
The Complement System as a Therapeutic Target in Retinal Disease




Abstract
1. Introduction
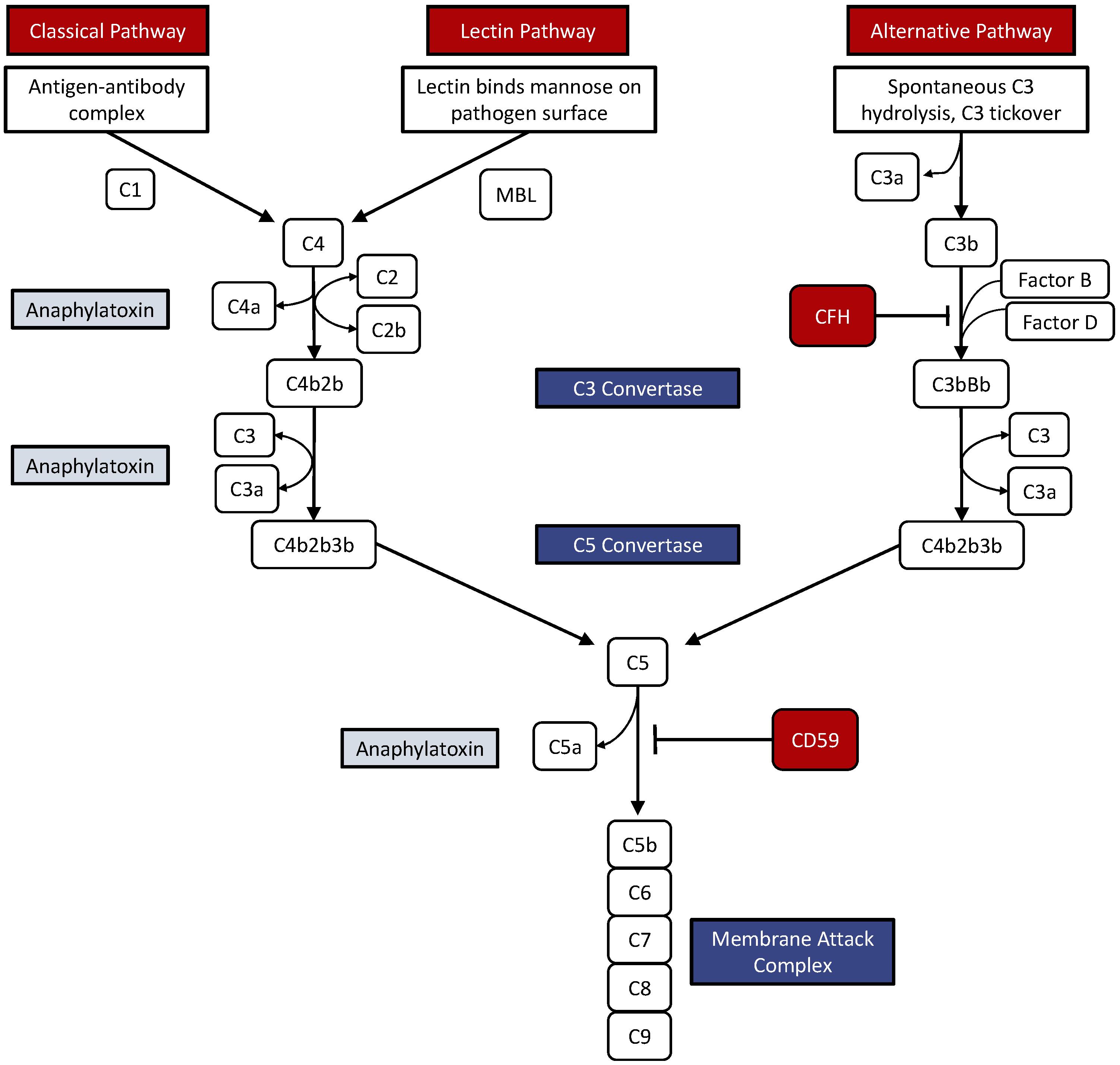
2. The Complement System
3. The Retina and the Complement System
4. Activation of the Complement System in Retinal Disease
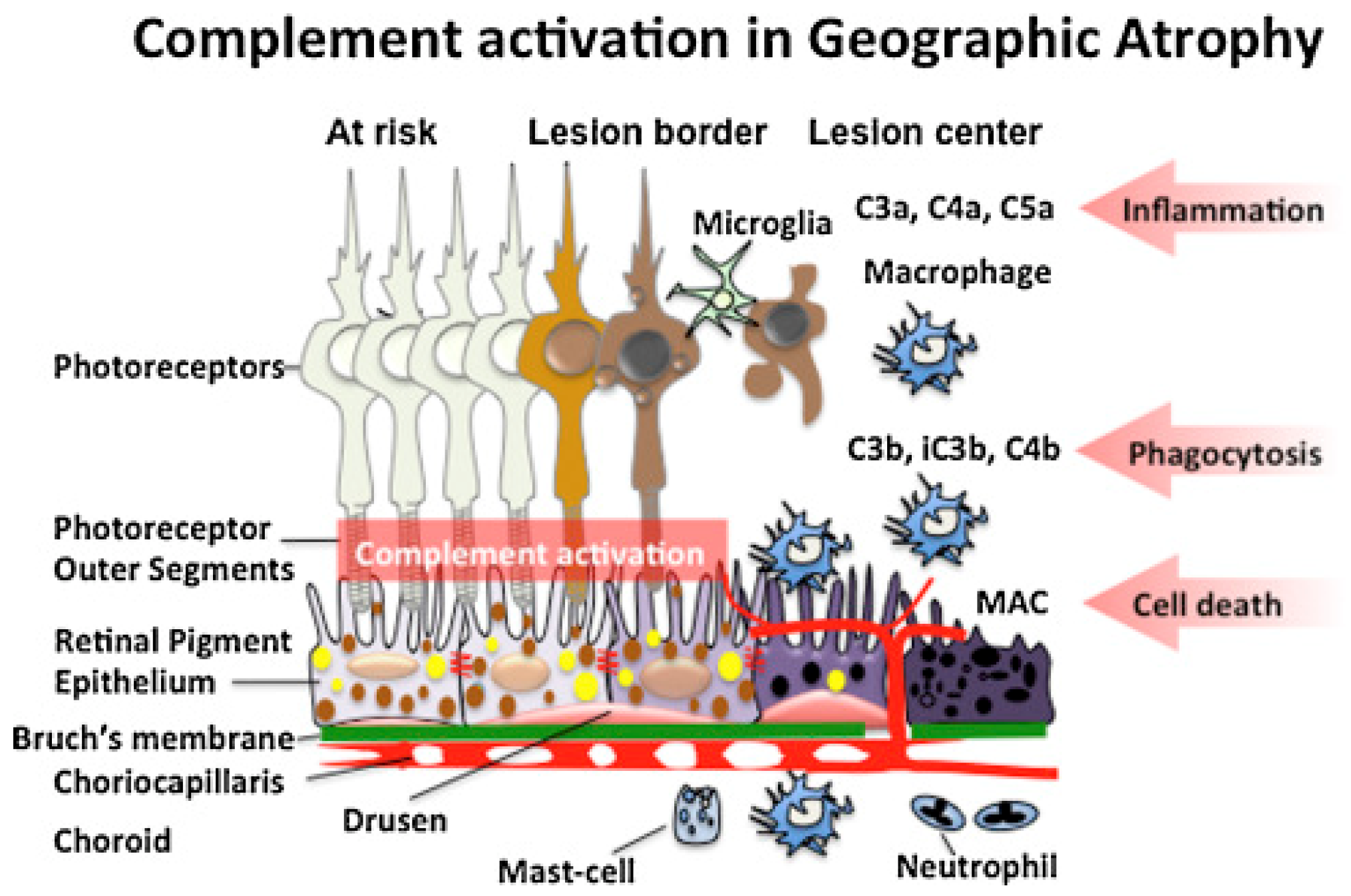
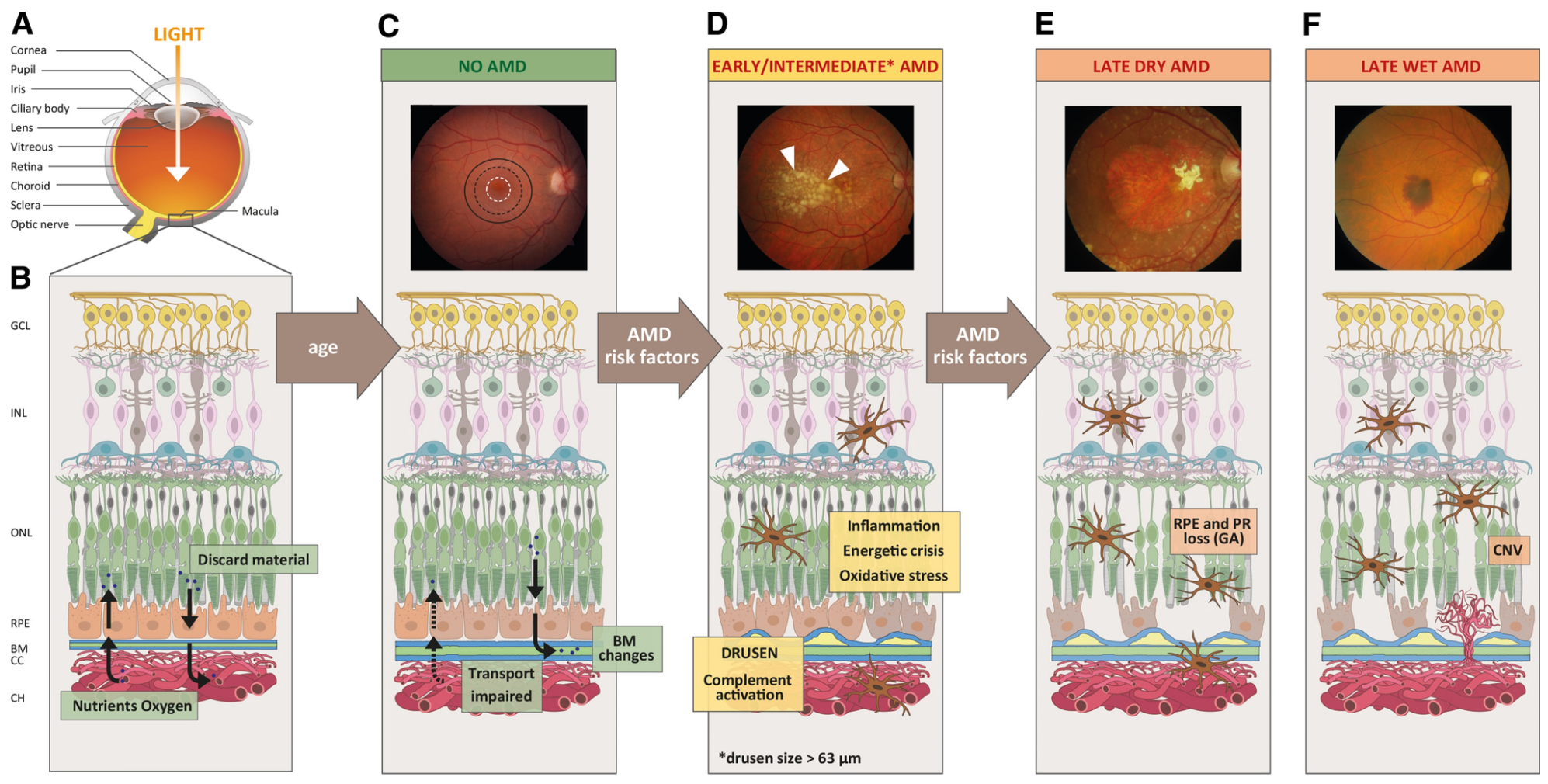
4.1. Age-Related Macular Degeneration
4.2. Complement in Uveitis
4.3. Complement in Diabetic Retinopathy
5. Targeting Complement Pathways in Retinal Disease
5.1. In vitro and Genetic Studies
5.2. Animal Studies
5.3. Clinical Trials
6. Clinically Available Inhibition Therapy in Geographic Atrophy
6.1. Imaging
6.2. Prior Treatments
7. Pegcetacoplan (SYFOVRE) for Geographic Atrophy
7.1. Mechanism of Action
7.2. Description, Dosing, and Administration
7.3. Phase 2 Clinical Trial (FILLY)
7.4. Phase 3 Clinical Trial (OAKS and DERBY)
7.5. Side Effects, Contradictions, and Reception
8. Avacincaptad Pegol (IZERVAY) for Geographic Atrophy
8.1. Mechanism of Action
8.2. Description, Dosing, and Administration
8.3. Phase 2/3 Clinical Trial (GATHER1)
8.4. Phase 3 Clinical Trial (GATHER2)
8.5. Side Effects, Contradictions, and Reception
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhou, R.; Caspi, R.R. Ocular immune privilege. F1000 Biol. Rep. 2010, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Chen, M. Targeting the complement system for the management of retinal inflammatory and degenerative diseases. Eur. J. Pharmacol. 2016, 787, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Desai, D.; Dugel, P.U. Complement cascade inhibition in geographic atrophy: A review. Eye 2022, 36, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Shahulhameed, S.; Vishwakarma, S.; Chhablani, J.; Tyagi, M.; Pappuru, R.R.; Jakati, S.; Chakrabarti, S.; Kaur, I. A Systematic Investigation on Complement Pathway Activation in Diabetic Retinopathy. Front. Immunol. 2020, 11, 154. [Google Scholar] [CrossRef] [PubMed]
- Afshar-Kharghan, V. The role of the complement system in cancer. J. Clin. Investig. 2017, 127, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; He, S. The complement system is also important in immunogenic cell death. Nat. Rev. Immunol. 2017, 17, 143. [Google Scholar] [CrossRef]
- Csuka, D.; Veszeli, N.; Varga, L.; Prohaszka, Z.; Farkas, H. The role of the complement system in hereditary angioedema. Mol. Immunol. 2017, 89, 59–68. [Google Scholar] [CrossRef]
- Hein, E.; Garred, P. The Lectin Pathway of Complement and Biocompatibility. Adv. Exp. Med. Biol. 2015, 865, 77–92. [Google Scholar] [CrossRef]
- Lintner, K.E.; Wu, Y.L.; Yang, Y.; Spencer, C.H.; Hauptmann, G.; Hebert, L.A.; Atkinson, J.P.; Yu, C.Y. Early Components of the Complement Classical Activation Pathway in Human Systemic Autoimmune Diseases. Front. Immunol. 2016, 7, 36. [Google Scholar] [CrossRef]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Travers, P.; Walport, M.; Shlomchik, M.J. The complement system and innate immunity. In Immunobiology: The Immune System in Health and Disease; Garland Science: New York, NY, USA, 2001. [Google Scholar]
- Janatova, J. C3, C5 components and C3a, C4a, and C5a fragments of the complement system. Methods Enzymol. 1988, 162, 579–625. [Google Scholar] [CrossRef] [PubMed]
- Markiewski, M.M.; Mastellos, D.; Tudoran, R.; DeAngelis, R.A.; Strey, C.W.; Franchini, S.; Wetsel, R.A.; Erdei, A.; Lambris, J.D. C3a and C3b activation products of the third component of complement (C3) are critical for normal liver recovery after toxic injury. J. Immunol. 2004, 173, 747–754. [Google Scholar] [CrossRef]
- Mead, B.; Tomarev, S. Evaluating retinal ganglion cell loss and dysfunction. Exp. Eye Res. 2016, 151, 96–106. [Google Scholar] [CrossRef]
- Sanes, J.R.; Masland, R.H. The types of retinal ganglion cells: Current status and implications for neuronal classification. Annu. Rev. Neurosci. 2015, 38, 221–246. [Google Scholar] [CrossRef]
- Chen, M.; Luo, C.; Zhao, J.; Devarajan, G.; Xu, H. Immune regulation in the aging retina. Prog. Retin. Eye Res. 2019, 69, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Medawar, P.B. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br. J. Exp. Pathol. 1948, 29, 58–69. [Google Scholar]
- Pauly, D.; Agarwal, D.; Dana, N.; Schäfer, N.; Biber, J.; Wunderlich, K.A.; Jabri, Y.; Straub, T.; Zhang, N.R.; Gautam, A.K.; et al. Cell-Type-Specific Complement Expression in the Healthy and Diseased Retina. Cell Rep. 2019, 29, 2835–2848.e2834. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Chen, M.; Xu, H. Complement gene expression and regulation in mouse retina and retinal pigment epithelium/choroid. Mol. Vis. 2011, 17, 1588–1597. [Google Scholar]
- Collier, R.J.; Wang, Y.; Smith, S.S.; Martin, E.; Ornberg, R.; Rhoades, K.; Romano, C. Complement deposition and microglial activation in the outer retina in light-induced retinopathy: Inhibition by a 5-HT1A agonist. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8108–8116. [Google Scholar] [CrossRef]
- Copland, D.A.; Hussain, K.; Baalasubramanian, S.; Hughes, T.R.; Morgan, B.P.; Xu, H.; Dick, A.D.; Nicholson, L.B. Systemic and local anti-C5 therapy reduces the disease severity in experimental autoimmune uveoretinitis. Clin. Exp. Immunol. 2010, 159, 303–314. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [PubMed]
- Mukai, R.; Okunuki, Y.; Husain, D.; Kim, C.B.; Lambris, J.D.; Connor, K.M. The Complement System Is Critical in Maintaining Retinal Integrity during Aging. Front. Aging Neurosci. 2018, 10, 15. [Google Scholar] [CrossRef] [PubMed]
- Hoh Kam, J.; Lenassi, E.; Malik, T.H.; Pickering, M.C.; Jeffery, G. Complement component C3 plays a critical role in protecting the aging retina in a murine model of age-related macular degeneration. Am. J. Pathol. 2013, 183, 480–492. [Google Scholar] [CrossRef] [PubMed]
- Stephan, A.H.; Madison, D.V.; Mateos, J.M.; Fraser, D.A.; Lovelett, E.A.; Coutellier, L.; Kim, L.; Tsai, H.H.; Huang, E.J.; Rowitch, D.H.; et al. A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci. 2013, 33, 13460–13474. [Google Scholar] [CrossRef] [PubMed]
- Nawash, B.; Ong, J.; Driban, M.; Hwang, J.; Chen, J.; Selvam, A.; Mohan, S.; Chhablani, J. Prognostic Optical Coherence Tomography Biomarkers in Neovascular Age-Related Macular Degeneration. J. Clin. Med. 2023, 12, 3049. [Google Scholar] [CrossRef] [PubMed]
- Selvam, A.; Shah, S.; Singh, S.R.; Sant, V.; Harihar, S.; Arora, S.; Patel, M.; Ong, J.; Yadav, S.; Ibrahim, M.N.; et al. Longitudinal changes in pigment epithelial detachment composition indices (PEDCI): New biomarkers in neovascular age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 2023, 262, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Pugazhendhi, A.; Hubbell, M.; Jairam, P.; Ambati, B. Neovascular Macular Degeneration: A Review of Etiology, Risk Factors, and Recent Advances in Research and Therapy. Int. J. Mol. Sci. 2021, 22, 1170. [Google Scholar] [CrossRef] [PubMed]
- Chhablani, J.; Jager, R.; Ong, J.; Lohrenz, R.; Hamilton, R.J.; Stea, B.; Drew, M.; Kokame, G.; Doheny Retina Study, G. Two-year outcomes of episcleral brachytherapy adjunct to anti-VEGF therapy for treatment-resistant nAMD. Graefes Arch. Clin. Exp. Ophthalmol. 2022, 260, 3791–3798. [Google Scholar] [CrossRef] [PubMed]
