

Novel Insight of N6-Methyladenosine in Cardiovascular System

 ,
,  ,
,
Abstract
:1. Introduction
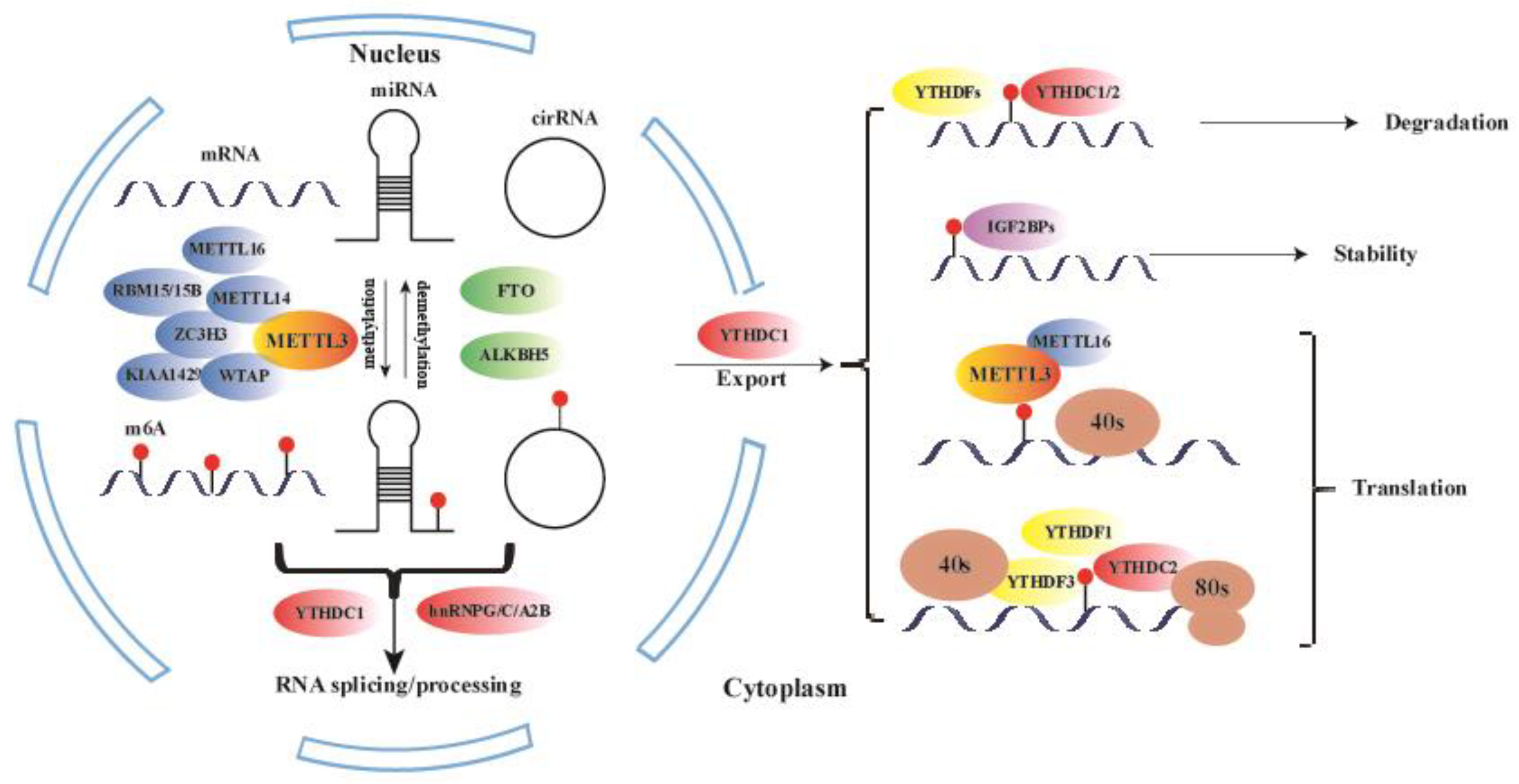
2. m6A RNA Methylation
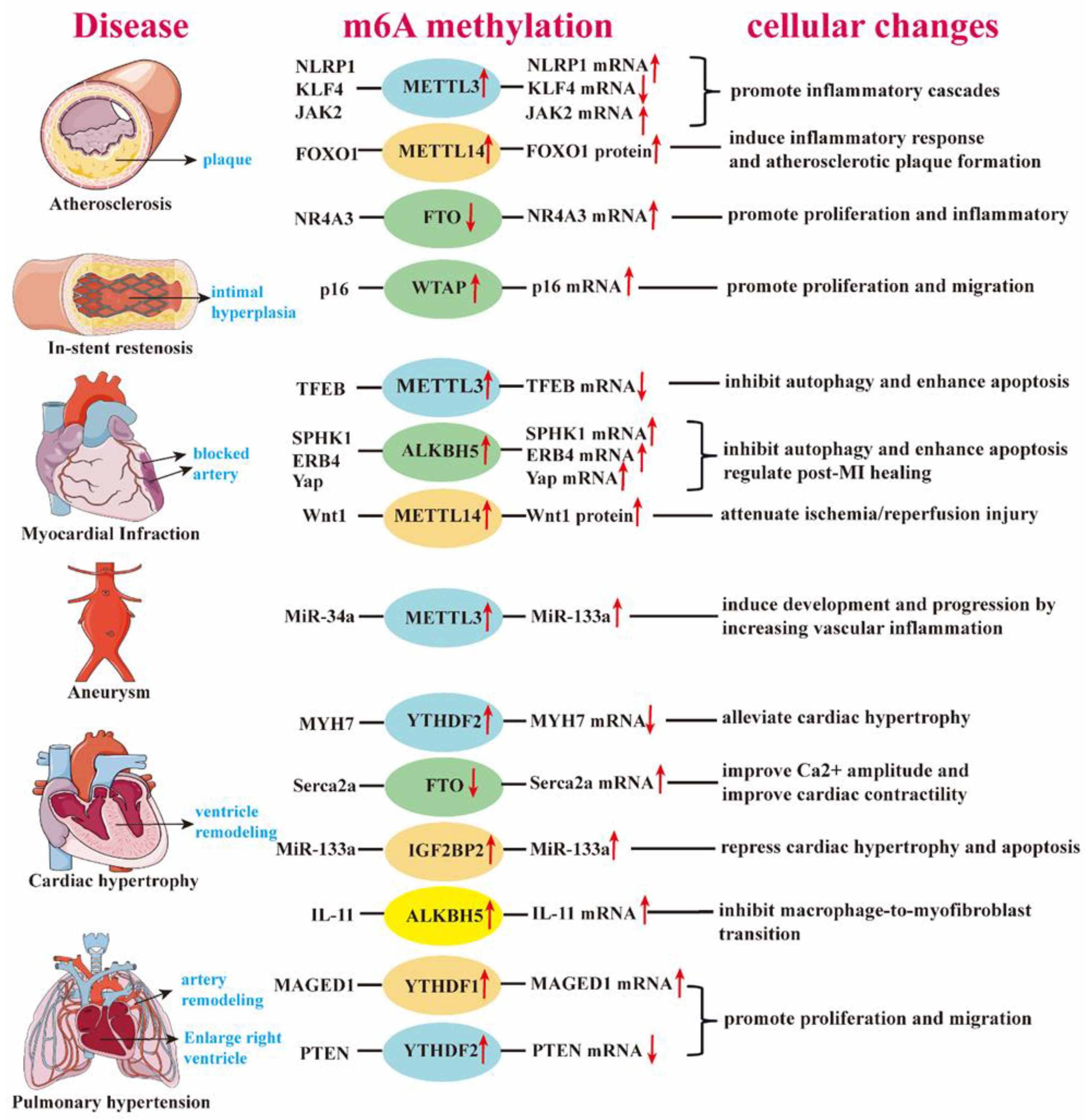
3. Role of m6A in Cardiovascular Disease
3.1. Risk Factors Associated with CVDs
3.1.1. Glucose Metabolism
3.1.2. Adipogenesis and Obesity
3.2. Function of m6A in CVDs
3.2.1. m6A and Ischemia–Hypoxia Injury
3.2.2. m6A and Atherosclerosis
m6A in Vascular Smooth Muscle Cell (VSMC) Differentiation and Angiogenesis
m6A and Calcification
m6A in Atherosclerosis
3.2.3. m6A and Acute Myocardial Infarction
3.2.4. m6A and Heart Failure (HF)
3.2.5. m6A and Other CVDs
3.2.6. m6A in CVDs in the Era of Artificial Intelligence and Machine Learning
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kottakis, F.; Nicolay, B.N.; Roumane, A.; Karnik, R.; Gu, H.; Nagle, J.M.; Boukhali, M.; Hayward, M.C.; Li, Y.Y.; Chen, T.; et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 2016, 539, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Hou, X.; Guan, Q.; Zhou, H.; Zhou, L.; Liu, L.; Liu, J.; Li, F.; Li, W.; Liu, H. RNA modification in cardiovascular disease: Implications for therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 412. [Google Scholar] [CrossRef] [PubMed]
- Motorin, Y.; Helm, M. RNA nucleotide methylation. Wiley Interdiscip. Rev. RNA 2011, 2, 611–631. [Google Scholar] [CrossRef]
- Desrosiers, R.; Friderici, K.; Rottman, F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc. Natl. Acad. Sci. USA 1974, 71, 3971–3975. [Google Scholar] [CrossRef]
- Davalos, V.; Blanco, S.; Esteller, M. SnapShot: Messenger RNA Modifications. Cell 2018, 174, 498. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Han, J.; Wang, J.Z.; Yang, X.; Yu, H.; Zhou, R.; Lu, H.C.; Yuan, W.B.; Lu, J.C.; Zhou, Z.J.; Lu, Q.; et al. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol. Cancer 2019, 18, 110. [Google Scholar] [CrossRef]
- Zhou, C.; Molinie, B.; Daneshvar, K.; Pondick, J.V.; Wang, J.; Van Wittenberghe, N.; Xing, Y.; Giallourakis, C.C.; Mullen, A.C. Genome-Wide Maps of m6A circRNAs Identify Widespread and Cell-Type-Specific Methylation Patterns that Are Distinct from mRNAs. Cell Rep. 2017, 20, 2262–2276. [Google Scholar] [CrossRef]
- Yang, D.; Qiao, J.; Wang, G.; Lan, Y.; Li, G.; Guo, X.; Xi, J.; Ye, D.; Zhu, S.; Chen, W.; et al. N6-Methyladenosine modification of lincRNA 1281 is critically required for mESC differentiation potential. Nucleic Acids Res. 2018, 46, 3906–3920. [Google Scholar] [CrossRef]
- Noale, M.; Limongi, F.; Maggi, S. Epidemiology of Cardiovascular Diseases in the Elderly. Adv. Exp. Med. Biol. 2020, 1216, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Liu, J.; He, C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes. Dev. 2015, 29, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Jaffrey, S.R. Rethinking m6A Readers, Writers, and Erasers. Annu. Rev. Cell Dev. Biol. 2017, 33, 319–342. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.; He, X.; Zhang, W.; Chu, D.; Feng, C. Alleviation Effect of Grape Seed Proanthocyanidins on Neuronal Apoptosis in Rats with Iron Overload. Biol. Trace Elem. Res. 2020, 194, 210–220. [Google Scholar] [CrossRef]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014, 10, 93–95. [Google Scholar] [CrossRef]
- Ping, X.L.; Sun, B.F.; Wang, L.; Xiao, W.; Yang, X.; Wang, W.J.; Adhikari, S.; Shi, Y.; Lv, Y.; Chen, Y.S.; et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014, 24, 177–189. [Google Scholar] [CrossRef]
- Schwartz, S.; Mumbach, M.R.; Jovanovic, M.; Wang, T.; Maciag, K.; Bushkin, G.G.; Mertins, P.; Ter-Ovanesyan, D.; Habib, N.; Cacchiarelli, D.; et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014, 8, 284–296. [Google Scholar] [CrossRef]
