
Natural History of Metabolic Dysfunction-Associated Steatotic Liver Disease: From Metabolic Syndrome to Hepatocellular Carcinoma

 , , , , , , , and
, , , , , , , and
Abstract
1. Introduction
2. Epidemiology of MASLD and Its Stages in the Adult Mexican Population and Globally
3. Etiology: From Visceral Fat to Liver Steatosis
3.1. Visceral Fat Anatomy
3.2. Development of Visceral Adiposity
3.2.1. Sexual Hormones
3.2.2. Insulin Resistance
3.2.3. Hepatic Fat Accumulation Pathways
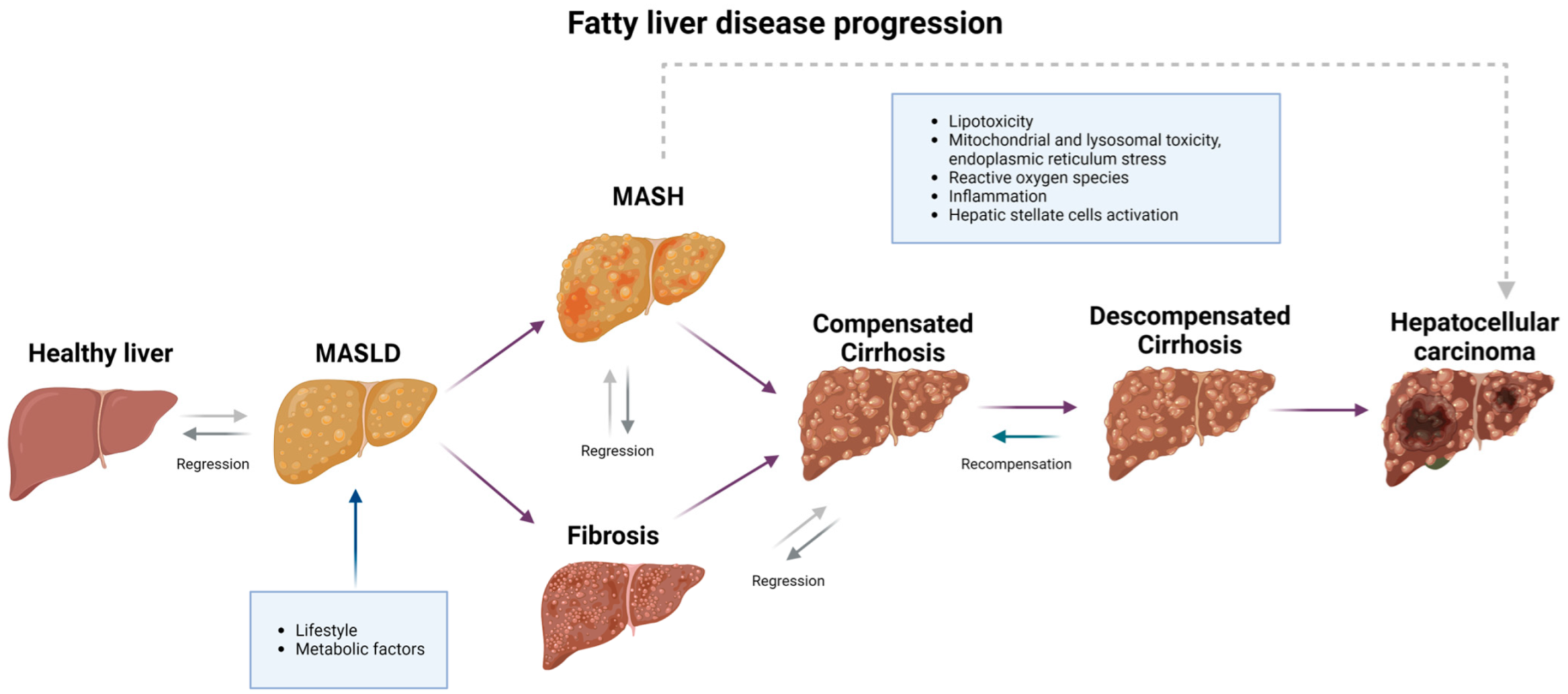
4. Pathophysiology: Progression from MASLD to HCC
4.1. First Step: From MASLD to MASH
4.2. Second Step: From MASH to Cirrhosis
4.3. Third Step: From Cirrhosis to HCC
5. Diagnosis
6. Complications
7. Discussion
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nguyen, P.; Leray, V.; Diez, M.; Serisier, S.; Le Bloc’h, J.; Siliart, B.; Dumon, H. Liver Lipid Metabolism. J. Anim. Physiol. Anim. Nutr. 2008, 92, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Horton, J.D. Molecular Mediators of Hepatic Steatosis and Liver Injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef]
- Jimba, S.; Nakagami, T.; Takahashi, M.; Wakamatsu, T.; Hirota, Y.; Iwamoto, Y.; Wasada, T. Prevalence of Non-alcoholic Fatty Liver Disease and Its Association with Impaired Glucose Metabolism in Japanese Adults. Diabet. Med. 2005, 22, 1141–1145. [Google Scholar] [CrossRef]
- Pagano, G.; Pacini, G.; Musso, G.; Gambino, R.; Mecca, F.; Depetris, N.; Cassader, M.; David, E.; Cavallo-Perin, P.; Rizzetto, M. Nonalcoholic Steatohepatitis, Insulin Resistance, and Metabolic Syndrome: Further Evidence for an Etiologic Association. Hepatology 2002, 35, 367–372. [Google Scholar] [CrossRef]
- Anderson, N.; Borlak, J. Molecular Mechanisms and Therapeutic Targets in Steatosis and Steatohepatitis. Pharmacol. Rev. 2008, 60, 311–357. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Wong, G.; Anstee, Q.M.; Henry, L. The Global Burden of Liver Disease. Clin. Gastroenterol. Hepatol. 2023, 21, 1978–1991. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A Multisociety Delphi Consensus Statement on New Fatty Liver Disease Nomenclature. J. Hepatol. 2023, 79, 1542–1556. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.-F.; Schattenberg, J.M.; et al. A New Definition for Metabolic Dysfunction-Associated Fatty Liver Disease: An International Expert Consensus Statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic Fatty Liver Disease. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Zou, B.; Yeo, Y.H.; Li, J.; Huang, D.Q.; Wu, Y.; Yang, H.; Liu, C.; Kam, L.Y.; Tan, X.X.E.; et al. Global Prevalence, Incidence, and Outcomes of Non-Obese or Lean Non-Alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Lancet Gastroenterol. Hepatol. 2020, 5, 739–752. [Google Scholar] [CrossRef]
- Zeng, J.; Yang, R.-X.; Sun, C.; Pan, Q.; Zhang, R.-N.; Chen, G.-Y.; Hu, Y.; Fan, J.-G. Prevalence, Clinical Characteristics, Risk Factors, and Indicators for Lean Chinese Adults with Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2020, 26, 1792–1804. [Google Scholar] [CrossRef] [PubMed]
- Campos-Nonato, I.; Galván-Valencia, Ó.; Hernández-Barrera, L.; Oviedo-Solís, C.; Barquera, S. Prevalencia de Obesidad y Factores de Riesgo Asociados En Adultos Mexicanos: Resultados de La Ensanut 2022. Salud Publica Mex. 2023, 65, s238–s247. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, M.; Kojima, T.; Takeda, N.; Nakagawa, T.; Taniguchi, H.; Fujii, K.; Omatsu, T.; Nakajima, T.; Sarui, H.; Shimazaki, M.; et al. The Metabolic Syndrome as a Predictor of Nonalcoholic Fatty Liver Disease. Ann. Intern. Med. 2005, 143, 722. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Martínez, R.; Aguilar-Salinas, C.A.; Romero-Martínez, M.; Castro-Porras, L.; Gómez-Velasco, D.; Mehta, R. Trends in the Prevalence of Metabolic Syndrome and Its Components in Mexican Adults, 2006–2018. Salud Publica Mex. 2021, 63, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Lizardi-Cervera, J.; Ivonne Becerra Laparra, D.; Chávez-Tapia, N.C.; Martha Ramos Ostos, D.E.; Uribe Esquivel, M. Prevalencia de Hígado Graso No Alcohólico y Síndrome Metabólico En Población Asintomática. Rev. Gastroenterol. Mex. 2006, 71, 453–459. [Google Scholar] [PubMed]