- Ferris, F.L.; Wilkinson, C.P.; Bird, A.; Chakravarthy, U.; Chew, E.; Csaky, K.; Sadda, S.R.; Beckman Initiative for Macular Research Classification Committee. Clinical classification of age-related macular degeneration. Ophthalmology 2013, 120, 844–851. [Google Scholar] [CrossRef]
- Bakri, S.J.; Bektas, M.; Sharp, D.; Luo, R.; Sarda, S.P.; Khan, S. Geographic atrophy: Mechanism of disease, pathophysiology, and role of the complement system. J. Manag. Care Spec. Pharm. 2023, 29, S2–S11. [Google Scholar] [CrossRef]
- Beatty, S.; Koh, H.-H.; Phil, M.; Henson, D.; Boulton, M. The Role of Oxidative Stress in the Pathogenesis of Age-Related Macular Degeneration. Surv. Ophthalmol. 2000, 45, 115–134. [Google Scholar] [CrossRef]
- Klein, R.; Klein, B.E.K.; Linton, K.L.P.; DeMets, D.L. The Beaver Dam Eye Study: The Relation of Age-related Maculopathy to Smoking. Am. J. Epidemiol. 1993, 137, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, U.; Wong, T.Y.; Fletcher, A.; Piault, E.; Evans, C.; Zlateva, G.; Buggage, R.; Pleil, A.; Mitchell, P. Clinical risk factors for age-related macular degeneration: A systematic review and meta-analysis. BMC Ophthalmol. 2010, 10, 31. [Google Scholar] [CrossRef]
- Choi, A.; Nawash, B.S.; Du, K.; Ong, J.; Chhablani, J. Barriers to care in neovascular age-related macular degeneration: Current understanding, developments, and future directions. Surv. Ophthalmol. 2024, 69, 160–164. [Google Scholar] [CrossRef]
- Boyer, D.S.; Schmidt-Erfurth, U.; van Lookeren Campagne, M.; Henry, E.C.; Brittain, C. The Pathophysiology of Geographic Atrophy Secondary to Age-Related Macular Degeneration and the Complement Pathway as a Therapeutic Target. Retina 2017, 37, 819–835. [Google Scholar] [CrossRef]
- Nadeem, A.; Malik, I.A.; Shariq, F.; Afridi, E.K.; Taha, M.; Raufi, N.; Naveed, A.K.; Iqbal, J.; Habte, A. Advancements in the treatment of geographic atrophy: Focus on pegcetacoplan in age-related macular degeneration. Ann. Med. Surg. 2023, 85, 6067–6077. [Google Scholar] [CrossRef]
- Anderson, D.H.; Radeke, M.J.; Gallo, N.B.; Chapin, E.A.; Johnson, P.T.; Curletti, C.R.; Hancox, L.S.; Hu, J.; Ebright, J.N.; Malek, G.; et al. The pivotal role of the complement system in aging and age-related macular degeneration: Hypothesis re-visited. Prog. Retin. Eye Res. 2010, 29, 95–112. [Google Scholar] [CrossRef]
- Sennlaub, F.; Auvynet, C.; Calippe, B.; Lavalette, S.; Poupel, L.; Hu, S.J.; Dominguez, E.; Camelo, S.; Levy, O.; Guyon, E.; et al. CCR2+ monocytes infiltrate atrophic lesions in age-related macular disease and mediate photoreceptor degeneration in experimental subretinal inflammation in Cx3cr1 deficient mice. EMBO Mol. Med. 2013, 5, 1775–1793. [Google Scholar] [CrossRef] [PubMed]
- Selvam, A.; Singh, S.R.; Arora, S.; Patel, M.; Kuchhal, A.; Shah, S.; Ong, J.; Rasheed, M.A.; Manne, S.R.; Ibrahim, M.N.; et al. Pigment epithelial detachment composition indices (PEDCI) in neovascular age-related macular degeneration. Sci. Rep. 2023, 13, 68. [Google Scholar] [CrossRef] [PubMed]
- Campagne, M.L.; Strauss, E.C.; Yaspan, B.L. Age-related macular degeneration: Complement in action. Immunobiology 2016, 6, 733–739. [Google Scholar] [CrossRef]
- Curcio, C.A.; Johnson, M.; Rudolf, M.; Huang, J.D. The oil spill in ageing Bruch membrane. Br. J. Ophthalmol. 2011, 95, 1638–1645. [Google Scholar] [CrossRef] [PubMed]
- Curcio, C.A.; Millican, C.L.; Bailey, T.; Kruth, H.S. Accumulation of cholesterol with age in human Bruch’s membrane. Investig. Ophthalmol. Vis. Sci. 2001, 42, 265–274. [Google Scholar]
- Pauleikhoff, D.; Harper, C.A.; Marshall, J.; Bird, A.C. Aging changes in Bruch’s membrane. A histochemical and morphologic study. Ophthalmology 1990, 97, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Ethier, C.R.; Johnson, M.; Ruberti, J. Ocular biomechanics and biotransport. Annu. Rev. Biomed. Eng. 2004, 6, 249–273. [Google Scholar] [CrossRef] [PubMed]
- Friedman, E. A hemodynamic model of the pathogenesis of age-related macular degeneration. Am. J. Ophthalmol. 1997, 124, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Pauleikhoff, D.; Chen, J.C.; Chisholm, I.H.; Bird, A.C. Choroidal perfusion abnormality with age-related Bruch’s membrane change. Am. J. Ophthalmol. 1990, 109, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Armento, A.; Ueffing, M.; Clark, S.J. The complement system in age-related macular degeneration. Cell Mol. Life Sci. 2021, 78, 4487–4505. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.; Bi, C.; Wang, C.; Zhou, H.; Takeuchi, M. The Relationship between Complements and Age-Related Macular Degeneration and Its Pathogenesis. J. Ophthalmol. 2024, 2024, 6416773. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef]
- Sofat, R.; Casas, J.P.; Webster, A.R.; Bird, A.C.; Mann, S.S.; Yates, J.R.; Moore, A.T.; Sepp, T.; Cipriani, V.; Bunce, C.; et al. Complement factor H genetic variant and age-related macular degeneration: Effect size, modifiers and relationship to disease subtype. Int. J. Epidemiol. 2012, 41, 250–262. [Google Scholar] [CrossRef]
- Seddon, J.M.; Gensler, G.; Rosner, B. C-reactive protein and CFH, ARMS2/HTRA1 gene variants are independently associated with risk of macular degeneration. Ophthalmology 2010, 117, 1560–1566. [Google Scholar] [CrossRef] [PubMed]
- Fagerness, J.A.; Maller, J.B.; Neale, B.M.; Reynolds, R.C.; Daly, M.J.; Seddon, J.M. Variation near complement factor I is associated with risk of advanced AMD. Eur. J. Hum. Genet. 2009, 17, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, Y.; Zhang, M.N. Complement factor B polymorphism (rs641153) and susceptibility to age-related macular degeneration: Evidence from published studies. Int. J. Ophthalmol. 2013, 6, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhang, J.; Ding, Y.; Huang, H.; Li, Y.; Chen, W. Predicting late-stage age-related macular degeneration by integrating marginally weak SNPs in GWA studies. Front. Genet. 2023, 14, 1075824. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, B.; Lukas, T.J.; Neufeld, A.H. The aged retinal pigment epithelium/choroid: A potential substratum for the pathogenesis of age-related macular degeneration. PLoS ONE 2008, 3, e2339. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Muckersie, E.; Forrester, J.V.; Xu, H. Immune activation in retinal aging: A gene expression study. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5888–5896. [Google Scholar] [CrossRef] [PubMed]
- Scholl, H.P.; Charbel Issa, P.; Walier, M.; Janzer, S.; Pollok-Kopp, B.; Börncke, F.; Fritsche, L.G.; Chong, N.V.; Fimmers, R.; Wienker, T.; et al. Systemic complement activation in age-related macular degeneration. PLoS ONE 2008, 3, e2593. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.J. Subretinal neovascularization. Natural history of an experimental model. Arch. Ophthalmol. 1982, 100, 1804–1809. [Google Scholar] [CrossRef] [PubMed]
- Bora, P.S.; Sohn, J.H.; Cruz, J.M.; Jha, P.; Nishihori, H.; Wang, Y.; Kaliappan, S.; Kaplan, H.J.; Bora, N.S. Role of complement and complement membrane attack complex in laser-induced choroidal neovascularization. J. Immunol. 2005, 174, 491–497. [Google Scholar] [CrossRef]
- Bora, N.S.; Kaliappan, S.; Jha, P.; Xu, Q.; Sohn, J.H.; Dhaulakhandi, D.B.; Kaplan, H.J.; Bora, P.S. Complement activation via alternative pathway is critical in the development of laser-induced choroidal neovascularization: Role of factor B and factor H. J. Immunol. 2006, 177, 1872–1878. [Google Scholar] [CrossRef]
