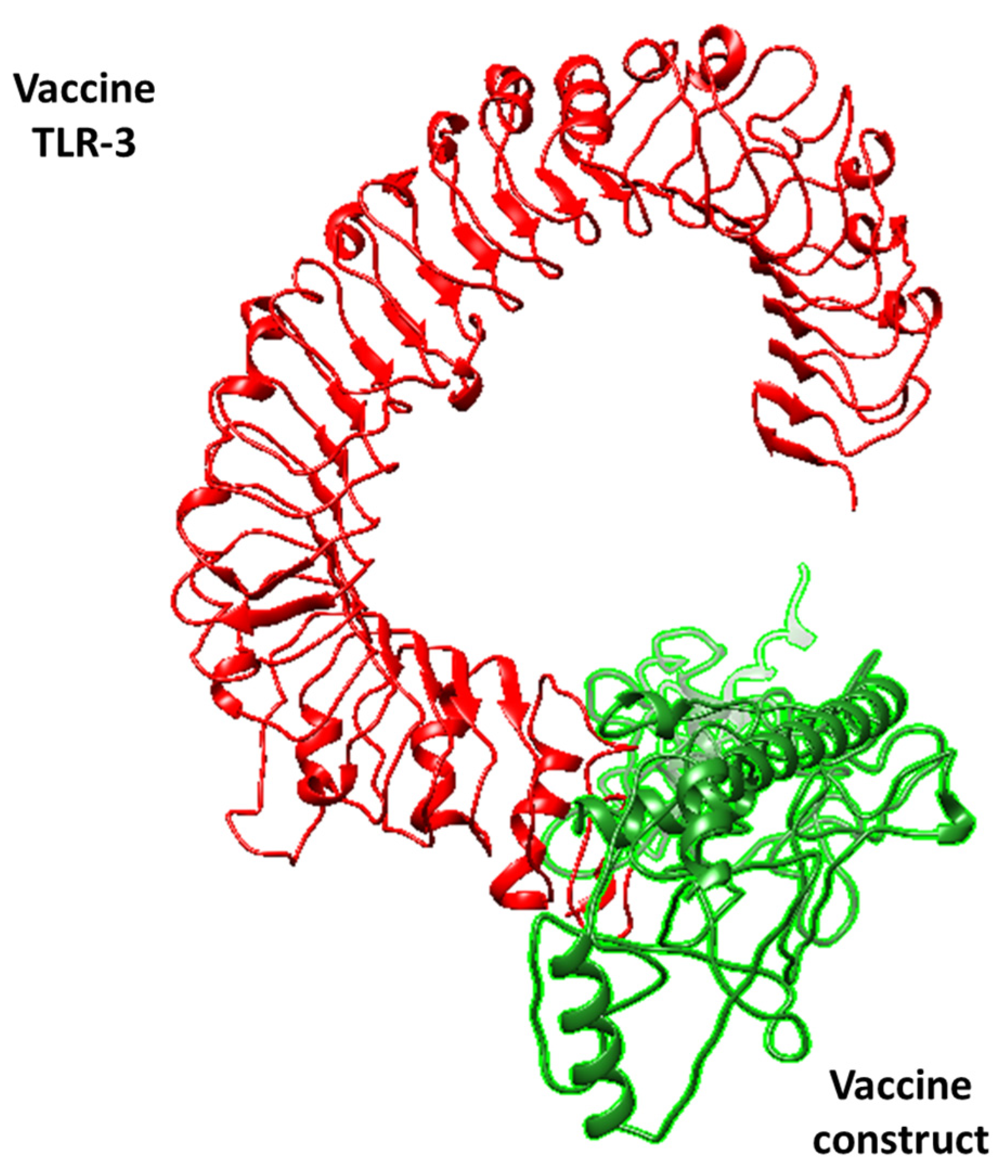
3.3. Molecular Docking Analysis
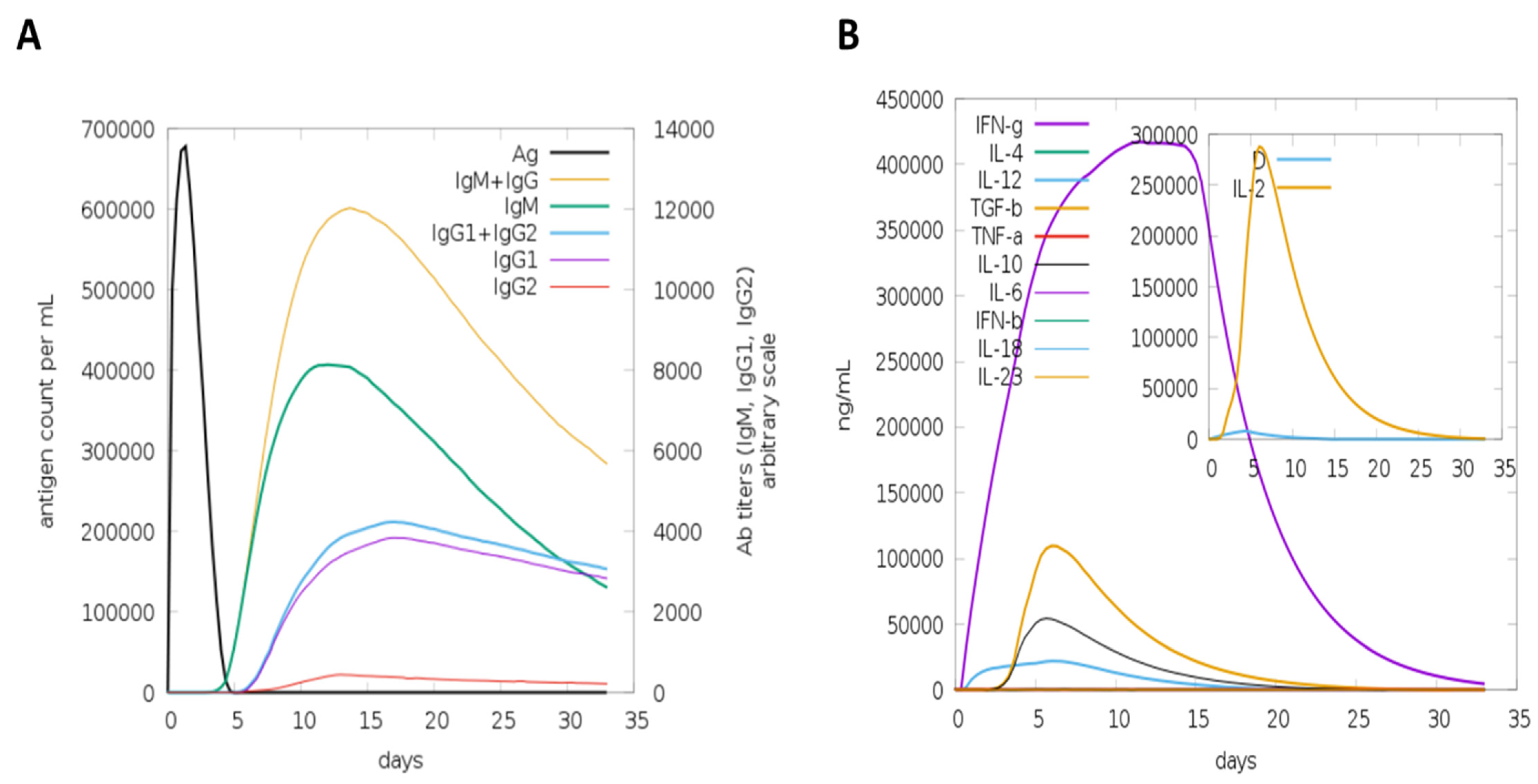
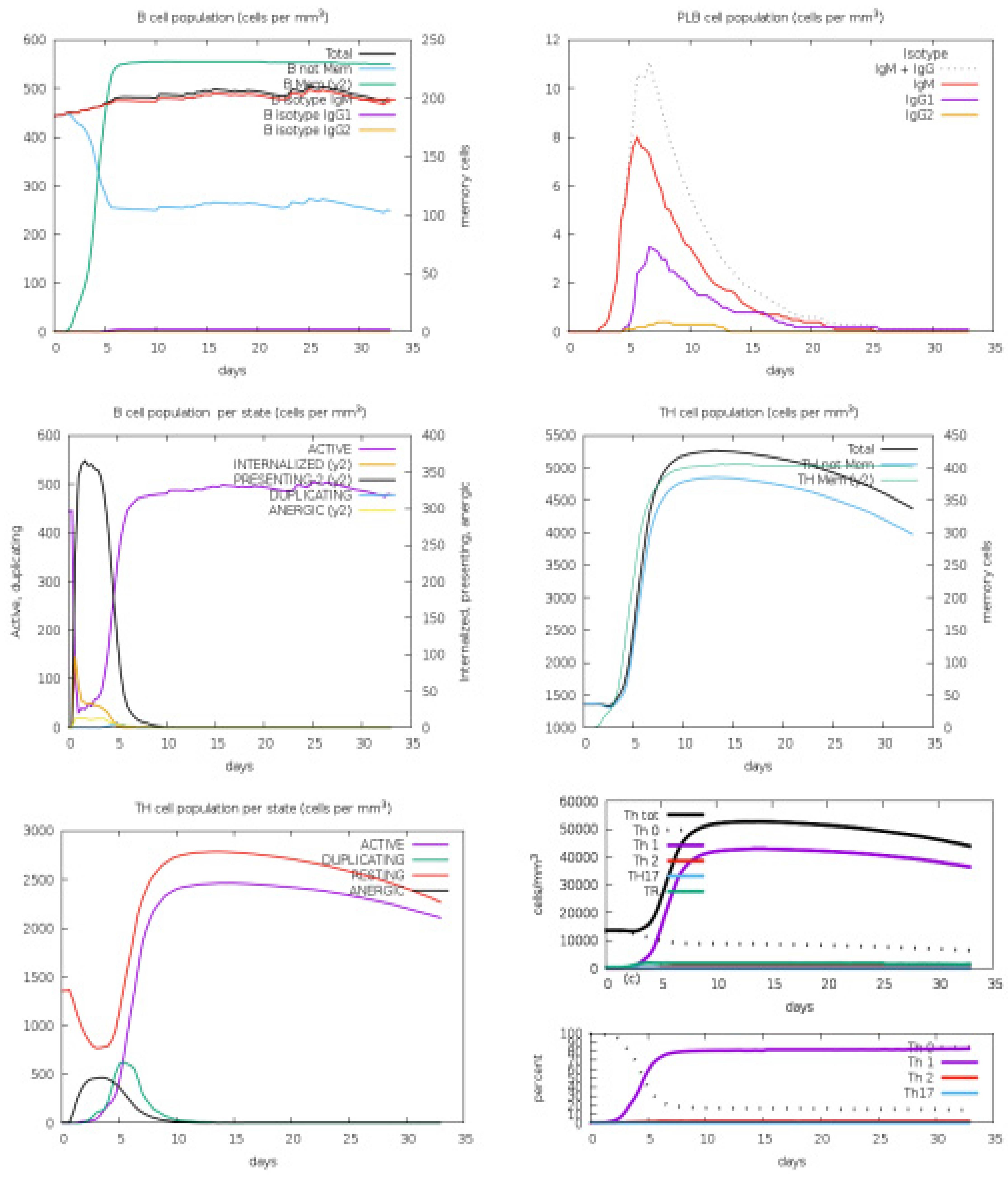
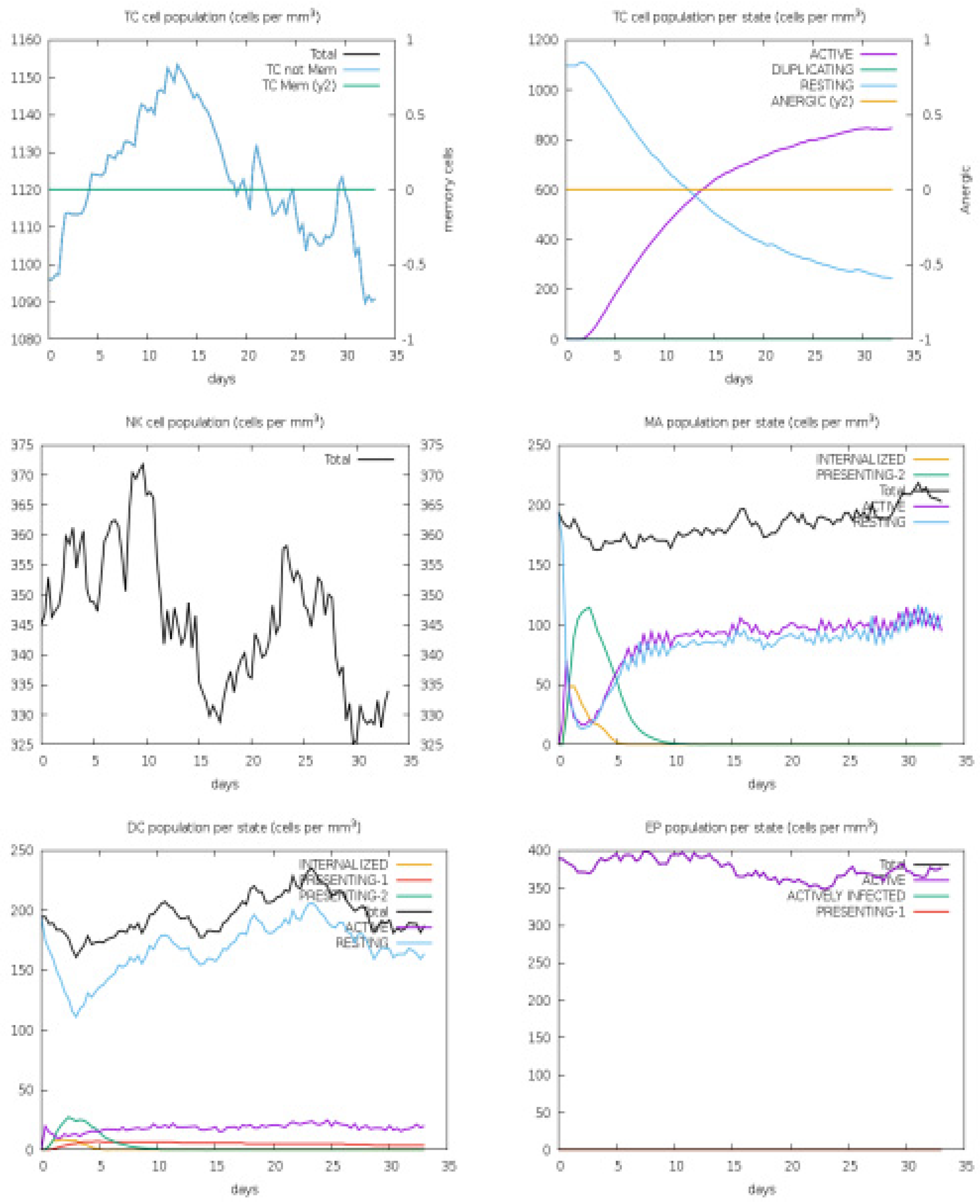
3.4. Host Immune Simulation
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gastanaduy, P.A. Update: Severe respiratory illness associated with Middle East respiratory syndrome coronavirus (MERS-CoV)—Worldwide, 2012–2013. Morb. Mortal. Wkly. Rep. 2013, 62, 480. [Google Scholar]
- Azhar, E.I.; Hui, D.S.C.; Memish, Z.A.; Drosten, C.; Zumla, A. The middle east respiratory syndrome (MERS). Infect. Dis. Clin. 2019, 33, 891–905. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.A.; Dunville, R.L.; Gerber, S.I.; Erdman, D.D.; Pesik, N.; Kuhar, D.; Mason, K.A.; Haynes, L.; Rotz, L.; St. Pierre, J. CDC’s early response to a novel viral disease, middle east respiratory syndrome coronavirus (MERS-CoV), September 2012–May 2014. Public Health Rep. 2015, 130, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, N.; Shaib, H. Middle East respiratory syndrome coronavirus (MERS-CoV): A review. Germs 2019, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Goldenthal, K.L.; Midthun, K.; Zoon, K.C. Control of Viral Infections and Diseases; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 2011. [Google Scholar]
- Bhattacharjee, M.; Banerjee, M.; Mukherjee, A. In silico designing of a novel polyvalent multi-subunit peptide vaccine leveraging cross-immunity against human visceral and cutaneous leishmaniasis: An immunoinformatics-based approach. J. Mol. Model. 2023, 29, 99. [Google Scholar] [CrossRef] [PubMed]
- Abraham Peele, K.; Srihansa, T.; Krupanidhi, S.; Ayyagari, V.S.; Venkateswarulu, T.C. Design of multi-epitope vaccine candidate against SARS-CoV-2: A in-silico study. J. Biomol. Struct. Dyn. 2021, 39, 3793–3801. [Google Scholar] [CrossRef]
- Gul, S.; Ahmad, S.; Ullah, A.; Ismail, S.; Khurram, M.; Tahir ul Qamar, M.; Hakami, A.R.; Alkhathami, A.G.; Alrumaihi, F.; Allemailem, K.S. Designing a Recombinant Vaccine against Providencia rettgeri Using Immunoinformatics Approach. Vaccines 2022, 10, 189. [Google Scholar] [CrossRef] [PubMed]
- Alshammari, A.; Alasmari, A.F.; Alharbi, M.; Ali, N.; Muhseen, Z.T.; Ashfaq, U.A.; Ud-Din, M.; Ullah, A.; Arshad, M.; Ahmad, S. Novel Chimeric Vaccine Candidate Development against Leptotrichia buccalis. Int. J. Environ. Res. Public. Health 2022, 19, 10742. [Google Scholar] [CrossRef]