- Briseño-Bass, P.; Chávez-Pérez, R.; López-Zendejas, M. Prevalencia y Relación de Esteatosis Hepática Con Perfil Lipídico y Hepático En Pacientes de Chequeo Médico. Rev. Gastroenterol. Mex. 2019, 84, 290–295. [Google Scholar] [CrossRef]
- Devarbhavi, H.; Asrani, S.K.; Arab, J.P.; Nartey, Y.A.; Pose, E.; Kamath, P.S. Global Burden of Liver Disease: 2023 Update. J. Hepatol. 2023, 79, 516–537. [Google Scholar] [CrossRef]
- Chan, K.E.; Koh, T.J.L.; Tang, A.S.P.; Quek, J.; Yong, J.N.; Tay, P.; Tan, D.J.H.; Lim, W.H.; Lin, S.Y.; Huang, D.; et al. Global Prevalence and Clinical Characteristics of Metabolic-Associated Fatty Liver Disease: A Meta-Analysis and Systematic Review of 10 739 607 Individuals. J. Clin. Endocrinol. Metab. 2022, 107, 2691–2700. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The Global Epidemiology of Nonalcoholic Fatty Liver Disease (NAFLD) and Nonalcoholic Steatohepatitis (NASH): A Systematic Review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef]
- Sepanlou, S.G.; Safiri, S.; Bisignano, C.; Ikuta, K.S.; Merat, S.; Saberifiroozi, M.; Poustchi, H.; Tsoi, D.; Colombara, D.V.; Abdoli, A.; et al. The Global, Regional, and National Burden of Cirrhosis by Cause in 195 Countries and Territories, 1990–2017: A Systematic Analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 245–266. [Google Scholar] [CrossRef]
- Gonzalez-Chagolla, A.; Olivas-Martinez, A.; Ruiz-Manriquez, J.; Servín-Rojas, M.; Kauffman-Ortega, E.; Chávez-García, L.C.; Juárez-León, O.; Cordova-Gallardo, J.; Díaz-García, J.D.; Gonzalez-Huezo, M.S.; et al. Cirrhosis Etiology Trends in Developing Countries: Transition from Infectious to Metabolic Conditions. Report from a Multicentric Cohort in Central Mexico. Lancet Reg. Health-Am. 2022, 7, 100151. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global Epidemiology of NAFLD-Related HCC: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef]
- Nauli, A.M.; Matin, S. Why Do Men Accumulate Abdominal Visceral Fat? Front. Physiol. 2019, 10, 1486. [Google Scholar] [CrossRef] [PubMed]
- Gavin, K.M.; Cooper, E.E.; Raymer, D.K.; Hickner, R.C. Estradiol Effects on Subcutaneous Adipose Tissue Lipolysis in Premenopausal Women Are Adipose Tissue Depot Specific and Treatment Dependent. Am. J. Physiol.-Endocrinol. Metab. 2013, 304, E1167–E1174. [Google Scholar] [CrossRef]
- Santosa, S.; Jensen, M.D. Why Are We Shaped Differently, and Why Does It Matter? Am. J. Physiol.-Endocrinol. Metab. 2008, 295, E531–E535. [Google Scholar] [CrossRef]
- Hodson, L.; Banerjee, R.; Rial, B.; Arlt, W.; Adiels, M.; Boren, J.; Marinou, K.; Fisher, C.; Mostad, I.L.; Stratton, I.M.; et al. Menopausal Status and Abdominal Obesity Are Significant Determinants of Hepatic Lipid Metabolism in Women. J. Am. Heart Assoc. 2015, 4, e002258. [Google Scholar] [CrossRef] [PubMed]
- Mårin, P.; Lönn, L.; Andersson, B.; Odén, B.; Olbe, L.; Bengtsson, B.A.; Björntorp, P. Assimilation of Triglycerides in Subcutaneous and Intraabdominal Adipose Tissues in Vivo in Men: Effects of Testosterone. J. Clin. Endocrinol. Metab. 1996, 81, 1018–1022. [Google Scholar] [CrossRef] [PubMed]
- Votruba, S.B.; Mattison, R.S.; Dumesic, D.A.; Koutsari, C.; Jensen, M.D. Meal Fatty Acid Uptake in Visceral Fat in Women. Diabetes 2007, 56, 2589–2597. [Google Scholar] [CrossRef]
- Salehi-sahlabadi, A.; Sadat, S.; Beigrezaei, S.; Pourmasomi, M.; Feizi, A.; Ghiasvand, R.; Hadi, A.; Clark, C.C.T.; Miraghajani, M. Dietary Patterns and Risk of Non-Alcoholic Fatty Liver Disease. BMC Gastroenterol. 2021, 21, 41. [Google Scholar] [CrossRef] [PubMed]
- Ushula, T.W.; Mamun, A.; Darssan, D.; Wang, W.Y.S.; Williams, G.M.; Whiting, S.J.; Najman, J.M. Dietary Patterns and the Risks of Metabolic Syndrome and Insulin Resistance among Young Adults: Evidence from a Longitudinal Study. Clin. Nutr. 2022, 41, 1523–1531. [Google Scholar] [CrossRef]