- Nozaki, M.; Raisler, B.J.; Sakurai, E.; Sarma, J.V.; Barnum, S.R.; Lambris, J.D.; Chen, Y.; Zhang, K.; Ambati, B.K.; Baffi, J.Z.; et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. USA 2006, 103, 2328–2333. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.; Aziz, A.A.; Sulahria, H.; Ahmed, A.; Choudhry, N.; Narayanan, R.; Danzig, C.; Khanani, A.M. Emerging Treatment Options for Geographic Atrophy (GA) Secondary to Age-Related Macular Degeneration. Clin. Ophthalmol. 2023, 17, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, G.J.; Westby, K.; Csaky, K.G.; Monés, J.; Pearlman, J.A.; Patel, S.S.; Joondeph, B.C.; Randolph, J.; Masonson, H.; Rezaei, K.A. C5 Inhibitor Avacincaptad Pegol for Geographic Atrophy Due to Age-Related Macular Degeneration: A Randomized Pivotal Phase 2/3 Trial. Ophthalmology 2021, 128, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Schalken, J.J.; Winkens, H.J.; van Vugt, A.H.; Bovée-Geurts, P.H.; de Grip, W.J.; Broekhuyse, R.M. Rhodopsin-induced experimental autoimmune uveoretinitis: Dose-dependent clinicopathological features. Exp. Eye Res. 1988, 47, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.K.; Caspi, R.R. Rodent models of experimental autoimmune uveitis. Methods Mol. Med. 2004, 102, 395–419. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Biswas, S.; Anand, R.; Agarwal, S.S. Experimental autoimmune uveitis as animal model for human posterior uveitis. Indian. J. Med. Res. 1998, 107, 53–67. [Google Scholar] [PubMed]
- Heeger, P.S.; Lalli, P.N.; Lin, F.; Valujskikh, A.; Liu, J.; Muqim, N.; Xu, Y.; Medof, M.E. Decay-accelerating factor modulates induction of T cell immunity. J. Exp. Med. 2005, 201, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Miwa, T.; Hilliard, B.; Chen, Y.; Lambris, J.D.; Wells, A.D.; Song, W.C. The complement inhibitory protein DAF (CD55) suppresses T cell immunity in vivo. J. Exp. Med. 2005, 201, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Read, R.W.; Szalai, A.J.; Vogt, S.D.; McGwin, G.; Barnum, S.R. Genetic deficiency of C3 as well as CNS-targeted expression of the complement inhibitor sCrry ameliorates experimental autoimmune uveoretinitis. Exp. Eye Res. 2006, 82, 389–394. [Google Scholar] [CrossRef]
- Kumar, B.; Cashman, S.M.; Kumar-Singh, R. Complement-Mediated Activation of the NLRP3 Inflammasome and Its Inhibition by AAV-Mediated Delivery of CD59 in a Model of Uveitis. Mol. Ther. 2018, 26, 1568–1580. [Google Scholar] [CrossRef]
- Lundeen, E.A.; Burke-Conte, Z.; Rein, D.B.; Wittenborn, J.S.; Saaddine, J.; Lee, A.Y.; Flaxman, A.D. Prevalence of Diabetic Retinopathy in the US in 2021. JAMA Ophthalmol. 2023, 141, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Gerl, V.B.; Bohl, J.; Pitz, S.; Stoffelns, B.; Pfeiffer, N.; Bhakdi, S. Extensive deposits of complement C3d and C5b-9 in the choriocapillaris of eyes of patients with diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1104–1108. [Google Scholar]
- Yanai, R.; Thanos, A.; Connor, K.M. Complement involvement in neovascular ocular diseases. Adv. Exp. Med. Biol. 2012, 946, 161–183. [Google Scholar] [CrossRef]
- García-Ramírez, M.; Canals, F.; Hernández, C.; Colomé, N.; Ferrer, C.; Carrasco, E.; García-Arumí, J.; Simó, R. Proteomic analysis of human vitreous fluid by fluorescence-based difference gel electrophoresis (DIGE): A new strategy for identifying potential candidates in the pathogenesis of proliferative diabetic retinopathy. Diabetologia 2007, 50, 1294–1303. [Google Scholar] [CrossRef]
- Huang, C.; Fisher, K.P.; Hammer, S.S.; Navitskaya, S.; Blanchard, G.J.; Busik, J.V. Plasma Exosomes Contribute to Microvascular Damage in Diabetic Retinopathy by Activating the Classical Complement Pathway. Diabetes 2018, 67, 1639–1649. [Google Scholar] [CrossRef]
- Ghosh, P.; Vaidya, A.; Sahoo, R.; Goldfine, A.; Herring, N.; Bry, L.; Chorev, M.; Halperin, J.A. Glycation of the complement regulatory protein CD59 is a novel biomarker for glucose handling in humans. J. Clin. Endocrinol. Metab. 2014, 99, E999–E1006. [Google Scholar] [CrossRef]
- Wang, J.; Yang, M.M.; Li, Y.B.; Liu, G.D.; Teng, Y.; Liu, X.M. Association of CFH and CFB gene polymorphisms with retinopathy in type 2 diabetic patients. Mediat. Inflamm. 2013, 2013, 748435. [Google Scholar] [CrossRef] [PubMed]
- Toni, M.; Hermida, J.; Toledo, E.; Goñi, M.J.; Díez Goñi, N. Role of CFH and ARMS2 polymorphisms in retinopathy and coronary artery disease in type 1 diabetes. An. Sist. Sanit. Navar. 2012, 35, 425–432. [Google Scholar] [CrossRef]
- Yang, M.M.; Wang, J.; Ren, H.; Sun, Y.D.; Fan, J.J.; Teng, Y.; Li, Y.B. Genetic Investigation of Complement Pathway Genes in Type 2 Diabetic Retinopathy: An Inflammatory Perspective. Mediat. Inflamm. 2016, 2016, 1313027. [Google Scholar] [CrossRef]
- Kishore, U.; Reid, K.B. C1q: Structure, function, and receptors. Immunopharmacology 2000, 49, 159–170. [Google Scholar] [CrossRef]
- Yednock, T.; Fong, D.S.; Lad, E.M. C1q and the classical complement cascade in geographic atrophy secondary to age-related macular degeneration. Int. J. Retin. Vitr. 2022, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.V.; Forest, D.L.; Banna, C.D.; Radeke, C.M.; Maloney, M.A.; Hu, J.; Spencer, C.N.; Walker, A.M.; Tsie, M.S.; Bok, D.; et al. Cell culture model that mimics drusen formation and triggers complement activation associated with age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 18277–18282. [Google Scholar] [CrossRef]
- Taylor, L.; Arnér, K.; Blom, A.M.; Ghosh, F. Complimentary action: C1q increases ganglion cell survival in an in vitro model of retinal degeneration. J. Neuroimmunol. 2016, 298, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M. Human C1-esterase inhibitor concentrate (Berinert). BioDrugs 2009, 23, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Panicker, S.; Shi, J.; Rose, E.; Hussain, S.; Tom, S.; Strober, W.; Sloan, S.R.; Parry, G.; Stagliano, N. TNT009, a Classical Complement Pathway Specific Inhibitor, Prevents Complement Dependent Hemolysis Induced by Cold Agglutinin Disease Patient Autoantibodies. Blood 2013, 122, 42. [Google Scholar] [CrossRef]
- Wahrmann, M.; Mühlbacher, J.; Marinova, L.; Regele, H.; Huttary, N.; Eskandary, F.; Cohen, G.; Fischer, G.F.; Parry, G.C.; Gilbert, J.C.; et al. Effect of the Anti-C1s Humanized Antibody TNT009 and Its Parental Mouse Variant TNT003 on HLA Antibody-Induced Complement Activation—A Preclinical In Vitro Study. Am. J. Transplant. 2017, 17, 2300–2311. [Google Scholar] [CrossRef]
- Jeon, B.; Kim, H.R.; Kim, H.; Chung, D.K. In vitro and in vivo downregulation of C3 by lipoteichoic acid isolated from Lactobacillus plantarum K8 suppressed cytokine-mediated complement system activation. FEMS Microbiol. Lett. 2016, 363, fnw140. [Google Scholar] [CrossRef] [PubMed]
- Schepers, A.; Vries, M.R.d.; Leuven, C.J.v.; Grimbergen, J.M.; Holers, V.M.; Daha, M.R.; Bockel, J.H.v.; Quax, P.H.A. Inhibition of Complement Component C3 Reduces Vein Graft Atherosclerosis in Apolipoprotein E3–Leiden Transgenic Mice. Circulation 2006, 114, 2831–2838. [Google Scholar] [CrossRef] [PubMed]
- Cerniauskas, E.; Kurzawa-Akanbi, M.; Xie, L.; Hallam, D.; Moya-Molina, M.; White, K.; Steel, D.; Doherty, M.; Whitfield, P.; Al-Aama, J.; et al. Complement modulation reverses pathology in Y402H-retinal pigment epithelium cell model of age-related macular degeneration by restoring lysosomal function. Stem Cells Transl. Med. 2020, 9, 1585–1603. [Google Scholar] [CrossRef]