- Patil, D.P.; Chen, C.K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef]
- Wen, J.; Lv, R.; Ma, H.; Shen, H.; He, C.; Wang, J.; Jiao, F.; Liu, H.; Yang, P.; Tan, L.; et al. Zc3h13 Regulates Nuclear RNA m6A Methylation and Mouse Embryonic Stem Cell Self-Renewal. Mol. Cell 2018, 69, 1028–1038.e6. [Google Scholar] [CrossRef]
- Yue, Y.; Liu, J.; Cui, X.; Cao, J.; Luo, G.; Zhang, Z.; Cheng, T.; Gao, M.; Shu, X.; Ma, H.; et al. VIRMA mediates preferential m6A mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.M.; Li, C.J.; Vagbo, C.B.; Shi, Y.; Wang, W.L.; Song, S.H.; et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N6-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [Google Scholar] [CrossRef]
- Shi, H.; Wang, X.; Lu, Z.; Zhao, B.S.; Ma, H.; Hsu, P.J.; Liu, C.; He, C. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res. 2017, 27, 315–328. [Google Scholar] [CrossRef]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Luo, G.Z.; Zhang, Z.; Wang, X.; Zhou, T.; Cui, Y.; Sha, J.; Huang, X.; Guerrero, L.; Xie, P.; et al. YTHDC1 mediates nuclear export of N6-methyladenosine methylated mRNAs. eLife 2017, 6, e31311. [Google Scholar] [CrossRef]
- Hsu, P.J.; Zhu, Y.; Ma, H.; Guo, Y.; Shi, X.; Liu, Y.; Qi, M.; Lu, Z.; Shi, H.; Wang, J.; et al. Ythdc2 is an N6-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017, 27, 1115–1127. [Google Scholar] [CrossRef]
- Degrauwe, N.; Suva, M.L.; Janiszewska, M.; Riggi, N.; Stamenkovic, I. IMPs: An RNA-binding protein family that provides a link between stem cell maintenance in normal development and cancer. Genes. Dev. 2016, 30, 2459–2474. [Google Scholar] [CrossRef]
- Wu, B.; Su, S.; Patil, D.P.; Liu, H.; Gan, J.; Jaffrey, S.R.; Ma, J. Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nat. Commun. 2018, 9, 420. [Google Scholar] [CrossRef]
- Tang, Y.; Chen, K.; Song, B.; Ma, J.; Wu, X.; Xu, Q.; Wei, Z.; Su, J.; Liu, G.; Rong, R.; et al. m6A-Atlas: A comprehensive knowledgebase for unraveling the N6-methyladenosine (m6A) epitranscriptome. Nucleic Acids Res. 2021, 49, D134–D143. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Gao, Y.; Jin, X.; Wang, H.; Lan, T.; Wei, M.; Yan, W.; Wang, G.; Li, Z.; Zhao, Z.; et al. Comprehensive analysis of N6-methylandenosine regulators and m6A-related RNAs as prognosis factors in colorectal cancer. Mol. Ther. Nucleic Acids 2022, 27, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Toth, J.I.; Petroski, M.D.; Zhang, Z.; Zhao, J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014, 16, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.S.; Hao, Y.J.; Sun, B.F.; Sun, H.Y.; Li, A.; Ping, X.L.; Lai, W.Y.; et al. Nuclear m6A Reader YTHDC1 Regulates mRNA Splicing. Mol. Cell 2016, 61, 507–519. [Google Scholar] [CrossRef]
- Yang, Y.; Shen, F.; Huang, W.; Qin, S.; Huang, J.T.; Sergi, C.; Yuan, B.F.; Liu, S.M. Glucose Is Involved in the Dynamic Regulation of m6A in Patients with Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2019, 104, 665–673. [Google Scholar] [CrossRef]
- De Jesus, D.F.; Zhang, Z.; Kahraman, S.; Brown, N.K.; Chen, M.; Hu, J.; Gupta, M.K.; He, C.; Kulkarni, R.N. m6A mRNA Methylation Regulates Human beta-Cell Biology in Physiological States and in Type 2 Diabetes. Nat. Metab. 2019, 1, 765–774. [Google Scholar] [CrossRef]
- Kruger, N.; Biwer, L.A.; Good, M.E.; Ruddiman, C.A.; Wolpe, A.G.; DeLalio, L.J.; Murphy, S.; Macal, E.H., Jr.; Ragolia, L.; Serbulea, V.; et al. Loss of Endothelial FTO Antagonizes Obesity-Induced Metabolic and Vascular Dysfunction. Circ. Res. 2020, 126, 232–242. [Google Scholar] [CrossRef]