- Demaria, T.M.; Crepaldi, L.D.; Costa-Bartuli, E.; Branco, J.R.; Zancan, P.; Sola-Penna, M. Once a Week Consumption of Western Diet over Twelve Weeks Promotes Sustained Insulin Resistance and Non-Alcoholic Fat Liver Disease in C57BL/6 J Mice. Sci. Rep. 2023, 13, 3058. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Chiang, S.-H.; Saltiel, A.R. Insulin Signaling and the Regulation of Glucose Transport. Mol. Med. 2004, 10, 65–71. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Kahn, C.R. Insulin Signalling and the Regulation of Glucose and Lipid Metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Bouskila, M.; Hirshman, M.F.; Jensen, J.; Goodyear, L.J.; Sakamoto, K. Insulin Promotes Glycogen Synthesis in the Absence of GSK3 Phosphorylation in Skeletal Muscle. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E28–E35. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D. Banting Lecture 2001. Diabetes 2002, 51, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.B.; Petersen, K.F.; Shulman, G.I. Mechanisms of Insulin Resistance in Humans and Possible Links With Inflammation. Hypertension 2005, 45, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Liu, Z.-X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of Hepatic Insulin Resistance in Non-Alcoholic Fatty Liver Disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef] [PubMed]
- Roden, M.; Price, T.B.; Perseghin, G.; Petersen, K.F.; Rothman, D.L.; Cline, G.W.; Shulman, G.I. Mechanism of Free Fatty Acid-Induced Insulin Resistance in Humans. J. Clin. Investig. 1996, 97, 2859–2865. [Google Scholar] [CrossRef] [PubMed]
- Allard, J.P.; Aghdassi, E.; Mohammed, S.; Raman, M.; Avand, G.; Arendt, B.M.; Jalali, P.; Kandasamy, T.; Prayitno, N.; Sherman, M.; et al. Nutritional Assessment and Hepatic Fatty Acid Composition in Non-Alcoholic Fatty Liver Disease (NAFLD): A Cross-Sectional Study. J. Hepatol. 2008, 48, 300–307. [Google Scholar] [CrossRef]
- Dulloo, A.G.; Mensi, N.; Seydoux, J.; Girardier, L. Differential Effects of High-Fat Diets Varying in Fatty Acid Composition on the Efficiency of Lean and Fat Tissue Deposition during Weight Recovery after Low Food Intake. Metabolism 1995, 44, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Rosqvist, F.; Iggman, D.; Kullberg, J.; Cedernaes, J.; Johansson, H.-E.; Larsson, A.; Johansson, L.; Ahlström, H.; Arner, P.; Dahlman, I.; et al. Overfeeding Polyunsaturated and Saturated Fat Causes Distinct Effects on Liver and Visceral Fat Accumulation in Humans. Diabetes 2014, 63, 2356–2368. [Google Scholar] [CrossRef]
- Wang, G.; Bonkovsky, H.L.; de Lemos, A.; Burczynski, F.J. Recent Insights into the Biological Functions of Liver Fatty Acid Binding Protein 1. J. Lipid Res. 2015, 56, 2238–2247. [Google Scholar] [CrossRef]
- Wolfrum, C.; Borrmann, C.M.; Börchers, T.; Spener, F. Fatty Acids and Hypolipidemic Drugs Regulate Peroxisome Proliferator-Activated Receptors α- and γ-Mediated Gene Expression via Liver Fatty Acid Binding Protein: A Signaling Path to the Nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 2323–2328. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, N.; Kato, M.; Tanaka, M.; Miyazaki, M.; Takao, S.; Kohjima, M.; Kotoh, K.; Enjoji, M.; Nakamuta, M.; Takayanagi, R. Effects of Insulin Resistance and Hepatic Lipid Accumulation on Hepatic MRNA Expression Levels of ApoB, MTP and L-FABP in Non-Alcoholic Fatty Liver Disease. Exp. Ther. Med. 2011, 2, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.E.; Ramos–Roman, M.A.; Browning, J.D.; Parks, E.J. Increased De Novo Lipogenesis Is a Distinct Characteristic of Individuals With Nonalcoholic Fatty Liver Disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Listenberger, L.L.; Han, X.; Lewis, S.E.; Cases, S.; Farese, R.V.; Ory, D.S.; Schaffer, J.E. Triglyceride Accumulation Protects against Fatty Acid-Induced Lipotoxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3077–3082. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of Fatty Acids Stored in Liver and Secreted via Lipoproteins in Patients with Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Uyeda, K.; Repa, J.J. Carbohydrate Response Element Binding Protein, ChREBP, a Transcription Factor Coupling Hepatic Glucose Utilization and Lipid Synthesis. Cell Metab. 2006, 4, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Berk, M.; McIntyre, T.M.; Gores, G.J.; Feldstein, A.E. The Lysosomal-Mitochondrial Axis in Free Fatty Acid–Induced Hepatic Lipotoxicity. Hepatology 2008, 47, 1495–1503. [Google Scholar] [CrossRef]