- Haynes, T.; Luz-Madrigal, A.; Reis, E.S.; Echeverri Ruiz, N.P.; Grajales-Esquivel, E.; Tzekou, A.; Tsonis, P.A.; Lambris, J.D.; Del Rio-Tsonis, K. Complement anaphylatoxin C3a is a potent inducer of embryonic chick retina regeneration. Nat. Commun. 2013, 4, 2312. [Google Scholar] [CrossRef]
- Fernandez-Godino, R.; Pierce, E.A. C3a triggers formation of sub-retinal pigment epithelium deposits via the ubiquitin proteasome pathway. Sci. Rep. 2018, 8, 9679. [Google Scholar] [CrossRef] [PubMed]
- Gorham, R.D., Jr.; Forest, D.L.; Tamamis, P.; López de Victoria, A.; Kraszni, M.; Kieslich, C.A.; Banna, C.D.; Bellows-Peterson, M.L.; Larive, C.K.; Floudas, C.A.; et al. Novel compstatin family peptides inhibit complement activation by drusen-like deposits in human retinal pigmented epithelial cell cultures. Exp. Eye Res. 2013, 116, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Halstead, S.K.; Humphreys, P.D.; Zitman, F.M.; Hamer, J.; Plomp, J.J.; Willison, H.J. C5 inhibitor rEV576 protects against neural injury in an in vitro mouse model of Miller Fisher syndrome. J. Peripher. Nerv. Syst. 2008, 13, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Brandstetter, C.; Holz, F.G.; Krohne, T.U. Complement Component C5a Primes Retinal Pigment Epithelial Cells for Inflammasome Activation by Lipofuscin-mediated Photooxidative Damage. J. Biol. Chem. 2015, 290, 31189–31198. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Xu, H.; Gong, X.; Shen, J.; Chen, X.; Wu, Z. The complement C3a-C3aR and C5a-C5aR pathways promote viability and inflammation of human retinal pigment epithelium cells by targeting NF-κB signaling. Exp. Ther. Med. 2022, 24, 493. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.J.; Efstathiou, N.E.; Konstantinou, E.K.; Maidana, D.E.; Miller, J.W.; Young, L.H.; Vavvas, D.G. AICAR suppresses TNF-α-induced complement factor B in RPE cells. Sci. Rep. 2017, 7, 17651. [Google Scholar] [CrossRef]
- Chen, M.; Muckersie, E.; Robertson, M.; Forrester, J.V.; Xu, H. Up-regulation of complement factor B in retinal pigment epithelial cells is accompanied by complement activation in the aged retina. Exp. Eye Res. 2008, 87, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Schubart, A.; Anderson, K.; Mainolfi, N.; Sellner, H.; Ehara, T.; Adams, C.M.; Mac Sweeney, A.; Liao, S.M.; Crowley, M.; Littlewood-Evans, A.; et al. Small-molecule factor B inhibitor for the treatment of complement-mediated diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 7926–7931. [Google Scholar] [CrossRef]
- Sultan, E.Y.; Rizk, D.E.; Kenawy, H.I.; Hassan, R. A small fragment of factor B as a potential inhibitor of complement alternative pathway activity. Immunobiology 2021, 226, 152106. [Google Scholar] [CrossRef]
- Barratt, J.; Weitz, I. Complement Factor D as a Strategic Target for Regulating the Alternative Complement Pathway. Front. Immunol. 2021, 12, 712572. [Google Scholar] [CrossRef]
- Harris, C.L. Expanding horizons in complement drug discovery: Challenges and emerging strategies. Semin. Immunopathol. 2018, 40, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Gavriilaki, E.; Thanassi, J.A.; Yang, G.; Baines, A.C.; Podos, S.D.; Huang, Y.; Huang, M.; Brodsky, R.A. Small-molecule factor D inhibitors selectively block the alternative pathway of complement in paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Haematologica 2017, 102, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Boyer, D.D.; Ko, Y.P.; Podos, S.D.; Cartwright, M.E.; Gao, X.; Wiles, J.A.; Huang, M. Danicopan, an Oral Complement Factor D Inhibitor, Exhibits High and Sustained Exposure in Ocular Tissues in Preclinical Studies. Transl. Vis. Sci. Technol. 2022, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, V.; Lorés-Motta, L.; He, F.; Fathalla, D.; Tilakaratna, V.; McHarg, S.; Bayatti, N.; Acar, İ.E.; Hoyng, C.B.; Fauser, S.; et al. Increased circulating levels of Factor H-Related Protein 4 are strongly associated with age-related macular degeneration. Nat. Commun. 2020, 11, 778. [Google Scholar] [CrossRef] [PubMed]
- Clermont, A.; Chilcote, T.J.; Kita, T.; Liu, J.; Riva, P.; Sinha, S.; Feener, E.P. Plasma kallikrein mediates retinal vascular dysfunction and induces retinal thickening in diabetic rats. Diabetes 2011, 60, 1590–1598. [Google Scholar] [CrossRef] [PubMed]
- Irmscher, S.; Döring, N.; Halder, L.D.; Jo, E.A.H.; Kopka, I.; Dunker, C.; Jacobsen, I.D.; Luo, S.; Slevogt, H.; Lorkowski, S.; et al. Kallikrein Cleaves C3 and Activates Complement. J. Innate Immun. 2018, 10, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Rutar, M.; Fernando, N.; Yednock, T.; Sankaranarayanan, S.; Aggio-Bruce, R.; Provis, J.; Natoli, R. Subretinal macrophages produce classical complement activator C1q leading to the progression of focal retinal degeneration. Mol. Neurodegener. 2018, 13, 45. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Sankaranarayanan, S.; Mathur, V.; Suri, P.; Qiu, H.; Andrews-Zwilling, Y.; Mease, K.; Taylor, L.K.; Cahir-McFarland, E.; Keswani, S.; et al. Pharmacokinetic and Target Engagement Measures of ANX007, an Anti-C1q Antibody Fragment, Following Intravitreal Administration in Nonhuman Primates. Investig. Ophthalmol. Vis. Sci. 2023, 64, 3. [Google Scholar] [CrossRef] [PubMed]
- Chi, Z.L.; Yoshida, T.; Lambris, J.D.; Iwata, T. Suppression of drusen formation by compstatin, a peptide inhibitor of complement C3 activation, on cynomolgus monkey with early-onset macular degeneration. Adv. Exp. Med. Biol. 2010, 703, 127–135. [Google Scholar] [CrossRef]
- Hughes, S.; Gumas, J.; Lee, R.; Rumano, M.; Berger, N.; Gautam, A.K.; Sfyroera, G.; Chan, A.L.; Gnanaguru, G.; Connor, K.M.; et al. Prolonged intraocular residence and retinal tissue distribution of a fourth-generation compstatin-based C3 inhibitor in non-human primates. Clin. Immunol. 2020, 214, 108391. [Google Scholar] [CrossRef]
- Natoli, R.; Fernando, N.; Jiao, H.; Racic, T.; Madigan, M.; Barnett, N.L.; Chu-Tan, J.A.; Valter, K.; Provis, J.; Rutar, M. Retinal Macrophages Synthesize C3 and Activate Complement in AMD and in Models of Focal Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2977–2990. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Peng, G.-H. Complement C3 deficiency alleviates alkylation-induced retinal degeneration in mice. Eye Vis. 2022, 9, 22. [Google Scholar] [CrossRef]
- Cashman, S.M.; Desai, A.; Ramo, K.; Kumar-Singh, R. Expression of Complement Component 3 (C3) from an Adenovirus Leads to Pathology in the Murine Retina. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3436–3445. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Mastellos, D.C.; Li, Y.; Dunaief, J.L.; Lambris, J.D. Targeting complement components C3 and C5 for the retina: Key concepts and lingering questions. Prog. Retin. Eye Res. 2021, 83, 100936. [Google Scholar] [CrossRef] [PubMed]
- Annamalai, B.; Parsons, N.; Nicholson, C.; Obert, E.; Jones, B.; Rohrer, B. Subretinal Rather Than Intravitreal Adeno-Associated Virus–Mediated Delivery of a Complement Alternative Pathway Inhibitor Is Effective in a Mouse Model of RPE Damage. Investig. Ophthalmol. Vis. Sci. 2021, 62, 11. [Google Scholar] [CrossRef] [PubMed]
