

GNB1 Encephalopathy: Clinical Case Report and Literature Review

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
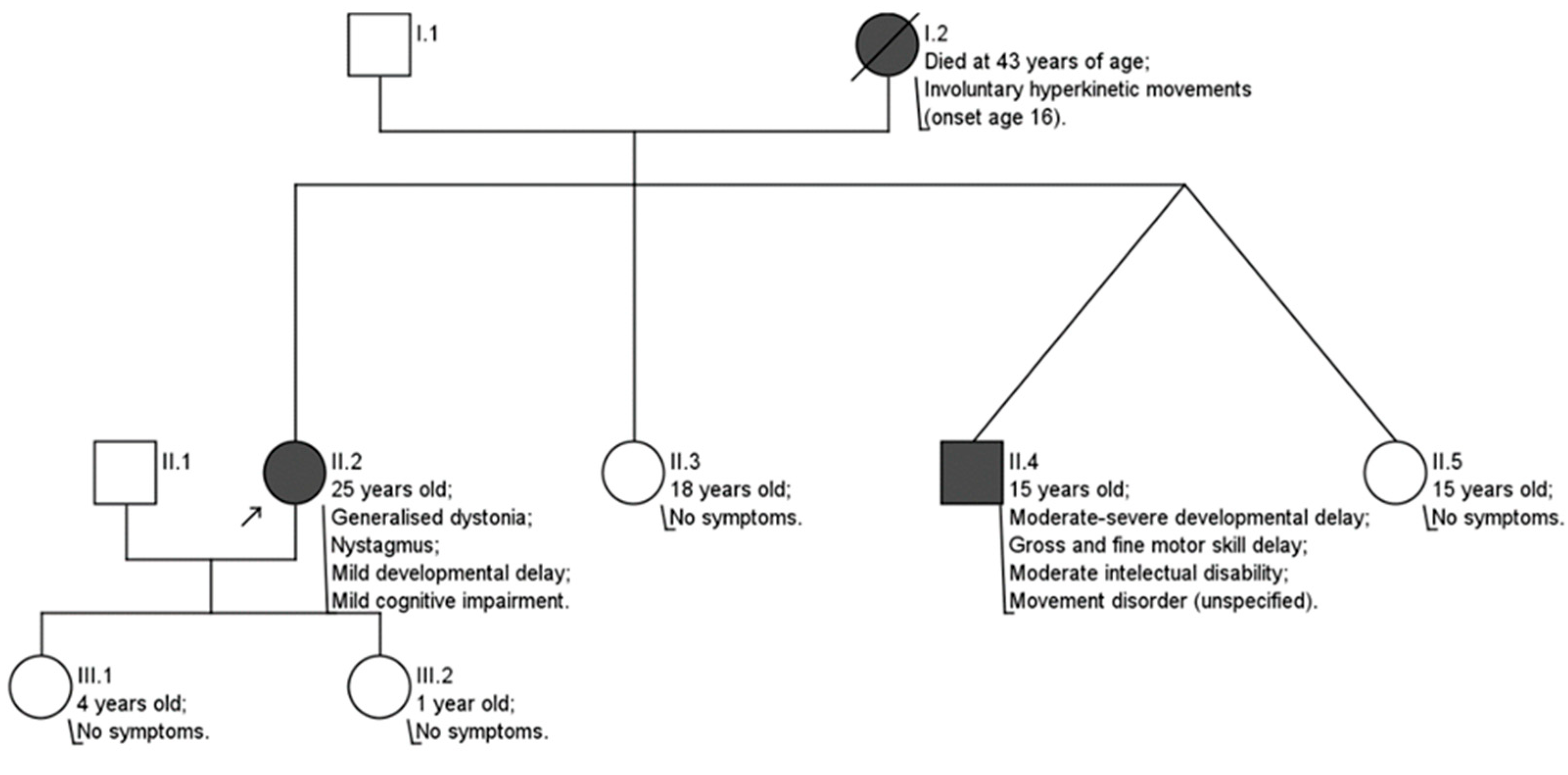
3. Clinical Case
4. Literature Review
4.1. Pathogenesis
4.2. Clinical Manifestations
4.2.1. Developmental Delay (DD)
4.2.2. Intellectual Disability (ID)
4.2.3. Neurological Symptoms
- Muscle tone disorders. Hypotonia is the most common clinical finding in infancy ranging from mild to severe, generalized forms that result in hypotonic quadriplegia and oropharyngeal dysphagia [6,8,14,16,17]. In rare cases, hypotonia persists with age [6]; however, usually, it transforms into spastic hypertonia. Some studies highlight the pattern of high limb and low axial muscle tone [6,13,14,16,17].