- Ud-Din, M.; Albutti, A.; Ullah, A.; Ismail, S.; Ahmad, S.; Naz, A.; Khurram, M.; Haq, M.U.; Afsheen, Z.; Bakri, Y.E.; et al. Vaccinomics to Design a Multi-Epitopes Vaccine for Acinetobacter baumannii. Int. J. Environ. Res. Public. Health 2022, 19, 5568. [Google Scholar] [CrossRef]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bansal, P.; Bridge, A.J.; Poux, S.; Bougueleret, L.; Xenarios, I. UniProtKB/Swiss-Prot, the manually annotated section of the UniProt KnowledgeBase: How to use the entry view. Plant Bioinform. Methods Protoc. 2016, 1374, 23–54. [Google Scholar]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; Greenbaum, J.A.; Marcatili, P. IEDB-AR: Immune epitope database—Analysis resource in 2019. Nucleic Acids Res. 2019, 47, W502–W506. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef]
- Blackwell, J.M.; Jamieson, S.E.; Burgner, D. HLA and infectious diseases. Clin. Microbiol. Rev. 2009, 22, 370–385. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Rida, T.; Ahmad, S.; Ullah, A.; Ismail, S.; Tahir ul Qamar, M.; Afsheen, Z.; Khurram, M.; Saqib Ishaq, M.; Alkhathami, A.G.; Alatawi, E.A. Pan-Genome Analysis of Oral Bacterial Pathogens to Predict a Potential Novel Multi-Epitopes Vaccine Candidate. Int. J. Environ. Res. Public. Health 2022, 19, 8408. [Google Scholar] [CrossRef]
- Ullah, A.; Rehman, B.; Khan, S.; Almanaa, T.N.; Waheed, Y.; Hassan, M.; Naz, T.; ul Haq, M.; Muhammad, R.; Sanami, S. An In Silico Multi-epitopes Vaccine Ensemble and Characterization Against Nosocomial Proteus penneri. Mol. Biotechnol. 2023, 1–16. [Google Scholar] [CrossRef]
- Ullah, A.; ul Haq, M.; Iqbal, M.; Irfan, M.; Khan, S.; Muhammad, R.; Ullah, A.; Khurram, M.; Alharbi, M.; Alasmari, A.F. A computational quest for identifying potential vaccine candidates against Moraxella lacunata: A multi-pronged approach. J. Biomol. Struct. Dyn. 2024, 42, 2976–2989. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, I.; Flower, D.R.; Doytchinova, I. AllerTOP-a server for in silico prediction of allergens. BMC Bioinform. 2013, 14, S4. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Naorem, L.D.; Jain, S.; Raghava, G.P.S. ToxinPred2: An improved method for predicting toxicity of proteins. Brief Bioinform. 2022, 23, bbac174. [Google Scholar] [CrossRef]
- Abbas, G.; Zafar, I.; Ahmad, S.; Azam, S.S. Staphylococcus hominis native mitral valve bacterial endocarditis (SBE) in a patient with hypertrophic obstructive cardiomyopathy. Heart Lung 2007, 36, 380–382. [Google Scholar] [CrossRef]
- Malik, M.; Khan, S.; Ullah, A.; Hassan, M.; Ahmad, S.; Al-Harbi, A.I.; Sanami, S.; Abideen, S.A.; Irfan, M.; Khurram, M. Proteome-wide Screening of Potential Vaccine Targets against Brucella melitensis. Vaccines 2023, 11, 263. [Google Scholar] [CrossRef]
- Schlake, T.; Thess, A.; Fotin-Mleczek, M.; Kallen, K.-J. Developing mRNA-vaccine technologies. RNA Biol. 2012, 9, 1319–1330. [Google Scholar] [CrossRef]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [PubMed]
- Bibi, S.; Ullah, I.; Zhu, B.; Adnan, M.; Liaqat, R.; Kong, W.B.; Niu, S. In silico analysis of epitope-based vaccine candidate against tuberculosis using reverse vaccinology. Sci. Rep. 2021, 11, 1149. [Google Scholar] [CrossRef] [PubMed]