- Wu, R.; Guo, G.; Bi, Z.; Liu, Y.; Zhao, Y.; Chen, N.; Wang, F.; Wang, Y.; Wang, X. m6A methylation modulates adipogenesis through JAK2-STAT3-C/EBPbeta signaling. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 796–806. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, Y.; Sun, B.F.; Shi, Y.; Yang, X.; Xiao, W.; Hao, Y.J.; Ping, X.L.; Chen, Y.S.; Wang, W.J.; et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014, 24, 1403–1419. [Google Scholar] [CrossRef]
- Wang, X.; Wu, R.; Liu, Y.; Zhao, Y.; Bi, Z.; Yao, Y.; Liu, Q.; Shi, H.; Wang, F.; Wang, Y. m6A mRNA methylation controls autophagy and adipogenesis by targeting Atg5 and Atg7. Autophagy 2020, 16, 1221–1235. [Google Scholar] [CrossRef]
- Zhang, B.; Jiang, H.; Wu, J.; Cai, Y.; Dong, Z.; Zhao, Y.; Hu, Q.; Hu, K.; Sun, A.; Ge, J. m6A demethylase FTO attenuates cardiac dysfunction by regulating glucose uptake and glycolysis in mice with pressure overload-induced heart failure. Signal Transduct. Target. Ther. 2021, 6, 377. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Chen, Z.; Friedman, J. Transcriptional regulation of adipogenesis by KLF4. Cell Metab. 2008, 7, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Song, T.; Yang, Y.; Wei, H.; Xie, X.; Lu, J.; Zeng, Q.; Peng, J.; Zhou, Y.; Jiang, S.; Peng, J. Zfp217 mediates m6A mRNA methylation to orchestrate transcriptional and post-transcriptional regulation to promote adipogenic differentiation. Nucleic Acids Res. 2019, 47, 6130–6144. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Han, D.; Liu, J.; Shi, J.; Zhu, P.; Wang, Y.; Dong, N. Factors influencing osteogenic differentiation of human aortic valve interstitial cells. J. Thorac. Cardiovasc. Surg. 2021, 161, e163–e185. [Google Scholar] [CrossRef]
- Chen, J.; Ning, Y.; Zhang, H.; Song, N.; Gu, Y.; Shi, Y.; Cai, J.; Ding, X.; Zhang, X. METTL14-dependent m6A regulates vascular calcification induced by indoxyl sulfate. Life Sci. 2019, 239, 117034. [Google Scholar] [CrossRef]
- Song, H.; Feng, X.; Zhang, H.; Luo, Y.; Huang, J.; Lin, M.; Jin, J.; Ding, X.; Wu, S.; Huang, H.; et al. METTL3 and ALKBH5 oppositely regulate m6A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy 2019, 15, 1419–1437. [Google Scholar] [CrossRef]
- Kumari, R.; Dutta, R.; Ranjan, P.; Suleiman, Z.G.; Goswami, S.K.; Li, J.; Pal, H.C.; Verma, S.K. ALKBH5 Regulates SPHK1-Dependent Endothelial Cell Angiogenesis Following Ischemic Stress. Front. Cardiovasc. Med. 2021, 8, 817304. [Google Scholar] [CrossRef]
- Han, Z.; Wang, X.; Xu, Z.; Cao, Y.; Gong, R.; Yu, Y.; Yu, Y.; Guo, X.; Liu, S.; Yu, M.; et al. ALKBH5 regulates cardiomyocyte proliferation and heart regeneration by demethylating the mRNA of YTHDF1. Theranostics 2021, 11, 3000–3016. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, J.; Sun, X.; Yang, K.; Yang, L.; Kong, L.; Zhang, B.; Li, F.; Li, C.; Shi, B.; et al. Loss of m6A demethylase ALKBH5 promotes post-ischemic angiogenesis via post-transcriptional stabilization of WNT5A. Clin. Transl. Med. 2021, 11, e402. [Google Scholar] [CrossRef]
- Shen, W.; Li, H.; Su, H.; Chen, K.; Yan, J. FTO overexpression inhibits apoptosis of hypoxia/reoxygenation-treated myocardial cells by regulating m6A modification of Mhrt. Mol. Cell Biochem. 2021, 476, 2171–2179. [Google Scholar] [CrossRef]
- Yin, T.; Wang, N.; Jia, F.; Wu, Y.; Gao, L.; Zhang, J.; Hou, R. Exosome-based WTAP siRNA delivery ameliorates myocardial ischemia-reperfusion injury. Eur. J. Pharm. Biopharm. 2024, 197, 114218. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.S.; Li, J.Y.; Chien, Y.; Wang, M.L.; Yarmishyn, A.A.; Tsai, P.H.; Juan, C.C.; Nguyen, P.; Cheng, H.M.; Huo, T.I.; et al. METTL3-dependent N6-methyladenosine RNA modification mediates the atherogenic inflammatory cascades in vascular endothelium. Proc. Natl. Acad. Sci. USA 2021, 118, e2025070118. [Google Scholar] [CrossRef] [PubMed]