- Movement disorders. The most commonly documented movement disorder is dystonia, varying from mild dystonic positioning of the fingers to generalized dystonia with age of onset as early as 2 years old and as late as 16 years old [3,8,13,14,15,16,17]. In some cases, myoclonus [3,15,16] or intermittent status dystonicus [15,17] were described. Other findings include tics [6], ataxia, or chorea [6,8,13].
- Epilepsy. Epilepsy manifests in some, but not all, cases (10/13 [6] and 6/18 [14]). Seizures can be focal or generalized, and several cases of status epilepticus were also documented [6,14]. Seizure morphology varies; tonic, atonic, myoclonic, tonic-clonic, absence seizures, and infantile spasms were documented [6,8,13,14,17]. GNB1 gene variants are also associated with West syndrome [14,17].
4.2.4. Other Symptoms
4.3. Prognosis
4.4. Diagnostics
4.4.1. Genetic Testing
4.4.2. Laboratory Testing
4.4.3. Electroencephalography
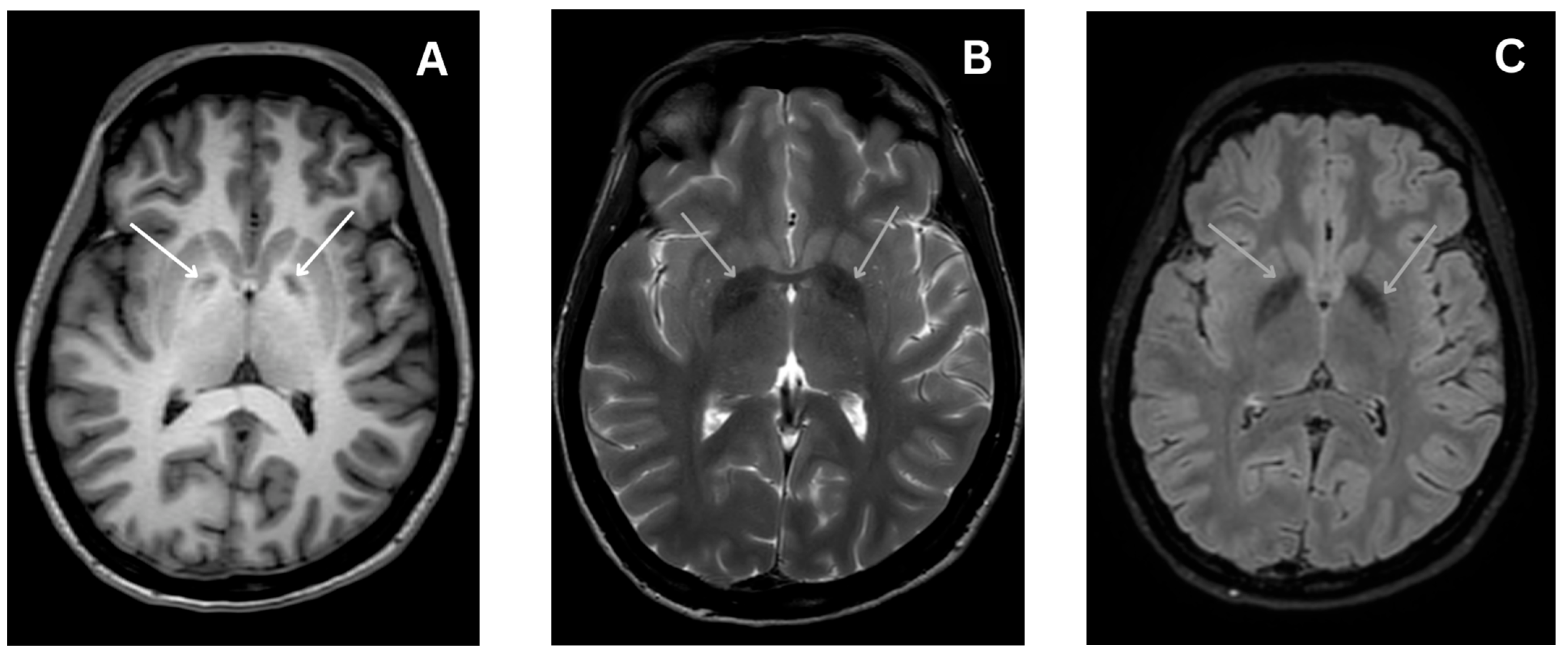
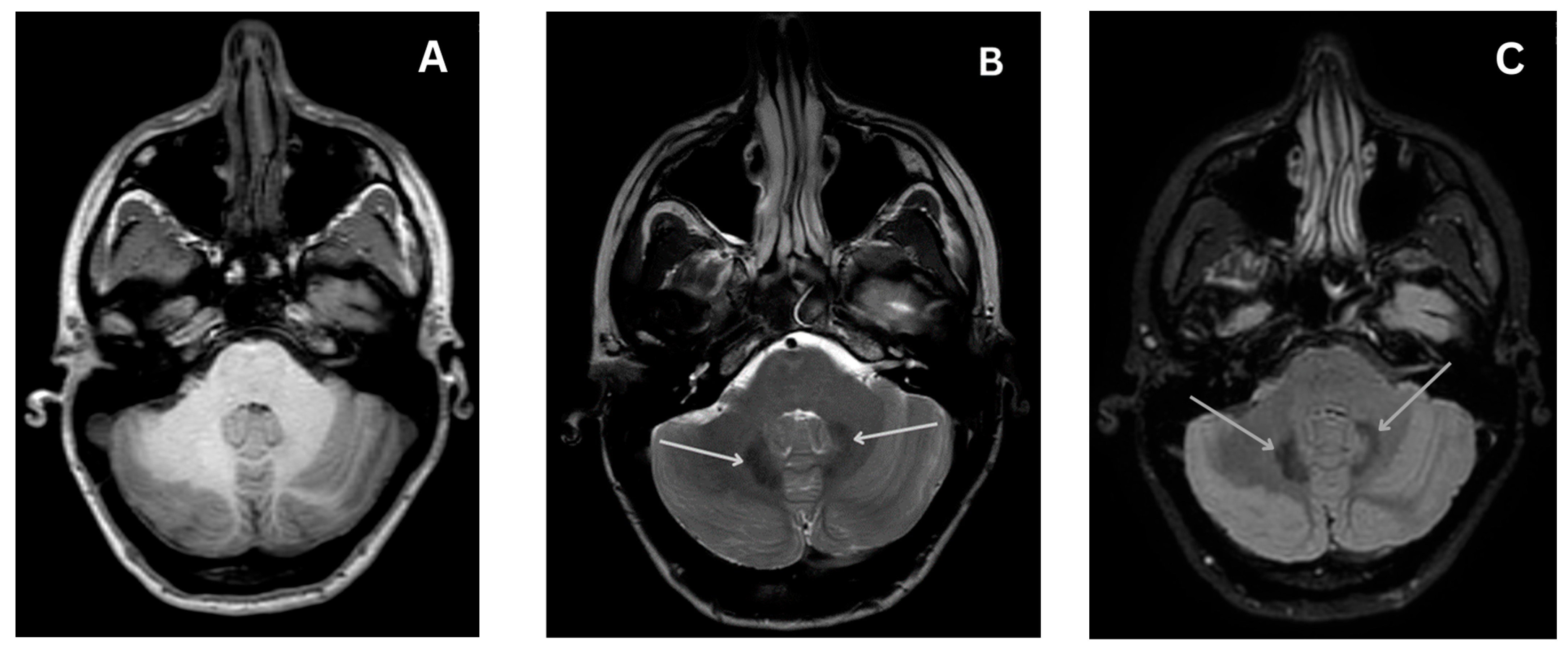
4.4.4. Magnetic Resonance Imaging
4.5. Treatment
5. Discussion
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Galosi, S.; Pollini, L.; Novelli, M.; Bernardi, K.; Di Rocco, M.; Martinelli, S.; Leuzzi, V. Motor, epileptic, and developmental phenotypes in genetic disorders affecting G protein coupled receptors-cAMP signaling. Front. Neurol. 2022, 13, 886751. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, M.; Ikeda, A.; Tsuyusaki, Y.; Iai, M.; Kurosawa, K.; Kosaki, K.; Goto, T. Atypical clinical course in two patients with GNB1 variants who developed acute encephalopathy. Brain Dev. 2023, 45, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Reyes, N.G.D.; Di Luca, D.G.; McNiven, V.; Lang, A.E. Dystonia with myoclonus and vertical supranuclear gaze palsy associated with a rare GNB1 variant. Park. Relat. Disord. 2022, 106, 105239. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.R.; Kassam, F.; Innes, A.M. Rod-cone dystrophy in an adult with GNB1-related disorder: An expansion of the phenotype and natural history. Am. J. Med. Genet. Part. C Semin. Med. Genet. 2023, 193, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Purves, D.; Augustine, G.J.; Fitzpatrick, D.; Katz, L.C.; LaMantia, A.S.; McNamara, J.O.; Williams, S.M. G-Proteins and Their Molecular Targets. In Neuroscience, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2001. [Google Scholar]
- Petrovski, S.; Küry, S.; Myers, C.T.; Anyane-Yeboa, K.; Cogné, B.; Bialer, M.; Xia, F.; Hemati, P.; Riviello, J.; Mehaffey, M.; et al. Germline De Novo Mutations in GNB1 Cause Severe Neurodevelopmental Disability, Hypotonia, and Seizures. Am. J. Hum. Genet. 2016, 98, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Smrcka, A.V. G protein βγ subunits: Central mediators of G protein-coupled receptor signaling. Cell. Mol. Life Sci. 2008, 65, 2191–2214. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, K.; Masuho, I.; Patil, D.N.; Baumann, H.; Hebert, E.; Steinrücke, S.; Trujillano, D.; Skamangas, N.K.; Dobricic, V.; Hüning, I.; et al. Novel GNB1 mutations disrupt assembly and function of G protein heterotrimers and cause global developmental delay in humans. Hum. Mol. Genet. 2017, 26, 1078–1086. [Google Scholar] [CrossRef]
- Reddy, H.P.; Yakubovich, D.; Keren-Raifman, T.; Tabak, G.; Tsemakhovich, V.A.; Pedersen, M.H.; Shalomov, B.; Colombo, S.; Goldstein, D.B.; Javitch, J.A.; et al. Encephalopathy-causing mutations in Gβ1 (GNB1) alter regulation of neuronal GIRK channels. iScience 2021, 24, 103018. [Google Scholar] [CrossRef]
- Isomoto, S.; Kondo, C.; Kurachi, Y. Inwardly Rectifying Potassium Channels: Their Molecular Heterogeneity and Function. Jpn. J. Physiol. 1997, 47, 11–39. [Google Scholar] [CrossRef]