- Jaydari, A.; Nazifi, N.; Forouharmehr, A. Computational design of a novel multi-epitope vaccine against Coxiella burnetii. Hum. Immunol. 2020, 81, 596–605. [Google Scholar] [CrossRef]
- Rapin, N.; Lund, O.; Castiglione, F. C-Immsim 10.1 server. PLoS Pathog. 2012, 8, 293. [Google Scholar]
- Ahmad, S.; Waheed, Y.; Ismail, S.; Abbasi, S.W.; Najmi, M.H. A computational study to disclose potential drugs and vaccine ensemble for COVID-19 conundrum. J. Mol. Liq. 2021, 324, 114734. [Google Scholar] [CrossRef]
- Asad, Y.; Ahmad, S.; Rungrotmongkol, T.; Ranaghan, K.E.; Azam, S.S. Immuno-informatics driven proteome-wide investigation revealed novel peptide-based vaccine targets against emerging multiple drug resistant Providencia stuartii. J. Mol. Graph. Model. 2018, 80, 238–250. [Google Scholar] [CrossRef]
- Reginald, K.; Chan, Y.; Plebanski, M.; Poh, C.L. Development of peptide vaccines in dengue. Curr. Pharm. Des. 2018, 24, 1157–1173. [Google Scholar] [CrossRef]
- Maleki, A.; Russo, G.; Parasiliti Palumbo, G.A.; Pappalardo, F. In silico design of recombinant multi-epitope vaccine against influenza A virus. BMC Bioinform. 2021, 22, 617. [Google Scholar] [CrossRef] [PubMed]
- Devi, A.; Chaitanya, N.S.N. In silico designing of multi-epitope vaccine construct against human coronavirus infections. J. Biomol. Struct. Dyn. 2021, 39, 6903–6917. [Google Scholar] [CrossRef] [PubMed]
- Arai, R.; Ueda, H.; Kitayama, A.; Kamiya, N.; Nagamune, T. Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng. 2001, 14, 529–532. [Google Scholar] [CrossRef]
- Ullah, A.; Shahid, F.A.; Haq, M.U.; Tahir ul Qamar, M.; Irfan, M.; Shaker, B.; Ahmad, S.; Alrumaihi, F.; Allemailem, K.S.; Almatroudi, A. An integrative reverse vaccinology, immunoinformatic, docking and simulation approaches towards designing of multi-epitopes based vaccine against monkeypox virus. J. Biomol. Struct. Dyn. 2023, 41, 7821–7834. [Google Scholar] [CrossRef]
- Alshammari, A.; Alharbi, M.; Alghamdi, A.; Alharbi, S.A.; Ashfaq, U.A.; Tahir ul Qamar, M.; Ullah, A.; Irfan, M.; Khan, A.; Ahmad, S. Computer-Aided Multi-Epitope Vaccine Design against Enterobacter xiangfangensis. Int. J. Environ. Res. Public. Health 2022, 19, 7723. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Irfan, M.; Hameed, A.R.; Ullah, A.; Abideen, S.A.; Ahmad, S.; Haq, M.U.; El Bakri, Y.; Al-Harbi, A.I.; Ali, M. Vaccinomics to design a multi-epitope-based vaccine against monkeypox virus using surface-associated proteins. J. Biomol. Struct. Dyn. 2023, 41, 10859–10868. [Google Scholar] [CrossRef]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [PubMed]
- Yousaf, M.; Ullah, A.; Sarosh, N.; Abbasi, S.W.; Ismail, S.; Bibi, S.; Hasan, M.M.; Albadrani, G.M.; Talaat Nouh, N.A.; Abdulhakim, J.A. Design of Multi-Epitope Vaccine for Staphylococcus saprophyticus: Pan-Genome and Reverse Vaccinology Approach. Vaccines 2022, 10, 1192. [Google Scholar] [CrossRef] [PubMed]