- Jian, D.; Wang, Y.; Jian, L.; Tang, H.; Rao, L.; Chen, K.; Jia, Z.; Zhang, W.; Liu, Y.; Chen, X.; et al. METTL14 aggravates endothelial inflammation and atherosclerosis by increasing FOXO1 N6-methyladeosine modifications. Theranostics 2020, 10, 8939–8956. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Yu, J.; Shan, G.; Su, L.; Yu, N.; Yang, S. N6-Methyladenosine Methyltransferase METTL3 Promotes Angiogenesis and Atherosclerosis by Upregulating the JAK2/STAT3 Pathway via m6A Reader IGF2BP1. Front. Cell Dev. Biol. 2021, 9, 731810. [Google Scholar] [CrossRef]
- Zhang, B.Y.; Han, L.; Tang, Y.F.; Zhang, G.X.; Fan, X.L.; Zhang, J.J.; Xue, Q.; Xu, Z.Y. METTL14 regulates M6A methylation-modified primary miR-19a to promote cardiovascular endothelial cell proliferation and invasion. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 7015–7023. [Google Scholar] [CrossRef]
- Huo, Y.B.; Gao, X.; Peng, Q.; Nie, Q.; Bi, W. Dihydroartemisinin alleviates AngII-induced vascular smooth muscle cell proliferation and inflammatory response by blocking the FTO/NR4A3 axis. Inflamm. Res. 2022, 71, 243–253. [Google Scholar] [CrossRef]
- Zhu, B.; Gong, Y.; Shen, L.; Li, J.; Han, J.; Song, B.; Hu, L.; Wang, Q.; Wang, Z. Total Panax notoginseng saponin inhibits vascular smooth muscle cell proliferation and migration and intimal hyperplasia by regulating WTAP/p16 signals via m6A modulation. Biomed. Pharmacother. 2020, 124, 109935. [Google Scholar] [CrossRef]
- Gong, R.; Wang, X.; Li, H.; Liu, S.; Jiang, Z.; Zhao, Y.; Yu, Y.; Han, Z.; Yu, Y.; Dong, C.; et al. Loss of m6A methyltransferase METTL3 promotes heart regeneration and repair after myocardial injury. Pharmacol. Res. 2021, 174, 105845. [Google Scholar] [CrossRef]
- Cheng, P.; Han, H.; Chen, F.; Cheng, L.; Ma, C.; Huang, H.; Chen, C.; Li, H.; Cai, H.; Huang, H.; et al. Amelioration of acute myocardial infarction injury through targeted ferritin nanocages loaded with an ALKBH5 inhibitor. Acta Biomater. 2022, 140, 481–491. [Google Scholar] [CrossRef]
- Yang, K.; Zhao, Y.; Hu, J.; Gao, R.; Shi, J.; Wei, X.; Chen, J.; Hu, K.; Sun, A.; Ge, J. ALKBH5 induces fibroblast-to-myofibroblast transformation during hypoxia to protect against cardiac rupture after myocardial infarction. J. Adv. Res. 2024, 61, 193–209. [Google Scholar] [CrossRef]
- Pang, P.; Qu, Z.; Yu, S.; Pang, X.; Li, X.; Gao, Y.; Liu, K.; Liu, Q.; Wang, X.; Bian, Y.; et al. Mettl14 Attenuates Cardiac Ischemia/Reperfusion Injury by Regulating Wnt1/beta-Catenin Signaling Pathway. Front. Cell Dev. Biol. 2021, 9, 762853. [Google Scholar] [CrossRef] [PubMed]
- Mathiyalagan, P.; Adamiak, M.; Mayourian, J.; Sassi, Y.; Liang, Y.; Agarwal, N.; Jha, D.; Zhang, S.; Kohlbrenner, E.; Chepurko, E.; et al. FTO-Dependent N6-Methyladenosine Regulates Cardiac Function During Remodeling and Repair. Circulation 2019, 139, 518–532. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Z.; Chen, M.; Zhao, W.; Tao, T.; Ma, L.; Ni, Y.; Li, W. YTHDF2 alleviates cardiac hypertrophy via regulating Myh7 mRNA decoy. Cell Biosci. 2021, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.; Wang, P.; Zhang, D.; Wu, L. m6A modification promotes miR-133a repression during cardiac development and hypertrophy via IGF2BP2. Cell Death Discov. 2021, 7, 157. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, T.; Chen, M.H.; Wu, R.X.; Wang, J.; Hu, X.D.; Meng, T.; Wu, A.H.; Li, Y.; Yang, Y.F.; Lei, Y.; et al. ALKBH5-mediated m6A modification of IL-11 drives macrophage-to-myofibroblast transition and pathological cardiac fibrosis in mice. Nat. Commun. 2024, 15, 1995. [Google Scholar] [CrossRef]
- Qin, Y.; Qiao, Y.; Li, L.; Luo, E.; Wang, D.; Yao, Y.; Tang, C.; Yan, G. The m6A methyltransferase METTL3 promotes hypoxic pulmonary arterial hypertension. Life Sci. 2021, 274, 119366. [Google Scholar] [CrossRef]