- Ford, C.E.; Skiba, N.P.; Bae, H.; Daaka, Y.; Reuveny, E.; Shekter, L.R.; Rosal, R.; Weng, G.; Yang, C.-S.; Iyengar, R.; et al. Molecular basis for interactions of G protein betagamma subunits with effectors. Science 1998, 280, 1271–1274. [Google Scholar] [CrossRef]
- Colombo, S.; Reddy, H.P.; Petri, S.; Williams, D.J.; Shalomov, B.; Dhindsa, R.S.; Gelfman, S.; Krizay, D.; Bera, A.K.; Yang, M.; et al. Epilepsy in a mouse model of GNB1 encepha-lopathy arises from altered potassium (GIRK) channel signaling and is alleviated by a GIRK inhibitor. Front. Cell Neurosci. 2023, 17, 1175895. [Google Scholar] [CrossRef] [PubMed]
- Revah-Politi, A.; Sands, T.T.; Colombo, S.; Goldstein, D.B.; Anyane-Yeboa, K. GNB1 Encephalopathy. In GeneReviews; University of Washington: Seattle, WA, USA, 2021. [Google Scholar]
- Hemati, P.; Revah-Politi, A.; Bassan, H.; Petrovski, S.; Bilancia, C.G.; Ramsey, K.; Griffin, N.G.; Bier, L.; Cho, M.T.; Rosello, M.; et al. Refining the phenotype associated with GNB1 mutations: Clinical data on 18 newly identified patients and review of the literature. Am. J. Med. Genet. Part. A 2018, 176, 2259–2275. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.F.; Morales-Briceño, H.; Barwick, K.; Lewis, J.; Sanchis-Juan, A.; Raymond, F.L.; Stewart, K.; Waugh, M.; Mahant, N.; Kurian, M.A.; et al. Myoclonus-dystonia caused by GNB1 mutation responsive to deep brain stimulation. Mov. Disord. 2019, 34, 1079–1080. [Google Scholar] [CrossRef] [PubMed]
- Steinrücke, S.; Lohmann, K.; Domingo, A.; Rolfs, A.; Bäumer, T.; Spiegler, J.; Hartmann, C.; Münchau, A. Novel GNB1 missense mutation in a patient with generalized dystonia, hypotonia, and intellectual disability. Neurol. Genet. 2016, 2, e106. [Google Scholar] [CrossRef]