- Yousaf, M.; Ismail, S.; Ullah, A.; Bibi, S. Immuno-informatics profiling of monkeypox virus cell surface binding protein for designing a next generation multi-valent peptide-based vaccine. Front. Immunol. 2022, 13, 1035924. [Google Scholar] [CrossRef]
- Marongiu, L.; Gornati, L.; Artuso, I.; Zanoni, I.; Granucci, F. Below the surface: The inner lives of TLR4 and TLR9. J. Leukoc. Biol. 2019, 106, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Jakhar, R.; Sehrawat, N. Designing spike protein (S-Protein) based multi-epitope peptide vaccine against SARS COVID-19 by immunoinformatics. Heliyon 2020, 6, e05528. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Selected Peptide | Type | Start and End Amino Acid | Antigenicity | Allergenicity | Toxicity | Homology | IFN-Gamma Peptide |
|---|---|---|---|---|---|---|---|
| FVSKDVKFENLTNLPPPLLN | Consensus | 101–119 | 1.1267 | Probable Non-Allergen | No | No | No |
| LDAGLNGDFNLTLLQVP | Consensus | 625–642 | 0.8180 | Probable Non-Allergen | No | No | Yes |
| CDNYDDYDFEPQKI | Consensus | 687–702 | 1.4164 | Probable Non-Allergen | No | No | Yes |
| CPMMDLGNSTITDLGV | Consensus | 135–151 | 1.2037 | Probable Non-Allergen | No | No | Yes |
| CCDYEEYDLEPHK | Consensus | 745–758 | 1.1076 | Probable Allergen | NA | NA | NA |
| ITTLNTRYVAPQVTF | Consensus | 514–528 | 1.0017 | Probable Non-Allergen | No | No | Yes |
| LKNRCCDRYEEYD | Consensus | 457–469 | 0.9942 | Probable Allergen | NA | NA | NA |
| ISPAAISSNCYS | Consensus | 320–333 | 0.9397 | Probable Allergen | NA | NA | NA |
| LFSVNDFTCSQISPAAIASN | Consensus | 945–965 | 0.8997 | Probable Allergen | No | No | Yes |
| EEYDLEPHKIHVH | Consensus | 638–650 | 0.8688 | Probable Non-Allergen | No | No | Yes |
| FMHVGYYPSNHIEVAS | Consensus | 637–652 | 0.8584 | Probable Non-Allergen | No | No | Yes |
| MTDNLQMAFVIS | Singleton | 661–673 | 0.821 | Probable Non-Allergen | No | No | Yes |
| IVDIQQTFFDKTWPRPIDVK | Consensus | 104–124 | 0.8024 | Probable Allergen | NA | NA | NA |
| GSSFGNFSANNKSGAYFN | Consensus | 639–657 | 0.7847 | Probable Allergen | NA | NA | NA |
| LLSNSTGTDFKD | Singleton | 388–400 | 0.7804 | Probable Non-Allergen | No | No | Yes |
| KNSQSSPIIPGF | Singleton | 398–410 | 0.7804 | Probable Non-Allergen | No | No | No |
| LALQEVVKALNESYIDLKELGNY | Consensus | 502–524 | 0.7702 | Probable Non-Allergen | No | No | No |
| FSDIKTHKSQPLNAG | Consensus | 601–615 | 0.7665 | Probable Non-Allergen | No | No | No |
| GFTTTNAFKVQ | Consensus | 362–373 | 0.7624 | Probable Allergen | NA | NA | NA |
| IIPGFGGNFNLKLLEPVS | Consensus | 302–320 | 0.7494 | Probable Non-Allergen | No | No | Yes |
| FNITEDEAEWFGITQNAQGVHL | Consensus | 111–133 | 0.7405 | Probable Non-Allergen | No | No | Yes |
| EHDEWFGITQDT | Singleton | 399–410 | 0.7036 | Probable Allergen | NA | NA | NA |