- Hu, L.; Wang, J.; Huang, H.; Yu, Y.; Ding, J.; Yu, Y.; Li, K.; Wei, D.; Ye, Q.; Wang, F.; et al. YTHDF1 Regulates Pulmonary Hypertension through Translational Control of MAGED1. Am. J. Respir. Crit. Care Med. 2021, 203, 1158–1172. [Google Scholar] [CrossRef]
- Zhong, L.; He, X.; Song, H.; Sun, Y.; Chen, G.; Si, X.; Sun, J.; Chen, X.; Liao, W.; Liao, Y.; et al. METTL3 Induces AAA Development and Progression by Modulating N6-Methyladenosine-Dependent Primary miR34a Processing. Mol. Ther. Nucleic Acids 2020, 21, 394–411. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Li, J.; Li, S.; Wang, F. Mechanism of METTL3-Mediated m6A Modification in Cardiomyocyte Pyroptosis and Myocardial Ischemia-Reperfusion Injury. Cardiovasc. Drugs Ther. 2023, 37, 435–448. [Google Scholar] [CrossRef]
- Kroemer, G. Autophagy: A druggable process that is deregulated in aging and human disease. J. Clin. Investig. 2015, 125, 1–4. [Google Scholar] [CrossRef]
- Jin, S.; Zhang, X.; Miao, Y.; Liang, P.; Zhu, K.; She, Y.; Wu, Y.; Liu, D.A.; Huang, J.; Ren, J.; et al. m6A RNA modification controls autophagy through upregulating ULK1 protein abundance. Cell Res. 2018, 28, 955–957. [Google Scholar] [CrossRef] [PubMed]
- Frismantiene, A.; Philippova, M.; Erne, P.; Resink, T.J. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity. Cell Signal 2018, 52, 48–64. [Google Scholar] [CrossRef]
- Lin, J.; Zhu, Q.; Huang, J.; Cai, R.; Kuang, Y. Hypoxia Promotes Vascular Smooth Muscle Cell (VSMC) Differentiation of Adipose-Derived Stem Cell (ADSC) by Regulating Mettl3 and Paracrine Factors. Stem Cells Int. 2020, 2020, 2830565. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Liu, Y.; Zhou, L.; Xue, Y.; Lu, Z.; Gan, J. YTHDC2-Mediated circYTHDC2 N6-Methyladenosine Modification Promotes Vascular Smooth Muscle Cells Dysfunction Through Inhibiting Ten-Eleven Translocation 2. Front. Cardiovasc. Med. 2021, 8, 686293. [Google Scholar] [CrossRef] [PubMed]
- Parial, R.; Li, H.; Li, J.; Archacki, S.; Yang, Z.; Wang, I.Z.; Chen, Q.; Xu, C.; Wang, Q.K. Role of epigenetic m6A RNA methylation in vascular development: Mettl3 regulates vascular development through PHLPP2/mTOR-AKT signaling. FASEB J. 2021, 35, e21465. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.D.; Jiang, Q.; Ma, Y.; Liu, C.; Zhu, C.Y.; Sun, Y.N.; Shan, K.; Ge, H.M.; Zhang, Q.Y.; Zhang, H.Y.; et al. Role of METTL3-Dependent N6-Methyladenosine mRNA Modification in the Promotion of Angiogenesis. Mol. Ther. 2020, 28, 2191–2202. [Google Scholar] [CrossRef]
- Geovanini, G.R.; Libby, P. Atherosclerosis and inflammation: Overview and updates. Clin. Sci. 2018, 132, 1243–1252. [Google Scholar] [CrossRef]
- Chiu, J.J.; Chien, S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiol. Rev. 2011, 91, 327–387. [Google Scholar] [CrossRef]
- Miano, J.M.; Fisher, E.A.; Majesky, M.W. Fate and State of Vascular Smooth Muscle Cells in Atherosclerosis. Circulation 2021, 143, 2110–2116. [Google Scholar] [CrossRef]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, Z.; Tang, H.; Shen, Y.; Gong, Z.; Xie, N.; Zhang, X.; Wang, W.; Kong, W.; Zhou, Y.; et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 facilitates M1 macrophage polarization through the methylation of STAT1 mRNA. Am. J. Physiol. Cell Physiol. 2019, 317, C762–C775. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Hu, X.; Huang, M.; Liu, J.; Gu, Y.; Ma, L.; Zhou, Q.; Cao, X. Mettl3-mediated mRNA m6A methylation promotes dendritic cell activation. Nat. Commun. 2019, 10, 1898. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Gan, X.; Jiang, X.; Diao, S.; Wu, H.; Hu, J. ALKBH5 inhibited autophagy of epithelial ovarian cancer through miR-7 and BCL-2. J. Exp. Clin. Cancer Res. 2019, 38, 163. [Google Scholar] [CrossRef]