- Ramos de Carvalho, J.E.; Klaassen, I.; Vogels, I.M.C.; Schipper-Krom, S.; van Noorden, C.J.F.; Reits, E.; Gorgels, T.G.M.F.; Bergen, A.A.B.; Schlingemann, R.O. Complement Factor C3a Alters Proteasome Function in Human RPE Cells and in an Animal Model of Age-Related RPE Degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6489–6501. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Du, L.; Yuan, S.; Peng, G.-H. Complement C3a receptor inactivation attenuates retinal degeneration induced by oxidative damage. Front. Neurosci. 2022, 16, 951491. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Sulewski, M.E.; Wang, C.; Song, J.; Bhuyan, R.; Sterling, J.; Clark, E.; Song, W.-C.; Dunaief, J.L. Complement C5a receptor knockout has diminished light-induced microglia/macrophage retinal migration. Mol. Vis. 2017, 23, 210–218. [Google Scholar] [PubMed]
- Yu, M.; Zou, W.; Peachey, N.S.; McIntyre, T.M.; Liu, J. A Novel Role of Complement in Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7684–7692. [Google Scholar] [CrossRef]
- Jo, D.H.; Kim, J.H.; Yang, W.; Kim, H.; Chang, S.; Kim, D.; Chang, M.; Lee, K.; Chung, J.; Kim, J.H. Anti-complement component 5 antibody targeting MG4 domain inhibits choroidal neovascularization. Oncotarget 2017, 8, 45506–45516. [Google Scholar] [CrossRef]
- Gassel, C.J.; Reinehr, S.; Gomes, S.C.; Dick, H.B.; Joachim, S.C. Preservation of optic nerve structure by complement inhibition in experimental glaucoma. Cell Tissue Res. 2020, 382, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, S.; Gomes, S.C.; Gassel, C.J.; Asaad, M.A.; Stute, G.; Schargus, M.; Dick, H.B.; Joachim, S.C. Intravitreal Therapy against the Complement Factor C5 Prevents Retinal Degeneration in an Experimental Autoimmune Glaucoma Model. Front. Pharmacol. 2019, 10, 1381. [Google Scholar] [CrossRef] [PubMed]
- Garland, D.L.; Pierce, E.A.; Fernandez-Godino, R. Complement C5 is not critical for the formation of sub-RPE deposits in Efemp1 mutant mice. Sci. Rep. 2021, 11, 10416. [Google Scholar] [CrossRef] [PubMed]
- Toomey, C.B.; Landowski, M.; Klingeborn, M.; Kelly, U.; Deans, J.; Dong, H.; Harrabi, O.; Van Blarcom, T.; Yeung, Y.A.; Grishanin, R.; et al. Effect of Anti-C5a Therapy in a Murine Model of Early/Intermediate Dry Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2018, 59, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Llorián-Salvador, M.; Byrne, E.M.; Szczepan, M.; Little, K.; Chen, M.; Xu, H. Complement activation contributes to subretinal fibrosis through the induction of epithelial-to-mesenchymal transition (EMT) in retinal pigment epithelial cells. J. Neuroinflamm. 2022, 19, 182. [Google Scholar] [CrossRef] [PubMed]
- Langer, H.F.; Chung, K.J.; Orlova, V.V.; Choi, E.Y.; Kaul, S.; Kruhlak, M.J.; Alatsatianos, M.; DeAngelis, R.A.; Roche, P.A.; Magotti, P.; et al. Complement-mediated inhibition of neovascularization reveals a point of convergence between innate immunity and angiogenesis. Blood 2010, 116, 4395–4403. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Kim, J.; Lee, J.; Cho, S.Y.; Kang, H.J.; Kim, K.Y.; Jin, D.K. Intravitreal human complement factor H in a rat model of laser-induced choroidal neovascularisation. Br. J. Ophthalmol. 2013, 97, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Bora, N.S.; Jha, P.; Lyzogubov, V.V.; Kaliappan, S.; Liu, J.; Tytarenko, R.G.; Fraser, D.A.; Morgan, B.P.; Bora, P.S. Recombinant membrane-targeted form of CD59 inhibits the growth of choroidal neovascular complex in mice. J. Biol. Chem. 2010, 285, 33826–33833. [Google Scholar] [CrossRef]
- Crowley, M.A.; Garland, D.L.; Sellner, H.; Banks, A.; Fan, L.; Rejtar, T.; Buchanan, N.; Delgado, O.; Xu, Y.Y.; Jose, S.; et al. Complement factor B is critical for sub-RPE deposit accumulation in a model of Doyne honeycomb retinal dystrophy with features of age-related macular degeneration. Hum. Mol. Genet. 2023, 32, 204–217. [Google Scholar] [CrossRef]
- Marin, A.I.; Poppelaars, F.; Wagner, B.D.; Palestine, A.G.; Patnaik, J.L.; Holers, V.M.; Frazer-Abel, A.A.; Mathias, M.T.; Manoharan, N.; Fonteh, C.N.; et al. Sex and Age-Related Differences in Complement Factors among Patients with Intermediate Age-Related Macular Degeneration. Transl. Vis. Sci. Technol. 2022, 11, 22. [Google Scholar] [CrossRef]
- Sweigard, J.H.; Yanai, R.; Gaissert, P.; Saint-Geniez, M.; Kataoka, K.; Thanos, A.; Stahl, G.L.; Lambris, J.D.; Connor, K.M. The alternative complement pathway regulates pathological angiogenesis in the retina. FASEB J. 2014, 28, 3171–3182. [Google Scholar] [CrossRef]
- Rohrer, B.; Guo, Y.; Kunchithapautham, K.; Gilkeson, G.S. Eliminating complement factor D reduces photoreceptor susceptibility to light-induced damage. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5282–5289. [Google Scholar] [CrossRef] [PubMed]
- Crowley, M.A.; Delgado, O.; Will-Orrego, A.; Buchanan, N.M.; Anderson, K.; Jaffee, B.D.; Dryja, T.P.; Liao, S.-M. Induction of Ocular Complement Activation by Inflammatory Stimuli and Intraocular Inhibition of Complement Factor D in Animal Models. Investig. Ophthalmol. Vis. Sci. 2018, 59, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Le, K.N.; Gibiansky, L.; Good, J.; Davancaze, T.; van Lookeren Campagne, M.; Loyet, K.M.; Morimoto, A.; Jin, J.; Damico-Beyer, L.A.; Hanley, W.D. A mechanistic pharmacokinetic/pharmacodynamic model of factor D inhibition in cynomolgus monkeys by lampalizumab for the treatment of geographic atrophy. J. Pharmacol. Exp. Ther. 2015, 355, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Loyet, K.M.; Good, J.; Davancaze, T.; Sturgeon, L.; Wang, X.; Yang, J.; Le, K.N.; Wong, M.; Hass, P.E.; van Lookeren Campagne, M.; et al. Complement inhibition in cynomolgus monkeys by anti-factor d antigen-binding fragment for the treatment of an advanced form of dry age-related macular degeneration. J. Pharmacol. Exp. Ther. 2014, 351, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Boyer, D.; Rivera, J.; Ko, Y.-P.; Podos, S.D.; Cartwright, M.; Gao, X.; Wiles, J.; Huang, M. Oral Administration of the Complement Factor D Inhibitor Danicopan (ALXN2040) in Preclinical Studies Demonstrates High and Sustained Drug Concentrations in Posterior Ocular Tissues for the Potential Treatment of Geographic Atrophy. Investig. Ophthalmol. Vis. Sci. 2021, 62, 187. [Google Scholar]
- Dreismann, A.K.; McClements, M.E.; Barnard, A.R.; Orhan, E.; Hughes, J.P.; Lachmann, P.J.; MacLaren, R.E. Functional expression of complement factor I following AAV-mediated gene delivery in the retina of mice and human cells. Gene Ther. 2021, 28, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Ellis, S.; Buchberger, A.; Holder, J.; Orhan, e.; Hughes, J. GT005, a gene therapy for the treatment of dry age-related macular degeneration (AMD). Investig. Ophthalmol. Vis. Sci. 2020, 61, 2295. [Google Scholar]
- Tradtrantip, L.; Asavapanumas, N.; Phuan, P.W.; Verkman, A.S. Potential therapeutic benefit of C1-esterase inhibitor in neuromyelitis optica evaluated in vitro and in an experimental rat model. PLoS ONE 2014, 9, e106824. [Google Scholar] [CrossRef] [PubMed]
- Simmons, K.T.; Chan, J.; Hussain, S.; Rose, E.L.; Markham, K.; Byun, T.S.; Paniker, S.; Parry, G.C.; Storek, M. Anti-C1s humanized monoclonal antibody SAR445088: A classical pathway complement inhibitor specific for the active form of C1s. Clin. Immunol. 2023, 251, 109629. [Google Scholar] [CrossRef]
- Levy, M.; Mealy, M.A. Purified human C1-esterase inhibitor is safe in acute relapses of neuromyelitis optica. Neurol. Neuroimmunol. Neuroinflamm. 2014, 1, e5. [Google Scholar] [CrossRef] [PubMed]