- Endo, W.; Ikemoto, S.; Togashi, N.; Miyabayashi, T.; Nakajima, E.; Hamano, S.-I.; Shibuya, M.; Sato, R.; Takezawa, Y.; Okubo, Y.; et al. Phenotype–genotype correlations in patients with GNB1 gene variants, including the first three reported Japanese patients to exhibit spastic diplegia, dyskinetic quadriplegia, and infantile spasms. Brain Dev. 2019, 42, 199–204. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Bledsoe, I.O.; Viser, A.C.; Luciano, M.S. Treatment of Dystonia: Medications, Neurotoxins, Neuromodulation, and Rehabilitation. Neurotherapeutics 2020, 17, 1622–1644. [Google Scholar] [CrossRef]
- Yang, M.; Kim, J.-W. Principles of Genetic Counseling in the Era of Next-Generation Sequencing. Ann. Lab. Med. 2018, 38, 291–295. [Google Scholar] [CrossRef]
- Farmer, J.P.; Mittal, S. Baclofen. Surg. Manag. Mov. Disord. 2022, 11, 257–276. [Google Scholar]
- Flotte, T.R.; Gessler, D.J. Gene Therapy for Rare Neurological Disorders. Clin. Pharmacol. Ther. 2022, 111, 743–757. [Google Scholar] [CrossRef]
- Jensen, T.L.; Gøtzsche, C.R.; Woldbye, D.P.D. Current and Future Prospects for Gene Therapy for Rare Genetic Diseases Affecting the Brain and Spinal Cord. Front. Mol. Neurosci. 2021, 14, 695937. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasvytis, M.; Čiauškaitė, J.; Jurkevičienė, G. GNB1 Encephalopathy: Clinical Case Report and Literature Review. Medicina 2024, 60, 589. https://doi.org/10.3390/medicina60040589
Nasvytis M, Čiauškaitė J, Jurkevičienė G. GNB1 Encephalopathy: Clinical Case Report and Literature Review. Medicina. 2024; 60(4):589. https://doi.org/10.3390/medicina60040589
Chicago/Turabian StyleNasvytis, Matas, Julija Čiauškaitė, and Giedrė Jurkevičienė. 2024. "GNB1 Encephalopathy: Clinical Case Report and Literature Review" Medicina 60, no. 4: 589. https://doi.org/10.3390/medicina60040589
APA StyleNasvytis, M., Čiauškaitė, J., & Jurkevičienė, G. (2024). GNB1 Encephalopathy: Clinical Case Report and Literature Review. Medicina, 60(4), 589. https://doi.org/10.3390/medicina60040589