| FQNISNLPPPLLN | Consensus | 1134–1147 | 0.7032 | Probable Non-Allergen | No | No | Yes |
| IDCGHDDLAQLRCS | Consensus | 456–471 | 0.6975 | Probable Allergen | NA | NA | NA |
| EQAEGVECDFSLLL | Consensus | 815–829 | 0.6827 | Probable Non-Allergen | No | No | Yes |
| DQLNSTYFKLSI | Singleton | 771–783 | 0.6751 | Probable Non-Allergen | No | No | Yes |
| DGIVIRIGQNANKTGSVI | Consensus | 823–841 | 0.6684 | Probable Allergen | NA | NA | NA |
| LAFSKVQEAVNA | Singleton | 697–709 | 0.668 | Probable Allergen | No | No | No |
| LPPPLLNNQTDL | Singleton | 1275–1287 | 0.658 | Probable Non-Allergen | No | No | Yes |
| MAATGVISSMTDNLQMAF | Consensus | 1134–1151 | 0.634 | Probable Allergen | NA | NA | NA |
| EQAEGFECDFSPLLSGTPPQV | Consensus | 786–807 | 0.6256 | Probable Non-Allergen | No | No | Yes |
| DKSWPRPIDPAAA | Consensus | 965–988 | 0.6222 | Probable Allergen | NA | NA | NA |
| IIPGFGGDFNLT | Singleton | 602–614 | 0.622 | Probable Non-Allergen | No | No | Yes |
| FMLGSSVGNFSNGS | Consensus | 236–250 | 0.6195 | Probable Non-Allergen | No | No | Yes |
| GYHPSQHIEVVA | Singleton | 404–416 | 0.6166 | Probable Non-Allergen | No | No | Yes |
| LFASVKSSQSSPII | Consensus | 345–359 | 0.6133 | Probable Allergen | NA | NA | NA |
| AGYKVLPPLYDPNMEAAYTSSL | Consensus | 928–950 | 0.6025 | Probable Non-Allergen | No | No | Yes |
| IIPHSIRSIQSDRKAWAAFYVYKLQPLTF | Consensus | 887–901 | 0.6015 | Probable Non-Allergen | No | No | Yes |
| STGSRSARSAIEDLLFKVTIADP | Consensus | 125–147 | 0.5925 | Probable Non-Allergen | No | No | Yes |
| IGAAANSTGTVIISP | Consensus | 625–639 | 0.5861 | Probable Allergen | NA | NA | NA |
| FVYDFDNIG | Consensus | 114–123 | 0.5855 | Probable Non-Allergen | No | No | Yes |
| GQSLCALPDTPSTLTPRSVRSVPGEMRLAS | Consensus | 732–760 | 0.5429 | Probable Non-Allergen | No | No | Yes |
| FSAYSGDIPHYVQPGQYTP | Consensus | 507–525 | 0.54 | Probable Non-Allergen | No | No | Yes |
| EFSCDGISPDAI | Singleton | 687–699 | 0.5325 | Probable Allergen | NA | NA | NA |
| ATDCSDGNYNRNASLNSF | Consensus | 514–531 | 0.5206 | Probable Allergen | NA | NA | NA |
| ACEHITTMMQFS | Consensus | 634–644 | 0.5155 | Probable Allergen | NA | NA | NA |
| GSSFYAPEPITSLNTKYVAPQ | Consensus | 333–355 | 0.4965 | Probable Allergen | NA | NA | NA |
| DGYIRRAIDCGFNDLSQLCSYE | Consensus | 367–386 | 0.4774 | Probable Non-Allergen | No | No | Yes |
| KITIADPGYMQGYDDCMQQGPASARDLICAQYVAG | Consensus | 896–931 | 0.4675 | Probable Non-Allergen | No | No | Yes |
| FAYPLSMKSYMQ | Singleton | 281–293 | 0.4593 | Probable Allergen | NA | NA | NA |
| KNVSSQGPNFQE | Singleton | 205–217 | 0.4589 | Probable Non-Allergen | No | No | Yes |
| IFATAPANLTISKPSSYS | Consensus | 976–994 | 0.4536 | Probable Non-Allergen | No | No | Yes |
| EPIDMNKADGVIYPGRTYS | Consensus | 231–248 | 0.4392 | Probable Allergen | NA | NA | NA |