- Wykoff, C.C.; Hershberger, V.; Eichenbaum, D.; Henry, E.; Younis, H.S.; Chandra, P.; Yuan, N.; Solloway, M.; DePaoli, A. Inhibition of Complement Factor 3 in Geographic Atrophy with NGM621: Phase 1 Dose-Escalation Study Results. Am. J. Ophthalmol. 2022, 235, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Mastellos, D.C.; Yancopoulou, D.; Kokkinos, P.; Huber-Lang, M.; Hajishengallis, G.; Biglarnia, A.R.; Lupu, F.; Nilsson, B.; Risitano, A.M.; Ricklin, D.; et al. Compstatin: A C3-targeted complement inhibitor reaching its prime for bedside intervention. Eur. J. Clin. Investig. 2015, 45, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.G.; Park, Y.S.; Kim, I.-B. Complement System and Potential Therapeutics in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2021, 22, 6851. [Google Scholar] [CrossRef] [PubMed]
- Kang, C. Avacincaptad Pegol: First Approval. Drugs 2023, 83, 1447–1453. [Google Scholar] [CrossRef] [PubMed]
- Yehoshua, Z.; de Amorim Garcia Filho, C.A.; Nunes, R.P.; Gregori, G.; Penha, F.M.; Moshfeghi, A.A.; Zhang, K.; Sadda, S.; Feuer, W.; Rosenfeld, P.J. Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: The COMPLETE study. Ophthalmology 2014, 121, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, J.I.; Ando, K.; Masuko, M.; Noji, H.; Ito, Y.; Mayer, J.; Griskevicius, L.; Bucher, C.; Müllershausen, F.; Gergely, P.; et al. Tesidolumab (LFG316) for treatment of C5-variant patients with paroxysmal nocturnal hemoglobinuria. Haematologica 2022, 107, 1483–1488. [Google Scholar] [CrossRef]
- Jaffe, G.J.; Sahni, J.; Fauser, S.; Geary, R.S.; Schneider, E.; McCaleb, M. Development of IONIS-FB-LRx to Treat Geographic Atrophy Associated with AMD. Investig. Ophthalmol. Vis. Sci. 2020, 61, 4305. [Google Scholar]
- Holz, F.G.; Sadda, S.R.; Busbee, B.; Chew, E.Y.; Mitchell, P.; Tufail, A.; Brittain, C.; Ferrara, D.; Gray, S.; Honigberg, L.; et al. Efficacy and Safety of Lampalizumab for Geographic Atrophy Due to Age-Related Macular Degeneration: Chroma and Spectri Phase 3 Randomized Clinical Trials. JAMA Ophthalmol. 2018, 136, 666–677. [Google Scholar] [CrossRef]
- Lee, J.W.; Griffin, M.; Kim, J.S.; Lee Lee, L.W.; Piatek, C.; Nishimura, J.I.; Carrillo Infante, C.; Jain, D.; Liu, P.; Filippov, G.; et al. Addition of danicopan to ravulizumab or eculizumab in patients with paroxysmal nocturnal haemoglobinuria and clinically significant extravascular haemolysis (ALPHA): A double-blind, randomised, phase 3 trial. Lancet Haematol. 2023, 10, e955–e965. [Google Scholar] [CrossRef]
- Kassa, E.; Ciulla, T.A.; Hussain, R.M.; Dugel, P.U. Complement inhibition as a therapeutic strategy in retinal disorders. Expert. Opin. Biol. Ther. 2019, 19, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Buonfiglio, F.; Korb, C.A.; Stoffelns, B.; Pfeiffer, N.; Gericke, A. Recent Advances in Our Understanding of Age-Related Macular Degeneration: Mitochondrial Dysfunction, Redox Signaling, and the Complement System. Aging Dis. 2024. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, M.; Mitchell, P.; Freund, K.B.; Sadda, S.; Holz, F.G.; Brittain, C.; Henry, E.C.; Ferrara, D. The Progression of Geographic Atrophy Secondary to Age-Related Macular Degeneration. Ophthalmology 2018, 125, 369–390. [Google Scholar] [CrossRef] [PubMed]
- Heesterbeek, T.J.; Lorés-Motta, L.; Hoyng, C.B.; Lechanteur, Y.T.E.; den Hollander, A.I. Risk factors for progression of age-related macular degeneration. Ophthalmic. Physiol. Opt. 2020, 40, 140–170. [Google Scholar] [CrossRef]
- Lejoyeux, R.; Benillouche, J.; Ong, J.; Errera, M.H.; Rossi, E.A.; Singh, S.R.; Dansingani, K.K.; da Silva, S.; Sinha, D.; Sahel, J.A.; et al. Choriocapillaris: Fundamentals and advancements. Prog. Retin. Eye Res. 2022, 87, 100997. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, U.; Bailey, C.C.; Johnston, R.L.; McKibbin, M.; Khan, R.S.; Mahmood, S.; Downey, L.; Dhingra, N.; Brand, C.; Brittain, C.J.; et al. Characterizing Disease Burden and Progression of Geographic Atrophy Secondary to Age-Related Macular Degeneration. Ophthalmology 2018, 125, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.G.; Klein, R.; Cheng, C.-Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [PubMed]
- Holekamp, N.; Wykoff, C.C.; Schmitz-Valckenberg, S.; Monés, J.; Souied, E.H.; Lin, H.; Rabena, M.D.; Cantrell, R.A.; Henry, E.C.; Tang, F.; et al. Natural History of Geographic Atrophy Secondary to Age-Related Macular Degeneration. Ophthalmology 2020, 127, 769–783. [Google Scholar] [CrossRef]
- Rudnicka, A.R.; Jarrar, Z.; Wormald, R.; Cook, D.G.; Fletcher, A.; Owen, C.G. Age and Gender Variations in Age-related Macular Degeneration Prevalence in Populations of European Ancestry: A Meta-analysis. Ophthalmology 2012, 119, 571–580. [Google Scholar] [CrossRef]
- Chakravarthy, U.; Augood, C.; Bentham, G.C.; de Jong, P.T.V.M.; Rahu, M.; Seland, J.; Soubrane, G.; Tomazzoli, L.; Topouzis, F.; Vingerling, J.R.; et al. Cigarette Smoking and Age-Related Macular Degeneration in the EUREYE Study. Ophthalmology 2007, 114, 1157–1163. [Google Scholar] [CrossRef]
- Risk factors associated with age-related macular degeneration. Ophthalmology 2000, 107, 2224–2232. [CrossRef] [PubMed]
- Sacconi, R.; Corbelli, E.; Querques, L.; Bandello, F.; Querques, G. A Review of Current and Future Management of Geographic Atrophy. Ophthalmol. Ther. 2017, 6, 69–77. [Google Scholar] [CrossRef]
- Sunness, J.S.; Margalit, E.; Srikumaran, D.; Applegate, C.A.; Tian, Y.; Perry, D.; Hawkins, B.S.; Bressler, N.M. The Long-term Natural History of Geographic Atrophy from Age-Related Macular Degeneration. Ophthalmology 2007, 114, 271–277. [Google Scholar] [CrossRef] [PubMed]
- The AREDS Research Group. Change in Area of Geographic Atrophy in the Age-Related Eye Disease Study. Arch. Ophthalmol. 2009, 127, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- Göbel, A.P.; Fleckenstein, M.; Schmitz-Valckenberg, S.; Brinkmann, C.K.; Holz, F.G. Imaging Geographic Atrophy in Age-Related Macular Degeneration. Ophthalmologica 2011, 226, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Scholl, H.P.N.; Peto, T.; Dandekar, S.; Bunce, C.; Xing, W.; Jenkins, S.; Bird, A.C. Inter- and intra-observer variability in grading lesions of age-related maculopathy and macular degeneration. Graefe’s Arch. Clin. Exp. Ophthalmol. 2003, 241, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Crincoli, E.; De Rosa, I.; Miere, A.; Colantuono, D.; Mehanna, C.J.; Souied, E.H. Comparison of Multimodal Imaging for the Characterization of Geographic Atrophy. Transl. Vis. Sci. Technol. 2022, 11, 21. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.R.; Boulton, M. RPE lipofuscin and its role in retinal pathobiology. Exp. Eye Res. 2005, 80, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Delori, F.C.; Dorey, C.K.; Staurenghi, G.; Arend, O.; Goger, D.G.; Weiter, J.J. In vivo fluorescence of the ocular fundus exhibits retinal pigment epithelium lipofuscin characteristics. Investig. Ophthalmol. Vis. Sci. 1995, 36, 718–729. [Google Scholar]
- Sadda, S.R.; Guymer, R.; Holz, F.G.; Schmitz-Valckenberg, S.; Curcio, C.A.; Bird, A.C.; Blodi, B.A.; Bottoni, F.; Chakravarthy, U.; Chew, E.Y.; et al. Consensus Definition for Atrophy Associated with Age-Related Macular Degeneration on OCT. Ophthalmology 2018, 125, 537–548. [Google Scholar] [CrossRef]