- Moore, M.P.; Cunningham, R.P.; Meers, G.M.; Johnson, S.A.; Wheeler, A.A.; Ganga, R.R.; Spencer, N.M.; Pitt, J.B.; Diaz-Arias, A.; Swi, A.I.A.; et al. Compromised Hepatic Mitochondrial Fatty Acid Oxidation and Reduced Markers of Mitochondrial Turnover in Human NAFLD. Hepatology 2022, 76, 1452–1465. [Google Scholar] [CrossRef]
- Yamakawa, T. The fate of branched-chain fatty acids in animal body. IV. A contribution to the mode of biological breakdown of naturally occurring fatty acids. J. Biochem. 1950, 37, 343–353. [Google Scholar] [CrossRef]
- Zhang, X.; Li, S.; Zhou, Y.; Su, W.; Ruan, X.; Wang, B.; Zheng, F.; Warner, M.; Gustafsson, J.-Å.; Guan, Y. Ablation of Cytochrome P450 Omega-Hydroxylase 4A14 Gene Attenuates Hepatic Steatosis and Fibrosis. Proc. Natl. Acad. Sci. USA 2017, 114, 3181–3185. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.K.; Doig, M.V.; Lewis, D.F.V.; Gibson, G.G. Role of Hepatic and Renal Cytochrome P-450 Iva1 in the Metabolism of Lipid Substrates. Biochem. Pharmacol. 1989, 38, 3621–3629. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Cao, Y.; Xia, H.; Zhu, X.; Jin, Y. CYP4A11 Is Involved in the Development of Nonalcoholic Fatty Liver Disease via ROS-induced Lipid Peroxidation and Inflammation. Int. J. Mol. Med. 2020, 45, 1121–1129. [Google Scholar] [CrossRef]
- Lake, A.D.; Novak, P.; Hardwick, R.N.; Flores-Keown, B.; Zhao, F.; Klimecki, W.T.; Cherrington, N.J. The Adaptive Endoplasmic Reticulum Stress Response to Lipotoxicity in Progressive Human Nonalcoholic Fatty Liver Disease. Toxicol. Sci. 2014, 137, 26–35. [Google Scholar] [CrossRef]
- Lei, N.; Song, H.; Zeng, L.; Ji, S.; Meng, X.; Zhu, X.; Li, X.; Feng, Q.; Liu, J.; Mu, J. Persistent Lipid Accumulation Leads to Persistent Exacerbation of Endoplasmic Reticulum Stress and Inflammation in Progressive NASH via the IRE1α/TRAF2 Complex. Molecules 2023, 28, 3185. [Google Scholar] [CrossRef]
- Hirsova, P.; Ibrahim, S.H.; Krishnan, A.; Verma, V.K.; Bronk, S.F.; Werneburg, N.W.; Charlton, M.R.; Shah, V.H.; Malhi, H.; Gores, G.J. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles From Hepatocytes. Gastroenterology 2016, 150, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.I.; Shankaran, M.; Yoshino, M.; Schweitzer, G.G.; Chondronikola, M.; Beals, J.W.; Okunade, A.L.; Patterson, B.W.; Nyangau, E.; Field, T.; et al. Insulin Resistance Drives Hepatic de Novo Lipogenesis in Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2020, 130, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Albert, V.; Woelnerhanssen, B.; Frei, I.C.; Weissenberger, D.; Meyer-Gerspach, A.C.; Clement, N.; Moes, S.; Colombi, M.; Meier, J.A.; et al. Insulin Resistance Causes Inflammation in Adipose Tissue. J. Clin. Investig. 2018, 128, 1538–1550. [Google Scholar] [CrossRef]
- Kirovski, G.; Dorn, C.; Huber, H.; Moleda, L.; Niessen, C.; Wobser, H.; Schacherer, D.; Buechler, C.; Wiest, R.; Hellerbrand, C. Elevated Systemic Monocyte Chemoattractrant Protein-1 in Hepatic Steatosis without Significant Hepatic Inflammation. Exp. Mol. Pathol. 2011, 91, 780–783. [Google Scholar] [CrossRef]
- Haukeland, J.W.; Damås, J.K.; Konopski, Z.; Løberg, E.M.; Haaland, T.; Goverud, I.; Torjesen, P.A.; Birkeland, K.; Bjøro, K.; Aukrust, P. Systemic Inflammation in Nonalcoholic Fatty Liver Disease Is Characterized by Elevated Levels of CCL2. J. Hepatol. 2006, 44, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Ferri, F.; Mischitelli, M.; Tozzi, G.; Messina, E.; Mignini, I.; Mazzuca, S.; Pellone, M.; Parisse, S.; Marrapodi, R.; Visentini, M.; et al. Reduced Lysosomal Acid Lipase Activity in Blood and Platelets Is Associated With Nonalcoholic Fatty Liver Disease. Clin. Transl. Gastroenterol. 2020, 11, e00116. [Google Scholar] [CrossRef]