| LFVEDCLPLGQSLCA | Consensus | 967–692 | 0.4392 | Probable Allergen | NA | NA | NA |
| EMCLASIAFNHPIQVDQLNSSYFK | Consensus | 850–873 | 0.4138 | Probable Non-Allergen | No | No | Yes |
| HFVYDAYNLVGYYSDDGNYYCV | Consensus | 233–253 | 0.412 | Probable Allergen | NA | NA | NA |
| GSSVGNYYNGYP | Singleton | 1041–1053 | 0.4007 | Probable Non-Allergen | No | No | Yes |
| GCSVGNFSDGKM | Singleton | 783–795 | 0.3891 | NA | NA | NA | NA |
| LILDYFSYPLSMKSDLSVSSS | Consensus | 364–384 | 0.388 | NA | NA | NA | NA |
| FATYHTPATDCSDGNY | Consensus | 532–549 | 0.3792 | NA | NA | NA | NA |
| ITTFMPQFSRMTQSALRMR | Consensus | 1041–1061 | 0.3727 | NA | NA | NA | NA |
| FGAISASIGDIIQRLDLEQDAQIDRLI | Consensus | 366–387 | 0.3635 | NA | NA | NA | NA |
| GNHCPAGNSYTSFATYHT | Consensus | 1265–1283 | 0.3452 | NA | NA | NA | NA |
| CPKEFANDTKIASQLGN | Consensus | 146–164 | 0.3352 | NA | NA | NA | NA |
| NAKADGIIYPTGKSYSNI | Consensus | 541–557 | 0.3262 | NA | NA | NA | NA |
| ILPPPLLSNST | Consensus | 442–452 | 0.3216 | NA | NA | NA | NA |
| LLGNSXGIDFQDELDEFFKNVSTSIPNFGS | Consensus | 951–981 | 0.3057 | NA | NA | NA | NA |
| CVLGLVNSSLVEDCK | Consensus | 712–728 | 0.3009 | NA | NA | NA | NA |
| GNMFRFASLPVY | Singleton | 463–475 | 0.2847 | NA | NA | NA | NA |
| DQLNSSYKLSIP | Singleton | 771–784 | 0.284 | NA | NA | NA | NA |
| CGISPDAIARGCYS | Consensus | 374–387 | 0.2786 | NA | NA | NA | NA |
| LDFKEELEEFFK | Singleton | 856–567 | 0.2785 | NA | NA | NA | NA |
| ASAYGLCDAANPTNCIAPVNG | Consensus | 784–805 | 0.2607 | NA | NA | NA | NA |
| DAVNNAQALSKLAS | Consensus | 459–463 | 0.2396 | NA | NA | NA | NA |
| AAIASNCYSSLID | Consensus | 931–943 | 0.2103 | NA | NA | NA | NA |
| LLGSIAGAGWTAGLSSFAAI | Consensus | 98–117 | 0.1785 | NA | NA | NA | NA |
| GYFIKTNNTIVDEWS | Consensus | 114–128 | 0.1375 | NA | NA | NA | NA |
| AANSTGNLIISSS | Consensus | 1187–1198 | 0.1231 | NA | NA | NA | NA |
| FDNIIGFHSDDGNYY | Consensus | 604–619 | 0.1103 | NA | NA | NA | NA |
| DVSKADGIIPQ | Consensus | 187–198 | 0.0638 | NA | NA | NA | NA |
| AKINQALHGANLRQDSVRNL | Consensus | 116–135 | 0.0607 | NA | NA | NA | NA |
| KLIANKFNQALGAMQTGF | Consensus | 254–271 | 0.0337 | NA | NA | NA | NA |
| HSDGNYYCVRPCVS | Consensus | 456–468 | −0.0086 | NA | NA | NA | NA |
| DLYGGNMFQFATPV | Consensus | 410–425 | −0.0297 | NA | NA | NA | NA |
| CNGFQKCEQLLREYGQFCA | Consensus | 435–448 | −0.0311 | NA | NA | NA | NA |
| ITKPLKYSYINKCSRLLSDDRTEVPQ | Consensus | 491–515 | −0.0554 | NA | NA | NA | NA |
| LMQDESVANLFSDIKTHKS | Consensus | 639–657 | −0.0873 | NA | NA | NA | NA |
| EKLLEQYGQFCS | Singleton | 523–534 | −0.0925 | NA | NA | NA | NA |
| AFVAQQLVRSEAAR | Consensus | 149–161 | −0.1895 | NA | NA | NA | NA |
| MYLYSAAHADPNRFILGKLY | Consensus | 84–104 | −0.3463 | NA | NA | NA | NA |
| GFAKCEKLLEQY | Singleton | 968–980 | −0.3673 | NA | NA | NA | NA |
| DQSFKDELEEFF | Singleton | 1214–1240 | −0.3752 | NA | NA | NA | NA |