- Sahoo, N.K.; Suresh, A.; Patil, A.; Ong, J.; Kazi, E.; Tyagi, M.; Narayanan, R.; Nayak, S.; Jacob, N.; Venkatesh, R.; et al. Novel En Face OCT-Based Closure Patterns in Idiopathic Macular Holes. Ophthalmol. Retin. 2023, 7, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, N.K.; Ong, J.; Selvam, A.; Avdalimov, M.; Gujar, R.; Lupidi, M.; Zur, D.; Chhablani, J. Ten-year follow-up and sequential evaluation of multifocal retinal pigment epithelium abnormalities in central serous chorioretinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 2023, 261, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Chhablani, J.; SOLS (Subthreshold Laser Ophthalmic Society) Writing Committee. Subthreshold laser therapy guidelines for retinal diseases. Eye 2022, 36, 2234–2235. [Google Scholar] [CrossRef] [PubMed]
- Ong, J.; Zarnegar, A.; Corradetti, G.; Singh, S.R.; Chhablani, J. Advances in Optical Coherence Tomography Imaging Technology and Techniques for Choroidal and Retinal Disorders. J. Clin. Med. 2022, 11, 5139. [Google Scholar] [CrossRef] [PubMed]
- Helb, H.M.; Issa, P.C.; Fleckenstein, M.; Schmitz-Valckenberg, S.; Scholl, H.P.N.; Meyer, C.H.; Eter, N.; Holz, F.G. Clinical evaluation of simultaneous confocal scanning laser ophthalmoscopy imaging combined with high-resolution, spectral-domain optical coherence tomography. Acta Ophthalmol. 2010, 88, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Wolf-Schnurrbusch, U.E.K.; Enzmann, V.; Brinkmann, C.K.; Wolf, S. Morphologic Changes in Patients with Geographic Atrophy Assessed with a Novel Spectral OCT–SLO Combination. Investig. Opthalmology Vis. Sci. 2008, 49, 3095–3099. [Google Scholar] [CrossRef] [PubMed]
- Guymer, R.H.; Rosenfeld, P.J.; Curcio, C.A.; Holz, F.G.; Staurenghi, G.; Freund, K.B.; Schmitz-Valckenberg, S.; Sparrow, J.; Spaide, R.F.; Tufail, A.; et al. Incomplete Retinal Pigment Epithelial and Outer Retinal Atrophy in Age-Related Macular Degeneration. Ophthalmology 2020, 127, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Sacconi, R.; Corbelli, E.; Carnevali, A.; Querques, L.; Bandello, F.; Querques, G. Optical Coherence Tomography Angiography in Geographic Atrophy. Retina 2018, 38, 2350–2355. [Google Scholar] [CrossRef]
- Thulliez, M.; Zhang, Q.; Shi, Y.; Zhou, H.; Chu, Z.; de Sisternes, L.; Durbin, M.K.; Feuer, W.; Gregori, G.; Wang, R.K.; et al. Correlations between Choriocapillaris Flow Deficits around Geographic Atrophy and Enlargement Rates Based on Swept-Source OCT Imaging. Ophthalmol. Retin. 2019, 3, 478–488. [Google Scholar] [CrossRef]
- Tan, A.C.S.; Fleckenstein, M.; Schmitz-Valckenberg, S.; Holz, F.G. Clinical Application of Multicolor Imaging Technology. Ophthalmologica 2016, 236, 8–18. [Google Scholar] [CrossRef]
- Singh, R.P.; Amoaku, W.; Bandello, F.; Chen, F.K.; Holz, F.G.; Kodjikian, L.; Ruiz-Moreno, J.M.; Joshi, P.; Wykoff, C.C. Diagnosis and Management of Patients with Geographic Atrophy Secondary to Age-Related Macular Degeneration: A Delphi Consensus Exercise. Ophthalmic Surg. Lasers Imaging Retin. 2023, 54, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.-J.; Chang, M.-L.; Zhang, F.F.; Li, T.; Gensler, G.; Schleicher, M.; Taylor, A. The Relationship of Major American Dietary Patterns to Age-Related Macular Degeneration. Am. J. Ophthalmol. 2014, 158, 118–127.e111. [Google Scholar] [CrossRef] [PubMed]
- Clemons, T.E.; Milton, R.C.; Klein, R.; Seddon, J.M.; Ferris, F.L., 3rd; Age-Related Eye Disease Study Research, G. Risk factors for the incidence of Advanced Age-Related Macular Degeneration in the Age-Related Eye Disease Study (AREDS) AREDS report no. 19. Ophthalmology 2005, 112, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Age-Related Eye Disease Study Research, G. A randomized, placebo-controlled, clinical trial of high-dose supplementation with vitamins C and E and beta carotene for age-related cataract and vision loss: AREDS report no. 9. Arch. Ophthalmol. 2001, 119, 1439–1452. [Google Scholar] [CrossRef] [PubMed]
- Freeman, W.R.; Bandello, F.; Souied, E.; Guymer, R.H.; Garg, S.J.; Chen, F.K.; Rich, R.; Holz, F.G.; Patel, S.S.; Kim, K.; et al. Randomized Phase IIb Study of Brimonidine Drug Delivery System Generation 2 for Geographic Atrophy in Age-Related Macular Degeneration. Ophthalmol. Retin. 2023, 7, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Kauper, K.; McGovern, C.; Sherman, S.; Heatherton, P.; Rapoza, R.; Stabila, P.; Dean, B.; Lee, A.; Borges, S.; Bouchard, B.; et al. Two-Year Intraocular Delivery of Ciliary Neurotrophic Factor by Encapsulated Cell Technology Implants in Patients with Chronic Retinal Degenerative Diseases. Investig. Opthalmology Vis. Sci. 2012, 53, 7484–7491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Hopkins, J.J.; Heier, J.S.; Birch, D.G.; Halperin, L.S.; Albini, T.A.; Brown, D.M.; Jaffe, G.J.; Tao, W.; Williams, G.A. Ciliary neurotrophic factor delivered by encapsulated cell intraocular implants for treatment of geographic atrophy in age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 6241–6245. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, M.; Lambiase, A.; Cerini, A.; Limoli, P.G.; La Cava, M.; Greco, A. Therapeutic Approaches with Intravitreal Injections in Geographic Atrophy Secondary to Age-Related Macular Degeneration: Current Drugs and Potential Molecules. Int. J. Mol. Sci. 2019, 20, 1693. [Google Scholar] [CrossRef] [PubMed]
- Augustin, A.J.; Diehm, C.; Grieger, F.; Bentz, J. Alprostadil infusion in patients with dry age related macular degeneration: A randomized controlled clinical trial. Expert Opin. Investig. Drugs 2013, 22, 803–812. [Google Scholar] [CrossRef]
- Wei, Y.; Liao, H.; Ye, J. Therapeutic effects of various therapeutic strategies on non-exudative age-related macular degeneration. Medicine 2018, 97, e10422. [Google Scholar] [CrossRef]
- Liao, D.S.; Grossi, F.V.; El Mehdi, D.; Gerber, M.R.; Brown, D.M.; Heier, J.S.; Wykoff, C.C.; Singerman, L.J.; Abraham, P.; Grassmann, F.; et al. Complement C3 Inhibitor Pegcetacoplan for Geographic Atrophy Secondary to Age-Related Macular Degeneration. Ophthalmology 2020, 127, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Weitz, I.C. Pegcetacoplan: A New Opportunity for Complement Inhibition in PNH. J. Blood Med. 2023, 14, 239–245. [Google Scholar] [CrossRef]
- Wilke, G.A.; Apte, R.S. Complement regulation in the eye: Implications for age-related macular degeneration. J. Clin. Investig. 2024, 134, e178296. [Google Scholar] [CrossRef]
- Heier, J.S.; Lad, E.M.; Holz, F.G.; Rosenfeld, P.J.; Guymer, R.H.; Boyer, D.; Grossi, F.; Baumal, C.R.; Korobelnik, J.-F.; Slakter, J.S.; et al. Pegcetacoplan for the treatment of geographic atrophy secondary to age-related macular degeneration (OAKS and DERBY): Two multicentre, randomised, double-masked, sham-controlled, phase 3 trials. Lancet 2023, 402, 1434–1448. [Google Scholar] [CrossRef]
- Wong, R.S.M.; Navarro-Cabrera, J.R.; Comia, N.S.; Goh, Y.T.; Idrobo, H.; Kongkabpan, D.; Gómez-Almaguer, D.; Al-Adhami, M.; Ajayi, T.; Alvarenga, P.; et al. Pegcetacoplan controls hemolysis in complement inhibitor–naive patients with paroxysmal nocturnal hemoglobinuria. Blood Adv. 2023, 7, 2468–2478. [Google Scholar] [CrossRef]
- Hoy, S.M. Pegcetacoplan: First Approval. Drugs 2021, 81, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Park, D.H.; Connor, K.M.; Lambris, J.D. The Challenges and Promise of Complement Therapeutics for Ocular Diseases. Front. Immunol. 2019, 10, 455062. [Google Scholar] [CrossRef] [PubMed]