- Writing Committee, M.; Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; et al. 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013, 128, e240–e327. [Google Scholar] [CrossRef]
- Roger, V.L.; Weston, S.A.; Redfield, M.M.; Hellermann-Homan, J.P.; Killian, J.; Yawn, B.P.; Jacobsen, S.J. Trends in heart failure incidence and survival in a community-based population. JAMA 2004, 292, 344–350. [Google Scholar] [CrossRef]
- Zhang, B.; Xu, Y.; Cui, X.; Jiang, H.; Luo, W.; Weng, X.; Wang, Y.; Zhao, Y.; Sun, A.; Ge, J. Alteration of m6A RNA Methylation in Heart Failure With Preserved Ejection Fraction. Front. Cardiovasc. Med. 2021, 8, 647806. [Google Scholar] [CrossRef]
- Berulava, T.; Buchholz, E.; Elerdashvili, V.; Pena, T.; Islam, M.R.; Lbik, D.; Mohamed, B.A.; Renner, A.; von Lewinski, D.; Sacherer, M.; et al. Changes in m6A RNA methylation contribute to heart failure progression by modulating translation. Eur. J. Heart Fail. 2020, 22, 54–66. [Google Scholar] [CrossRef]
- Hinger, S.A.; Wei, J.; Dorn, L.E.; Whitson, B.A.; Janssen, P.M.L.; He, C.; Accornero, F. Remodeling of the m6A landscape in the heart reveals few conserved post-transcriptional events underlying cardiomyocyte hypertrophy. J. Mol. Cell Cardiol. 2021, 151, 46–55. [Google Scholar] [CrossRef]
- Li, T.; Zhuang, Y.; Yang, W.; Xie, Y.; Shang, W.; Su, S.; Dong, X.; Wu, J.; Jiang, W.; Zhou, Y.; et al. Silencing of METTL3 attenuates cardiac fibrosis induced by myocardial infarction via inhibiting the activation of cardiac fibroblasts. FASEB J. 2021, 35, e21162. [Google Scholar] [CrossRef]
- Fang, M.; Deng, J.; Zhou, Q.; Hu, Z.; Yang, L. Maslinic acid protects against pressure-overload-induced cardiac hypertrophy by blocking METTL3-mediated m6A methylation. Aging 2022, 14, 2548–2557. [Google Scholar] [CrossRef] [PubMed]
- Dorn, L.E.; Lasman, L.; Chen, J.; Xu, X.; Hund, T.J.; Medvedovic, M.; Hanna, J.H.; van Berlo, J.H.; Accornero, F. The N6-Methyladenosine mRNA Methylase METTL3 Controls Cardiac Homeostasis and Hypertrophy. Circulation 2019, 139, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Kmietczyk, V.; Riechert, E.; Kalinski, L.; Boileau, E.; Malovrh, E.; Malone, B.; Gorska, A.; Hofmann, C.; Varma, E.; Jurgensen, L.; et al. m6A-mRNA methylation regulates cardiac gene expression and cellular growth. Life Sci. Alliance 2019, 2, e201800233. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Hu, Y.; Hou, L.; Ju, J.; Li, X.; Du, N.; Guan, X.; Liu, Z.; Zhang, T.; Qin, W.; et al. beta-Blocker carvedilol protects cardiomyocytes against oxidative stress-induced apoptosis by up-regulating miR-133 expression. J. Mol. Cell Cardiol. 2014, 75, 111–121. [Google Scholar] [CrossRef]
- Liu, N.; Bezprozvannaya, S.; Williams, A.H.; Qi, X.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes. Dev. 2008, 22, 3242–3254. [Google Scholar] [CrossRef]
- Marcadenti, A.; Fuchs, F.D.; Matte, U.; Sperb, F.; Moreira, L.B.; Fuchs, S.C. Effects of FTO RS9939906 and MC4R RS17782313 on obesity, type 2 diabetes mellitus and blood pressure in patients with hypertension. Cardiovasc. Diabetol. 2013, 12, 103. [Google Scholar] [CrossRef]
- Mo, X.B.; Lei, S.F.; Zhang, Y.H.; Zhang, H. Examination of the associations between m6A-associated single-nucleotide polymorphisms and blood pressure. Hypertens. Res. 2019, 42, 1582–1589. [Google Scholar] [CrossRef]
- Su, H.; Wang, G.; Wu, L.; Ma, X.; Ying, K.; Zhang, R. Transcriptome-wide map of m6A circRNAs identified in a rat model of hypoxia mediated pulmonary hypertension. BMC Genom. 2020, 21, 39. [Google Scholar] [CrossRef]
- Ma, D.; Liu, X.; Zhang, J.J.; Zhao, J.J.; Xiong, Y.J.; Chang, Q.; Wang, H.Y.; Su, P.; Meng, J.; Zhao, Y.B. Vascular Smooth Muscle FTO Promotes Aortic Dissecting Aneurysms via m6A Modification of Klf5. Front. Cardiovasc. Med. 2020, 7, 592550. [Google Scholar] [CrossRef]