- Leopold, C.; Duta-Mare, M.; Sachdev, V.; Goeritzer, M.; Maresch, L.K.; Kolb, D.; Reicher, H.; Wagner, B.; Stojakovic, T.; Ruelicke, T.; et al. Hepatocyte-Specific Lysosomal Acid Lipase Deficiency Protects Mice from Diet-Induced Obesity but Promotes Hepatic Inflammation. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2019, 1864, 500–511. [Google Scholar] [CrossRef]
- Thoen, R.U.; Longo, L.; Neto, S.C.; Álvares-da-Silva, M.R. Low Levels of Lysosomal Acid Lipase (LAL) Activity Increases Necroinflammation in Adult Patients with Biopsy-Proven Metabolic Associated Fatty Liver Disease. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101638. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, Y.; Tanaka, M.; Itoh, M.; Ochi, K.; Ito, A.; Hidaka, I.; Sakaida, I.; Ogawa, Y.; Suganami, T. Iron-Rich Kupffer Cells Exhibit Phenotypic Changes during the Development of Liver Fibrosis in NASH. iScience 2021, 24, 102032. [Google Scholar] [CrossRef] [PubMed]
- Senoo, H.; Sato, M.; Imai, K. Hepatic Stellate Cells--from the Viewpoint of Retinoid Handling and Function of the Extracellular Matrix. Kaibogaku Zasshi 1997, 72, 79–94. [Google Scholar]
- Zhang, J.; Jiang, N.; Ping, J.; Xu, L. TGF-β1-induced Autophagy Activates Hepatic Stellate Cells via the ERK and JNK Signaling Pathways. Int. J. Mol. Med. 2020, 47, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Hernández–Gea, V.; Ghiassi–Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy Releases Lipid That Promotes Fibrogenesis by Activated Hepatic Stellate Cells in Mice and in Human Tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The Diagnosis and Management of Nonalcoholic Fatty Liver Disease: Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis Progression in Nonalcoholic Fatty Liver vs Nonalcoholic Steatohepatitis: A Systematic Review and Meta-Analysis of Paired-Biopsy Studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654.e9. [Google Scholar] [CrossRef]
- Schuppan, D.; Afdhal, N.H. Liver Cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Adams, L.A. The 20% Rule of NASH Progression: The Natural History of Advanced Fibrosis and Cirrhosis Caused by NASH. Hepatology 2019, 70, 1885–1888. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Kumar, S.; Prakash, S.S. Compensated Liver Cirrhosis: Natural Course and Disease-Modifying Strategies. World J. Methodol. 2023, 13, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Anstee, Q.M.; Trauner, M.; Lawitz, E.J.; Abdelmalek, M.F.; Ding, D.; Han, L.; Jia, C.; Huss, R.S.; Chung, C.; et al. Cirrhosis Regression Is Associated with Improved Clinical Outcomes in Patients with Nonalcoholic Steatohepatitis. Hepatology 2022, 75, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Hohenester, S.; Christiansen, S.; Nagel, J.; Wimmer, R.; Artmann, R.; Denk, G.; Bischoff, M.; Bischoff, G.; Rust, C. Lifestyle Intervention for Morbid Obesity: Effects on Liver Steatosis, Inflammation, and Fibrosis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2018, 315, G329–G338. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.K.; Yim, H.J. Reversal of Liver Cirrhosis: Current Evidence and Expectations. Korean J. Intern. Med. 2017, 32, 213–228. [Google Scholar] [CrossRef]
- Xu, X.-Y.; Ding, H.-G.; Li, W.-G.; Xu, J.-H.; Han, Y.; Jia, J.-D.; Wei, L.; Duan, Z.-P.; Ling-Hu, E.-Q.; Zhuang, H. Chinese Guidelines on the Management of Liver Cirrhosis (Abbreviated Version). World J. Gastroenterol. 2020, 26, 7088–7103. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J. A Review of Liver Fibrosis and Cirrhosis Regression. J. Pathol. Transl. Med. 2023, 57, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.R.; McLerran, D.; Marsh, T.; Parikh, N.; Roberts, L.R.; Schwartz, M.; Nguyen, M.H.; Befeler, A.; Page-Lester, S.; Tang, R.; et al. Incidence and Risk Factors for Hepatocellular Carcinoma in Cirrhosis: The Multicenter Hepatocellular Carcinoma Early Detection Strategy (HEDS) Study. Gastroenterology 2023, 165, 1053–1063.e6. [Google Scholar] [CrossRef]
- Schults, M.A.; Nagle, P.W.; Rensen, S.S.; Godschalk, R.W.; Munnia, A.; Peluso, M.; Claessen, S.M.; Greve, J.W.; Driessen, A.; Verdam, F.J.; et al. Decreased Nucleotide Excision Repair in Steatotic Livers Associates with Myeloperoxidase-Immunoreactivity. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2012, 736, 75–81. [Google Scholar] [CrossRef]