- Apellis-Pharmaceuticals. Syfovre—Full Prescribing Information; Drugs@FDA: FDA-Approved Drugs; Apellis-Pharmaceuticals: Waltham, MA, USA, 2023; pp. 1–17. [Google Scholar]
- Wykoff, C.C.; Rosenfeld, P.J.; Waheed, N.K.; Singh, R.P.; Ronca, N.; Slakter, J.S.; Staurenghi, G.; Monés, J.; Baumal, C.R.; Saroj, N.; et al. Characterizing New-Onset Exudation in the Randomized Phase 2 FILLY Trial of Complement Inhibitor Pegcetacoplan for Geographic Atrophy. Ophthalmology 2021, 128, 1325–1336. [Google Scholar] [CrossRef]
- Nittala, M.G.; Metlapally, R.; Ip, M.; Chakravarthy, U.; Holz, F.G.; Staurenghi, G.; Waheed, N.; Velaga, S.B.; Lindenberg, S.; Karamat, A.; et al. Association of Pegcetacoplan with Progression of Incomplete Retinal Pigment Epithelium and Outer Retinal Atrophy in Age-Related Macular Degeneration. JAMA Ophthalmol. 2022, 140, 243–249. [Google Scholar] [CrossRef]
- Cruz-Pimentel, M.; Wu, L. Complement Inhibitors for Advanced Dry Age-Related Macular Degeneration (Geographic Atrophy): Some Light at the End of the Tunnel? J. Clin. Med. 2023, 12, 5131. [Google Scholar] [CrossRef]
- Schachar, I.H. Concerning Syfovre Approval for Geographic Atrophy. JAMA Ophthalmol. 2023, 142, 85–86. [Google Scholar] [CrossRef] [PubMed]
- Khanani, A.M.; Patel, S.S.; Staurenghi, G.; Tadayoni, R.; Danzig, C.J.; Eichenbaum, D.A.; Hsu, J.; Wykoff, C.C.; Heier, J.S.; Lally, D.R.; et al. Efficacy and safety of avacincaptad pegol in patients with geographic atrophy (GATHER2): 12-month results from a randomised, double-masked, phase 3 trial. Lancet 2023, 402, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chen, S.; Ma, M.; Qian, J.; Ma, X. Complement 5b-9 complex-induced alterations in human RPE cell physiology. Med. Sci. Monit. 2010, 16, BR17–BR23. [Google Scholar] [PubMed]
- Silverman, S.M.; Ma, W.; Wang, X.; Zhao, L.; Wong, W.T. C3- and CR3-dependent microglial clearance protects photoreceptors in retinitis pigmentosa. J. Exp. Med. 2019, 216, 1925–1943. [Google Scholar] [CrossRef]
- IVERIC. Izervay—Full Prescribing Information; IVERIC: Parsippany, NJ, USA, 2023. [Google Scholar]
- Patel, S.S.; Lally, D.R.; Hsu, J.; Wykoff, C.C.; Eichenbaum, D.; Heier, J.S.; Jaffe, G.J.; Westby, K.; Desai, D.; Zhu, L.; et al. Avacincaptad pegol for geographic atrophy secondary to age-related macular degeneration: 18-month findings from the GATHER1 trial. Eye 2023, 37, 3551–3557. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Investigation | Experimental Model | Main Findings | Representative Reference |
|---|---|---|---|
| C1-esterase inhibition | Chinese hamster ovary in vitro model; rat model | Modest complement inhibition with concentration-dependent cytotoxicity; decreased retinal vascular permeability | [106,140] |
| Anti-C1s antibody (TNT009) | In vitro alloserum from dialysis patient; in vitro RBCs | Decreased activation of complement pathway | [87,141] |
| Kallikrein inhibition | Rat model | Decreased retinal vascular permeability | [106] |
| C3 inhibition/absence | In vitro RPE cells; Primate/murine model | Restored lysosomal function and reversed disease phenotype; suppressed drusen formation; decreased retinal degeneration | [90,110,112] |
| C3a/C3a receptor inhibition | In vitro embryonal chick cells; in vitro human fetal RPE cells; murine model | Retinal regeneration; C3ar blockage prevents C3a-induced sub-RPE deposit formation; supplementation associated with increased proteasome activity; alleviated neuroinflammation and restored visual function with receptor blockade | [91,117] |
| C5 inhibition | In vitro RPE cells; murine model | Suppressed priming for inflammasome activation; inhibited choroidal neovascularization after laser; improved retinal function, decreased loss of retinal ganglion cells, cones, and photoreceptors; preserved optic nerve function | [95,118,121,122,123] |
| C5a/C5a receptor inhibition | Murine model | C5a inhibition insufficient to block development of AMD-like pathology; C5ar-mediated signaling key to normal retinal function and structure; C5ar-deficient mice have increased neovascularization | [120,125,127] |
| CD59 | Murine model | Inhibited growth and reduced size of choroidal neovascularization | [60] |
| Factor B inhibition/absence | In vitro RPE cells; In vitro erythrocytes; Murine model | Blocking TNF-α can block factor B production; factor B inhibition inhibits alternative pathway; normalized dysregulated complement pathway, reduced sub-RPE deposits with inhibition | [97,99,130] |
| Factor D inhibition/absence | In vitro erythrocytes; Murine model | Alternative pathway blockade preserves bacterial clearance; photoreceptor protection | [133] |
| Factor H | In vitro purified C3b; Rat/murine models | Factor H competition results in complement dysregulation; complete knockout associated with increased retinal inflammation and amyloid β deposition | [25,105,128] |
| Factor I | Murine model | Reduction in choroidal neovascularization | [138,139] |
| Pharmaceutical | Target | Status | Reference/Trial Identifier |
|---|---|---|---|
| Cinryze | C1 esterase inhibitor | Phase 1b completed (2014), current status unknown | 144; NCT01759602 |
| NGM621 | C3 inhibitor | Phase 2 completed (2023) | NCT04465955 |
| POT-4 | C3 inhibitor | Phase 2 terminated | NCT01603043 |
| Avacincaptad Pegol/ARC1905/Zimura | C5 inhibitor | Various stages | NCT03374670; NCT03364153; NCT03362190; NCT05536297 |
| Eculizumab | C5 inhibitor | Phase 2 completed | NCT00935883 |
| LFG316/Tesidolumab | C5 inhibitor | Phase 2 completed | NCT01255462; NCT01526889 |
| IONIS-FB-LRx | Factor B inhibitor | Phase 2 ongoing | NCT03815825 |
| FCFD4514S/Lampalizumab | Factor D inhibitor | Phase 2 completed, terminated | NCT02288559 |
| ALXN2040/Danicopan | Factor D inhibitor | Phase 2 ongoing | NCT05019521 |
| CLG516 | Properidin inhibitor | Phase 2 completed (2019) | NCT02515942 |
| GT005 | AAV-based Factor I gene therapy | Phase 2 terminated | NCT04566445 |
| Reduction in GA Growth Compared to Combined Sham Group | |||
|---|---|---|---|
| Treatment | FILLY (Phase 2) | OAKS (Phase 3) | DERBY (Phase 3) |
| 12 Months | |||
| Monthly Injection | 30% | 21% | 12% |
| EOM Injection | 20% | 16% | 11% |
| 24 Months | |||
| Monthly Injections | - | 22% | 19% |
| EOM Injections | - | 18% | 16% |
| Reduction in GA Growth Compared to Sham Group | ||
|---|---|---|
| Treatment | GATHER1 (Phase 2/3) | GATHER2 (Phase 3) |
| 12 Months | ||
| 2 mg | 30.5% | 18% |
| 4 mg | 25.6% | - |
| 18 Months | ||
| 2 mg | 32.2% | - |
| 4 mg | 29.4% | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ong, J.; Zarnegar, A.; Selvam, A.; Driban, M.; Chhablani, J. The Complement System as a Therapeutic Target in Retinal Disease. Medicina 2024, 60, 945. https://doi.org/10.3390/medicina60060945
Ong J, Zarnegar A, Selvam A, Driban M, Chhablani J. The Complement System as a Therapeutic Target in Retinal Disease. Medicina. 2024; 60(6):945. https://doi.org/10.3390/medicina60060945
Chicago/Turabian StyleOng, Joshua, Arman Zarnegar, Amrish Selvam, Matthew Driban, and Jay Chhablani. 2024. "The Complement System as a Therapeutic Target in Retinal Disease" Medicina 60, no. 6: 945. https://doi.org/10.3390/medicina60060945
APA StyleOng, J., Zarnegar, A., Selvam, A., Driban, M., & Chhablani, J. (2024). The Complement System as a Therapeutic Target in Retinal Disease. Medicina, 60(6), 945. https://doi.org/10.3390/medicina60060945