
Wilms’ Tumor: A Review of Clinical Characteristics, Treatment Advances, and Research Opportunities

 , ,
, ,
Abstract
:1. Introduction
2. Prevalence and Epidemiology of Nephroblastomas
3. Characteristics and Classification of Pediatric Nephroblastoma
4. Biomarkers in the Diagnosis of Nephroblastoma
Early Detection Screening of Wilms’ Tumor
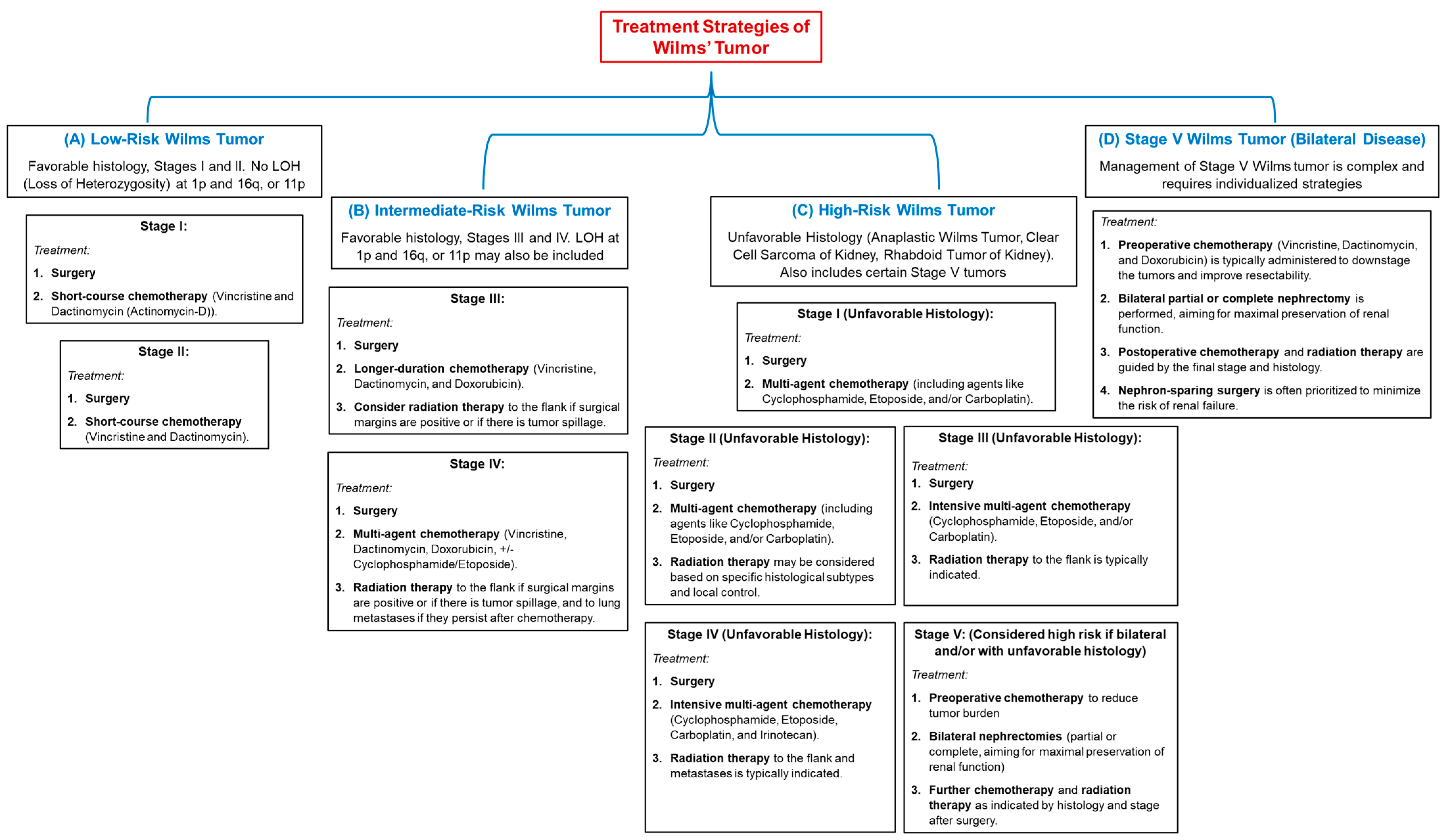
5. Current Treatment Therapies for Wilms’ Tumor
6. Alternative Treatment Therapies
7. Future Research Directions
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BWS | Beckwith-Wiedemann syndrome |
| CCRS | Clear-cell renal sarcoma |
| CMN | Congenital mesoblastic nephroma |
| CT | Computer tomograph |
| DAWT | Diffuse Anaplastic Wilms’ Tumor |
| DDS | Denys-Drash syndrome |
| ISPO/SIOP | International Society of Pediatric Oncology |
| lncRNAs | long non-coding RNAs |
| MAL | Myelin and lymphocyte myelin protein |
| MRTK | Malignant rhabdoid tumor of the kidney |
| NCAMs | Neural cell adhesion molecules |
| NSS | Nephron-sparing surgery |
| NWRT | Non-Wilms renal tumors |
| NWTS | National Wilms Tumor Study |
| PNET | Primitive neuroectodermal tumor |
| PPGGL | Polish Pediatric Solid Tumor Treatment Group |
| RCC | Renal cell carcinoma |
References
- Hong, B.; Dong, R. Research advances in the targeted therapy and immunotherapy of Wilms tumor: A narrative review. Transl. Cancer Res. 2021, 10, 1559–1567. [Google Scholar] [CrossRef]
- Tian, X.; Xiang, B.; Jin, L.; Mi, T.; Wang, J.; Zhanghuang, C.; Zhang, Z.X.; Chen, M.L.; Shi, Q.L.; Liu, F.; et al. Immune-related gene signature associates with immune landscape and predicts prognosis accurately in patients with Wilms tumor. Front. Immunol. 2022, 13, 01–18. [Google Scholar]
- Percicote, A.P.; Mardegan, G.L.; Gugelmim, E.S.; Ioshii, S.O.; Kuczynski, A.P.; Nagashima, S.; de Noronha, L. Tissue expression of retinoic acid receptor alpha and CRABP2 in metastatic nephroblastomas. Diagn. Pathol. 2018, 13, 9. [Google Scholar] [CrossRef]
- Wu, Y.; Chu, C.; Zhang, J.; Nitish, B.; Ni, J.; Xu, X. A case of ovarian Teratoma with nephroblastoma presenting abdomen metastasis. J. Clin. Lab. Anal. 2022, 36, e24364. [Google Scholar] [CrossRef] [PubMed]
- Kanyamuhunga, A.; Tuyisenge, L.; Stefan, D.C. Treating childhood cancer in Rwanda: The nephroblastoma example. Pan Afr. Med. J. 2015, 21, 326. [Google Scholar] [CrossRef] [PubMed]
- Turcas, A.; Gheara, C.; Galatan, V.; Blag, C.; Cernea, D. Second, peculiar recurrence of a wilms tumor—Pleural and late. J. Med. Radiat. Oncol. 2022, 2, 46–51. [Google Scholar] [CrossRef]
- Emeka, C.K.; Chikaodili, E.T.; Patrick, A.L. Nephrectomy in children: A single center experience. ASOAJ 2022, 3, 27–30. [Google Scholar] [CrossRef]