- Hagiwara, S.; Nishida, N.; Ueshima, K.; Minami, Y.; Komeda, Y.; Aoki, T.; Takita, M.; Morita, M.; Chishina, H.; Yoshida, A.; et al. Accumulation of Genetic and Epigenetic Alterations in the Background Liver and Emergence of Hepatocellular Carcinoma in Patients with Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 3257. [Google Scholar] [CrossRef] [PubMed]
- Demirtas, C.O.; Tolu, T.; Keklikkiran, C.; Ozdogan, O.C.; Gunduz, F. Hepatocellular Carcinoma in Non-Cirrhotic Liver Arises with a More Advanced Tumoral Appearance: A Single-Center Cohort Study. Turk. J. Gastroenterol. 2021, 32, 685–693. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of Hepatocellular Cancer in Patients With Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1828–1837.e2. [Google Scholar] [CrossRef]
- Peiseler, M.; Tacke, F. Inflammatory Mechanisms Underlying Nonalcoholic Steatohepatitis and the Transition to Hepatocellular Carcinoma. Cancers 2021, 13, 730. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Qian, H.; He, X.; Zhang, J.; Xue, S.; Wu, Y.; Chen, J.; Wu, X.; Zhang, S. Screening of the Key Genes for the Progression of Liver Cirrhosis to Hepatocellular Carcinoma Based on Bioinformatics. J. Oncol. 2022, 2022, 2515513. [Google Scholar] [CrossRef] [PubMed]
- Motalebzadeh, J.; Eskandari, E. Transcription Factors Linked to the Molecular Signatures in the Development of Hepatocellular Carcinoma on a Cirrhotic Background. Med. Oncol. 2021, 38, 121. [Google Scholar] [CrossRef]
- Bravo, A.A.; Sheth, S.G.; Chopra, S. Liver Biopsy. N. Engl. J. Med. 2001, 344, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Cadranel, J.-F.; Rufat, P.; Degos, F. Practices of Liver Biopsy in France: Results of a Prospective Nationwide Survey. Hepatology 2000, 32, 477–481. [Google Scholar] [CrossRef]
- Carrión, J.A. Utilidad Del Fibroscan® Para Evaluar La Fibrosis Hepática. Gastroenterol. Hepatol. 2009, 32, 415–423. [Google Scholar] [CrossRef]
- Afdhal, N.H. Diagnosing Fibrosis in Hepatitis C: Is the Pendulum Swinging From Biopsy to Blood Tests? Hepatology 2003, 37, 972–974. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S. Radiologic Evaluation of Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2014, 20, 7392. [Google Scholar] [CrossRef]
- Koplay, M. Importance of Imaging and Recent Developments in Diagnosis of Nonalcoholic Fatty Liver Disease. World J. Hepatol. 2015, 7, 769. [Google Scholar] [CrossRef] [PubMed]
- Palmentieri, B.; Desio, I.; Lamura, V.; Masarone, M.; Vecchione, R.; Bruno, S.; Torella, R.; Persico, M. The Role of Bright Liver Echo Pattern on Ultrasound B-Mode Examination in the Diagnosis of Liver Steatosis. Dig. Liver Dis. 2006, 38, 485–489. [Google Scholar] [CrossRef]
- Hernaez, R.; Lazo, M.; Bonekamp, S.; Kamel, I.; Brancati, F.L.; Guallar, E.; Clark, J.M. Diagnostic Accuracy and Reliability of Ultrasonography for the Detection of Fatty Liver: A Meta-Analysis. Hepatology 2011, 54, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Berzigotti, A.; Abraldes, J.G.; Tandon, P.; Erice, E.; Gilabert, R.; García-Pagan, J.C.; Bosch, J. Ultrasonographic Evaluation of Liver Surface and Transient Elastography in Clinically Doubtful Cirrhosis. J. Hepatol. 2010, 52, 846–853. [Google Scholar] [CrossRef]
- Idilman, I.S.; Keskin, O.; Celik, A.; Savas, B.; Halil Elhan, A.; Idilman, R.; Karcaaltincaba, M. A Comparison of Liver Fat Content as Determined by Magnetic Resonance Imaging-Proton Density Fat Fraction and MRS versus Liver Histology in Non-Alcoholic Fatty Liver Disease. Acta Radiol. 2016, 57, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Yokoo, T.; Serai, S.D.; Pirasteh, A.; Bashir, M.R.; Hamilton, G.; Hernando, D.; Hu, H.H.; Hetterich, H.; Kühn, J.-P.; Kukuk, G.M.; et al. Linearity, Bias, and Precision of Hepatic Proton Density Fat Fraction Measurements by Using MR Imaging: A Meta-Analysis. Radiology 2018, 286, 486–498. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Clark, J.M.; Bass, N.M.; Van Natta, M.L.; Unalp-Arida, A.; Tonascia, J.; Zein, C.O.; Brunt, E.M.; Kleiner, D.E.; McCullough, A.J.; et al. Clinical, Laboratory and Histological Associations in Adults with Nonalcoholic Fatty Liver Disease†. Hepatology 2010, 52, 913–924. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Isaacs, S.; Barb, D.; Basu, R.; Caprio, S.; Garvey, W.T.; Kashyap, S.; Mechanick, J.I.; Mouzaki, M.; Nadolsky, K.; et al. American Association of Clinical Endocrinology Clinical Practice Guideline for the Diagnosis and Management of Nonalcoholic Fatty Liver Disease in Primary Care and Endocrinology Clinical Settings. Endocr. Pract. 2022, 28, 528–562. [Google Scholar] [CrossRef]