- Li, T.; Wu, Y.; Yang, J.; Jing, J.; Ma, C.; Sun, L. N6-methyladenosine-associated genetic variants in NECTIN2 and HPCAL1 are risk factors for abdominal aortic aneurysm. iScience 2024, 27, 109419. [Google Scholar] [CrossRef]
- Huang, Y.; Yan, J.; Li, Q.; Li, J.; Gong, S.; Zhou, H.; Gan, J.; Jiang, H.; Jia, G.F.; Luo, C.; et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015, 43, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Ambale-Venkatesh, B.; Yang, X.; Wu, C.O.; Liu, K.; Hundley, W.G.; McClelland, R.; Gomes, A.S.; Folsom, A.R.; Shea, S.; Guallar, E.; et al. Cardiovascular Event Prediction by Machine Learning: The Multi-Ethnic Study of Atherosclerosis. Circ. Res. 2017, 121, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Hou, L.; Park, Y.P.; Molinie, B.; Consortium, G.T.; Gregory, R.I.; Kellis, M. Genetic drivers of m6A methylation in human brain, lung, heart and muscle. Nat. Genet. 2021, 53, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, K.; Fang, Q.; Shi, S.; Nan, J.; He, J.; Yin, Y.; Li, X.; Li, J.; Hou, L.; et al. Crosstalk between epitranscriptomic and epigenomic modifications and its implication in human diseases. Cell Genom. 2024, 4, 100605. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Type | Regulator | Function of RNA Modification | Reference |
|---|---|---|---|
| writers | METTL3 | main catalytic subunit of m6A | [16] |
| METTL14/16 | activate METTL3 through allosteric and RNA substrate recognition | [16] | |
| WTAP | the third subunit of the METTL3-METTL14 complex | [17,18] | |
| ZC3H3 | assist the localization of the methyltransferase complex in nuclear speckles and U-rich regions adjacent to the m6A sites in mRNAs | [19] | |
| RBM15/15B | [20] | ||
| KIAA1429 | [21] | ||
| erasers | FTO | demethylation of m6a | [2,22,23] |
| ALKBH5 | |||
| readers | YTHDF1 | promote mRNA translation | [24,25] |
| YTHDF2 | accelerate the decay of m6A-modified transcripts | ||
| YTHDF3 | promote mRNA translation or enhance RNA decay | ||
| YTHDC1 | promote mRNA translation and splicing and nuclear export | [26,27] | |
| YTHDC2 | enhance translation | [28] | |
| IGF2BP1/2/3 | regulate RNA localization, translation, and stability | [29] | |
| hnRNPG/C/A2B | promote RNA stability and mediate RNA splicing and microRNA process | [30] |
| Risk Factors | Regulators | Cell | Regulation | Signaling | Function | Reference |
|---|---|---|---|---|---|---|
| glucose metabolism | FTO↑ | hepatocellular cell | up-regulate mRNA | FOXO1/FASN/ G6PC/DGAT2 | improve the production of serum glucose and lipids | [35] |
| diabetes | METTL14↓ | pancreatic β-cell | promote mRNA translation | AKT/PDX1 | induce cell-cycle arrest and impair insulin secretion | [36] |
| obesity | FTO↑ | endothelial cell | down-regulate mRNA | AKT/ prostaglandinD2 | aggravate vascular dysfunction | [37] |
| FTO↑ | preadipocyte | up-regulate mRNA | JAK2-STAT3-C/EBPβ | promote adipogenesis | [38] | |
| FTO↑ | preadipocyte | control exonic splicing | RUNX1T1 | modulate differentiation to promote adipogenesis | [39] | |
| FTO↑ | preadipocytes | improve mRNA stability | Atg5/Atg7 | promote autophagy and adipogenesis | [40] |
| Diseases | Regulators | Cell | Regulation | Signaling | Function | Reference |
|---|---|---|---|---|---|---|
| calcification | METTL3↑ | valve interstitial cell | up-regulate mRNA | TWIST1 | promote osteogenic differentiation process | [44] |
| calcification | METTL14↑ | smooth muscle cell | down-regulate mRNA | Klotho | promote calcification | [45] |
| Hypoxia-reoxygenation | METTL3↑ | cardiomyocyte | down-regulate mRNA | TFEB | inhibit autophagy and enhance apoptosis | [46] |
| ischemic injury | ALKBH5↑ | endothelial cell | up-regulate mRNA | SPHK1/eNOS-AKT | maintain angiogenesis | [47] |