| EQEVQIDRLING | Singleton | 987–997 | −0.4307 | NA | NA | NA | NA |
| Model | GDT-HA | RMSD | MolProbity | Clash Score | Poor | Model |
|---|---|---|---|---|---|---|
| Initial | 1.0000 | 0.000 | 3.802 | 124.4 | 6.8 | 87.3 |
| MODEL 1 | 0.9110 | 0.502 | 2.197 | 17.6 | 0.3 | 92.9 |
| MODEL 2 | 0.9047 | 0.530 | 2.336 | 20.8 | 1.3 | 93.4 |
| MODEL 3 | 0.9085 | 0.520 | 2.260 | 21.1 | 1.0 | 93.1 |
| MODEL 4 | 0.9097 | 0.512 | 2.175 | 19.9 | 1.0 | 94.4 |
| MODEL 5 | 0.9066 | 0.513 | 2.225 | 19.4 | 1.0 | 93.1 |
| Residues1 Seq # | Residues1 AA | Residues2 Seq # | Residues2 AA | Chi3 | Energy |
|---|---|---|---|---|---|
| 3 | PRO | 40 | ILE | 101.55 | 4.59 |
| 8 | ASP | 14 | HIS | 126.98 | 3.65 |
| 17 | GLN | 20 | THR | 91.37 | 0.67 |
| 40 | ILE | 45 | ASN | 119.1 | 6.36 |
| 89 | TRP | 94 | PRO | 113.14 | 4.49 |
| 94 | PRO | 108 | ALA | −67.22 | 1.94 |
| 100 | ILE | 104 | ASN | 119.75 | 1.75 |
| 114 | ASP | 117 | TYR | −115.58 | 5.25 |
| 117 | TYR | 119 | GLN | −104.25 | 3.99 |
| 135 | PHE | 139 | GLY | −88.2 | 3.57 |
| 140 | PRO | 158 | ASN | −71.39 | 5.81 |
| 144 | VAL | 150 | SER | 77.6 | 3.93 |
| 146 | GLN | 149 | SER | 86.9 | 3.39 |
| 161 | TYR | 170 | PRO | 101.78 | 4.59 |
| 163 | LYS | 167 | GLY | 97.48 | 3.31 |
| 164 | LEU | 167 | GLY | 114.04 | 1.57 |
| 166 | ILE | 189 | PRO | −78.73 | 6.1 |
| 168 | PRO | 177 | ASN | 108.77 | 2.71 |
| 171 | GLY | 174 | ASN | −111.44 | 7.95 |
| 175 | ILE | 178 | LEU | −92.5 | 4 |
| 183 | PRO | 217 | LEU | −103.94 | 5.86 |
| 218 | ASN | 237 | ASN | −116.83 | 6.08 |
| 228 | GLY | 232 | PRO | −68.03 | 2.7 |
| 234 | PHE | 238 | PHE | −63.92 | 6.41 |
| 247 | PRO | 257 | PRO | 121.42 | 3.06 |
| 250 | GLY | 290 | GLY | −86.8 | 3.77 |
| 253 | PHE | 256 | GLY | −91.29 | 5.01 |
| 260 | GLY | 263 | ILE | 121.84 | 6.03 |
| 295 | ASP | 321 | PHE | −88.18 | 2.56 |
| 309 | CYS | 321 | PHE | −77.33 | 1.61 |
| 310 | ASP | 313 | LEU | 107.72 | 5.77 |
| 313 | LEU | 318 | GLY | 108.9 | 2.53 |
| 322 | MET | 336 | ARG | −61.42 | 5.7 |
| 324 | GLY | 335 | GLY | −86.84 | 4.62 |
| 325 | SER | 328 | GLY | 102.97 | 4.66 |
| 339 | ILE | 360 | TRP | −65.91 | 2.85 |
| 348 | GLY | 353 | ILE | 84.25 | 3.34 |
| 362 | PRO | 366 | MET | 80.19 | 6.16 |
| 378 | GLY | 381 | ASP | −112.52 | 3.12 |
| Cluster | Members | Representative | Weighted Score |
|---|---|---|---|
| 0 | 47 | Center | −1000.4 |
| Lowest Energy | −1033.2 | ||
| 1 | 37 | Center | −835.7 |
| Lowest Energy | −835.7 | ||
| 2 | 30 | Center | −890.0 |
| Lowest Energy | −975.4 | ||
| 3 | 27 | Center | −813.8 |
| Lowest Energy | −903.8 | ||
| 4 | 24 | Center | −801.0 |
| Lowest Energy | −888.1 | ||
| 5 | 19 | Center | −906.6 |
| Lowest Energy | −946.3 | ||
| 6 | 19 | Center | −902.1 |
| Lowest Energy | −902.1 | ||
| 7 | 18 | Center | −881.3 |
| Lowest Energy | −886.3 | ||
| 8 | 18 | Center | −960.8 |
| Lowest Energy | −960.8 | ||