- Priego-Parra, B.A.; Triana-Romero, A.; Bernal-Reyes, R.; Icaza-Chávez, M.E.; Martínez-Vázquez, S.E.; Amieva-Balmori, M.; Cano-Contreras, A.D.; Vivanco-Cid, H.; Remes-Troche, J.M. Evaluación Comparativa de APRI, FIB-4, HFS y NFS: Herramientas de Puntuación Para La Fibrosis Hepática En La Población Mexicana Con MASLD. Rev. Gastroenterol. Mex. 2024, 89, 498–505. [Google Scholar] [CrossRef]
- Castera, L.; Forns, X.; Alberti, A. Non-Invasive Evaluation of Liver Fibrosis Using Transient Elastography. J. Hepatol. 2008, 48, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Sandrin, L.; Fourquet, B.; Hasquenoph, J.-M.; Yon, S.; Fournier, C.; Mal, F.; Christidis, C.; Ziol, M.; Poulet, B.; Kazemi, F.; et al. Transient Elastography: A New Noninvasive Method for Assessment of Hepatic Fibrosis. Ultrasound Med. Biol. 2003, 29, 1705–1713. [Google Scholar] [CrossRef]
- Sasso, M.; Beaugrand, M.; de Ledinghen, V.; Douvin, C.; Marcellin, P.; Poupon, R.; Sandrin, L.; Miette, V. Controlled Attenuation Parameter (CAP): A Novel VCTETM Guided Ultrasonic Attenuation Measurement for the Evaluation of Hepatic Steatosis: Preliminary Study and Validation in a Cohort of Patients with Chronic Liver Disease from Various Causes. Ultrasound Med. Biol. 2010, 36, 1825–1835. [Google Scholar] [CrossRef]
- Eddowes, P.J.; Sasso, M.; Allison, M.; Tsochatzis, E.; Anstee, Q.M.; Sheridan, D.; Guha, I.N.; Cobbold, J.F.; Deeks, J.J.; Paradis, V.; et al. Accuracy of FibroScan Controlled Attenuation Parameter and Liver Stiffness Measurement in Assessing Steatosis and Fibrosis in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2019, 156, 1717–1730. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and Validation of a Histological Scoring System for Nonalcoholic Fatty Liver Disease†. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Petroff, D.; Blank, V.; Newsome, P.N.; Shalimar; Voican, C.S.; Thiele, M.; de Lédinghen, V.; Baumeler, S.; Chan, W.K.; Perlemuter, G.; et al. Assessment of Hepatic Steatosis by Controlled Attenuation Parameter Using the M and XL Probes: An Individual Patient Data Meta-Analysis. Lancet Gastroenterol. Hepatol. 2021, 6, 185–198. [Google Scholar] [CrossRef]
- Fraquelli, M.; Rigamonti, C.; Casazza, G.; Conte, D.; Donato, M.F.; Ronchi, G.; Colombo, M. Reproducibility of Transient Elastography in the Evaluation of Liver Fibrosis in Patients with Chronic Liver Disease. Gut 2007, 56, 968–973. [Google Scholar] [CrossRef]
- Clark, J.M. The Epidemiology of Nonalcoholic Fatty Liver Disease in Adults. J. Clin. Gastroenterol. 2006, 40, S5–S10. [Google Scholar] [CrossRef]
- Mantovani, A.; Ballestri, S.; Lonardo, A.; Targher, G. Cardiovascular Disease and Myocardial Abnormalities in Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1246–1267. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Reyes, R.; Castro-Narro, G.; Malé-Velázquez, R.; Carmona-Sánchez, R.; González-Huezo, M.S.; García-Juárez, I.; Chávez-Tapia, N.; Aguilar-Salinas, C.; Aiza-Haddad, I.; Ballesteros-Amozurrutia, M.A.; et al. Consenso Mexicano de La Enfermedad Por Hígado Graso No Alcohólico. Rev. Gastroenterol. Mex. 2019, 84, 69–99. [Google Scholar] [CrossRef]
- Méndez-Sánchez, N.; Díaz-Orozco, L.; Córdova-Gallardo, J. Redefinition of Fatty Liver Disease from NAFLD to MAFLD Raised Disease Awareness: Mexican Experience. J. Hepatol. 2021, 75, 221–222. [Google Scholar] [CrossRef] [PubMed]