| heart regeneration | ALKBH5↑ | cardiomyocyte | improve mRNA stability | YAP | promote proliferation | [48] |
| post-ischemic | ALKBH5↑ | endothelial cell | decrease mRNA stability | WNT5A | exacerbate dysfunction of CMECs | [49] |
| Hypoxia-reoxygenation | FTO↑ | cardiomyocyte | up-regulate mRNA | Mhrt | inhibit apoptosis | [50] |
| Hypoxia-reoxygenation | WTAP↑ | cardiomyocyte | up-regulate mRNA | TXNIP | enhance apoptosis | [51] |
| atherosclerosis | METTL3↑ | endothelial cell | up/down-regulate mRNA | NLRP1/KLF4 | promote inflammatory cascades | [52] |
| atherosclerosis | METTL14↑ | endothelial cell | promote translation | FOXO1/VCAM-1/ICAM-1 | induce inflammatory response and promote atherosclerotic plaque formation | [53] |
| atherosclerosis | METTL3↑ | endothelial cell | up-regulate mRNA | JAK2/STAT3 | promote atherosclerosis progression | [54] |
| atherosclerosis | METTL14↑ | endothelial cell | up-regulate miRNA | pri-miR-19a /DGCR8 | promote proliferation and invasion of ASVEC | [55] |
| atherosclerosis | FTO↓ | smooth muscle cell | up-regulate mRNA | NR4A3 | promote proliferation and inflammation | [56] |
| intimal hyperplasia | WTAP↓ | smooth muscle cell | up-regulate mRNA | p16 | promote proliferation and migration of VSMC | [57] |
| heart regeneration | METTL3↓ | cardiomyocyte | up-regulate miRNA | miR-143 /Yap/Ctnnd1 | inhibit heart regeneration | [58] |
| AML | ALKBH5↑ | cardiomyocyte | not mention | TCA cycle | affect cell metabolism and survival | [59] |
| AML | ALKBH5↑ | fibroblast | improve mRNA stability | ErbB4 | regulate post-MI healing | [60] |
| Ischemia-reperfusion injury | METTL14↑ | cardiomyocyte | promote translation efficiency | Wnt1 | attenuate ischemia–reperfusion Injury | [61] |
| heart failure | FTO↓ | cardiomyocyte | improve mRNA stability | Serca2a | improve Ca2+ amplitude | [62] |
| heart failure | YTHDF2↑ | cardiomyocyte | promote mRNA degradation | Myh7 | alleviate cardiac hypertrophy | [63] |
| heart failure | IGF2BP2↑ | cardiomyocyte | promote miRNA accumulation on its target site | miR-133a | repress cardiac hypertrophy and apoptosis | [64] |
| heart failure | FTO↓ | cardiomyocytes | improve mRNA stability | Pgam2 | regulate glucose uptake | [41] |
| heart failure | ALKBH5↑ | macrophage | improve mRNA stability | IL-11 | inhibit macrophage-to-myofibroblast transition | [65] |
| pulmonary hypertension | YTHDF2↑ | smooth muscle cell | promote mRNA degradation | PTEN/PI3K/Akt | enhance proliferation | [66] |
| pulmonary hypertension | YTHDF1↑ | smooth muscle cell | promote translation efficiency | MAGED1 | promote proliferation | [67] |
| aneurysm | METTL3↑ | smooth muscle cell | up-regulate miRNA | DGCR8/ miR-34a | induce development and progression | [68] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Lithuanian University of Health Sciences. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Lu, W.; Tang, H.; Chen, A.; Gao, X.; Zhu, C.; Zhang, J. Novel Insight of N6-Methyladenosine in Cardiovascular System. Medicina 2025, 61, 222. https://doi.org/10.3390/medicina61020222
Zhang H, Lu W, Tang H, Chen A, Gao X, Zhu C, Zhang J. Novel Insight of N6-Methyladenosine in Cardiovascular System. Medicina. 2025; 61(2):222. https://doi.org/10.3390/medicina61020222
Chicago/Turabian StyleZhang, Huan, Wei Lu, Haoyue Tang, Aiqun Chen, Xiaofei Gao, Congfei Zhu, and Junjie Zhang. 2025. "Novel Insight of N6-Methyladenosine in Cardiovascular System" Medicina 61, no. 2: 222. https://doi.org/10.3390/medicina61020222
APA StyleZhang, H., Lu, W., Tang, H., Chen, A., Gao, X., Zhu, C., & Zhang, J. (2025). Novel Insight of N6-Methyladenosine in Cardiovascular System. Medicina, 61(2), 222. https://doi.org/10.3390/medicina61020222