| 9 | 17 | Center | −900.9 |
| Lowest Energy | −918.8 | ||
| 10 | 16 | Center | −853.9 |
| Lowest Energy | −940.1 | ||
| 11 | 15 | Center | −839.4 |
| Lowest Energy | −951.9 | ||
| 12 | 14 | Center | −792.0 |
| Lowest Energy | −911.9 | ||
| 13 | 13 | Center | −943.0 |
| Lowest Energy | −943.0 | ||
| 14 | 12 | Center | −905.4 |
| Lowest Energy | −905.4 | ||
| 15 | 12 | Center | −882.5 |
| Lowest Energy | −883.4 | ||
| 16 | 12 | Center | −851.5 |
| Lowest Energy | −851.5 | ||
| 17 | 12 | Center | −837.8 |
| Lowest Energy | −1040.0 | ||
| 18 | 11 | Center | −1033.1 |
| Lowest Energy | −1033.1 | ||
| 19 | 11 | Center | −881.3 |
| Lowest Energy | −881.3 | ||
| 20 | 11 | Center | −871.7 |
| Lowest Energy | −871.7 |
| Cluster | Members | Representative | Weighted Score |
|---|---|---|---|
| 0 | 30 | Center | −1036.7 |
| Lowest Energy | −1203.3 | ||
| 1 | 24 | Center | −1318.0 |
| Lowest Energy | −1318.0 | ||
| 2 | 23 | Center | −1068.5 |
| Lowest Energy | −1228.9 | ||
| 3 | 22 | Center | −995.4 |
| Lowest Energy | −1147.7 | ||
| 4 | 21 | Center | −1068.5 |
| Lowest Energy | −1298.6 | ||
| 5 | 21 | Center | −1055.6 |
| Lowest Energy | −1172.7 | ||
| 6 | 21 | Center | −1009.1 |
| Lowest Energy | −1295.4 | ||
| 7 | 20 | Center | −1212.1 |
| Lowest Energy | −1242.1 | ||
| 8 | 19 | Center | −991.7 |
| Lowest Energy | −1338.5 | ||
| 9 | 18 | Center | −1016.1 |
| Lowest Energy | −1206.8 | ||
| 10 | 18 | Center | −1140.1 |
| Lowest Energy | −1217.3 | ||
| 11 | 16 | Center | −1030.0 |
| Lowest Energy | −1424.7 | ||
| 12 | 16 | Center | −1199.1 |
| Lowest Energy | −1199.1 | ||
| 13 | 16 | Center | −1150.4 |
| Lowest Energy | −1300.8 | ||
| 14 | 16 | Center | −1125.0 |
| Lowest Energy | −1155.1 | ||
| 15 | 14 | Center | −1138.0 |
| Lowest Energy | −1138.0 | ||
| 16 | 14 | Center | −1084.5 |
| Lowest Energy | −1122.5 | ||
| 17 | 13 | Center | −1108.6 |
| Lowest Energy | −1151.3 | ||
| 18 | 13 | Center | −1215.2 |
| Lowest Energy | −1215.2 | ||
| 19 | 13 | Center | −1047.6 |
| Lowest Energy | −1095.5 | ||
| 20 | 13 | Center | −1003.2 |
| Lowest Energy | −1218.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Published by MDPI on behalf of the Lithuanian University of Health Sciences. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almanaa, T.N. Design of an Epitope-Based Vaccine Against MERS-CoV. Medicina 2024, 60, 1632. https://doi.org/10.3390/medicina60101632
Almanaa TN. Design of an Epitope-Based Vaccine Against MERS-CoV. Medicina. 2024; 60(10):1632. https://doi.org/10.3390/medicina60101632
Chicago/Turabian StyleAlmanaa, Taghreed N. 2024. "Design of an Epitope-Based Vaccine Against MERS-CoV" Medicina 60, no. 10: 1632. https://doi.org/10.3390/medicina60101632
APA StyleAlmanaa, T. N. (2024). Design of an Epitope-Based Vaccine Against MERS-CoV. Medicina, 60(10), 1632. https://doi.org/10.3390/medicina60101632