- Hagström, H.; Shang, Y.; Hegmar, H.; Nasr, P. Natural History and Progression of Metabolic Dysfunction-Associated Steatotic Liver Disease. Lancet Gastroenterol. Hepatol. 2024, 9, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Lekakis, V.; Papatheodoridis, G.V. Natural History of Metabolic Dysfunction-Associated Steatotic Liver Disease. Eur. J. Intern. Med. 2024, 122, 3–10. [Google Scholar] [CrossRef]
- Gallardo-Rincón, H.; Ríos-Blancas, M.J.; Montoya, A.; Saucedo-Martínez, R.; Morales-Juárez, L.; Mujica, R.; Cantoral, A.; Idueta, L.S.; Lozano, R.; Tapia-Conyer, R. Evaluation of Effective Coverage for Type 2 Diabetes in Mexican Primary Care Health Information Systems: A Retrospective Registry Analysis. Int. J. Equity Health 2023, 22, 61. [Google Scholar] [CrossRef]

{kind=link}
| Liver Stage | Evolution Time for Develop | Clinical Manifestations | References |
|---|---|---|---|
| MASLD | Not determined in this review. The degree of metabolic damage and lipotoxicity may accelerate its onset. | Necroinflammation and metabolic alterations perpetuate a vicious cycle, triggering subsequent stages. | [57,65] |
| MASH | Not determined in this review. The degree of inflammation and lipotoxicity may accelerate its onset. | Inflammation and/or liver lesions ballooning, with or without fibrosis. | [69] |
| Liver fibrosis | Progression from MASLD, one stage, every 14.3 years; from MASH, one stage, every 7.1 years. | Usually asymptomatic, although liver morphology continues to evolve due to chronic inflammation. | [70] |
| Compensated cirrhosis | Progression from MASH and advanced fibrosis in 22% of patients, 2 years. | Usually asymptomatic. Ascites, encephalopathy, and esophageal varices develop during the transition to decompensated cirrhosis. | [72] |
| Decompensated cirrhosis | Progression from compensated cirrhosis in 19% of patients, 2 years. | Chronic hemodynamic changes, systemic inflammation, portal hypertension, and metabolic alterations. | [72] |
| Hepatocellular carcinoma | Not determined in this review. The development time depends on the previous stage and can occur from MASLD, although it is less likely. | Genetic mutations and chronic inflammation damage DNA via ROS. | [79,80,81] |
| Steatosis Grade | CAP Score (dB/m) |
|---|---|
| S0 | <294 |
| S1 | 294–309 |
| S2 | 310–330 |
| S3 | ≥331 |
| Fibrosis Level | LSM Cutoff (kPa) |
|---|---|
| F0–F1 | <8.1 |
| F2 | 8.2–9.6 |
| F3 | 9.7–13.5 |
| F4 | ≥13.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Lithuanian University of Health Sciences. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alpízar Salazar, M.; Olguín Reyes, S.E.; Medina Estévez, A.; Saturno Lobos, J.A.; De Aldecoa Castillo, J.M.; Carrera Aguas, J.C.; Alaniz Monreal, S.; Navarro Rodríguez, J.A.; Alpízar Sánchez, D.M.F. Natural History of Metabolic Dysfunction-Associated Steatotic Liver Disease: From Metabolic Syndrome to Hepatocellular Carcinoma. Medicina 2025, 61, 88. https://doi.org/10.3390/medicina61010088
Alpízar Salazar M, Olguín Reyes SE, Medina Estévez A, Saturno Lobos JA, De Aldecoa Castillo JM, Carrera Aguas JC, Alaniz Monreal S, Navarro Rodríguez JA, Alpízar Sánchez DMF. Natural History of Metabolic Dysfunction-Associated Steatotic Liver Disease: From Metabolic Syndrome to Hepatocellular Carcinoma. Medicina. 2025; 61(1):88. https://doi.org/10.3390/medicina61010088
Chicago/Turabian StyleAlpízar Salazar, Melchor, Samantha Estefanía Olguín Reyes, Andrea Medina Estévez, Julieta Alejandra Saturno Lobos, Jesús Manuel De Aldecoa Castillo, Juan Carlos Carrera Aguas, Samary Alaniz Monreal, José Antonio Navarro Rodríguez, and Dulce María Fernanda Alpízar Sánchez. 2025. "Natural History of Metabolic Dysfunction-Associated Steatotic Liver Disease: From Metabolic Syndrome to Hepatocellular Carcinoma" Medicina 61, no. 1: 88. https://doi.org/10.3390/medicina61010088
APA StyleAlpízar Salazar, M., Olguín Reyes, S. E., Medina Estévez, A., Saturno Lobos, J. A., De Aldecoa Castillo, J. M., Carrera Aguas, J. C., Alaniz Monreal, S., Navarro Rodríguez, J. A., & Alpízar Sánchez, D. M. F. (2025). Natural History of Metabolic Dysfunction-Associated Steatotic Liver Disease: From Metabolic Syndrome to Hepatocellular Carcinoma. Medicina, 61(1), 88. https://doi.org/10.3390/medicina61010088