- Littooij, A.S.; Nikkels, P.G.; Hulsbergen-van de Kaa, C.A.; van de Ven, C.P.; van den Heuvel-Eibrink, M.M.; Olsen, Ø.E. Apparent diffusion coefficient as it relates to histopathology findings in post-chemotherapy nephroblastoma: A feasibility study. Pediatr. Radiol. 2017, 47, 1608–1614. [Google Scholar] [CrossRef]
- Grill, C.; Sunitsch, S.; Hatz, M.; Hauser-Kronberger, C.; Leuschner, I.; Hoefler, G.; Guertl, B. Activation of beta-catenin is a late event in the pathogenesis of nephroblastomas and rarely correlated with genetic changes of the APC gene. Pathology 2011, 43, 702–706. [Google Scholar] [CrossRef]
- Saula, P.W.; Hadley, G.P. Pediatric non-Wilms’ renal tumors: A third world experience. World J. Surg. 2012, 36, 565–572. [Google Scholar] [CrossRef]
- Karlsson, J.; Valind, A.; Jansson, C.; O’Sullivan, M.J.; Holmquist Mengelbier, L.; Gisselsson, D. Aberrant epigenetic regulation in clear cell sarcoma of the kidney featuring distinct DNA hypermethylation and EZH2 overexpression. Oncotarget 2016, 7, 11127–11136. [Google Scholar] [CrossRef]
- Megison, M.L.; Gillory, L.A.; Stewart, J.E.; Nabers, H.C.; Mrozcek-Musulman, E.; Beierle, E.A. FAK inhibition abrogates the malignant phenotype in aggressive pediatric renal tumors. Mol. Cancer Res. 2014, 12, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.; Nayak, G.; Mohanty, M.; Mohanty, S.; Senapati, U. Aggressive uncommon pediatric renal tumor: Rhabdoid tumor of kidney. Indian. J. Pathol. Oncol. 2021, 8, 299–301. [Google Scholar] [CrossRef]
- Zhuge, Y.; Cheung, M.C.; Yang, R.; Perez, E.A.; Koniaris, L.G.; Sola, J.E. Pediatric non-Wilms renal tumors: Subtypes, survival, and prognostic indicators. J. Surg. Res. 2010, 163, 257–263. [Google Scholar] [CrossRef]
- Narui, R.; Yagasaki, H.; Takahashi, Y.; Hama, A.; Nishio, N.; Muramatsu, H.; Shimoyama, Y.; Kojima, S. Concurrent Langerhans cell histiocytosis and nephroblastoma. Pediatr. Blood Cancer 2009, 52, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Hajzler, W.; Kopera, J.; Kosek, K.; Mazur, D.; Rurańska, I.; Szczepański, T.; Pobudejska-Pieniążek, A. Characteristics of patients with blastemal-type wilms’ tumour. Pediatr. Pol. 2021, 96, 134–138. [Google Scholar] [CrossRef]
- Li, P.; Zhang, K.; Tang, S.; Tang, W. Knockdown of lncRNA HAGLROS inhibits metastasis and promotes apoptosis in nephroblastoma cells by inhibition of autophagy. Bioengineered 2022, 13, 7552–7562. [Google Scholar] [CrossRef]
- Nofech-Mozes, R.; Khella, H.W.; Scorilas, A.; Youssef, L.; Krylov, S.N.; Lianidou, E.; Sidiropoulos, K.G.; Gabril, M.; Evans, A.; Yousef, G.M. MicroRNA-194 is a Marker for Good Prognosis in Clear Cell Renal Cell Carcinoma. Cancer Med. 2016, 5, 656–664. [Google Scholar] [CrossRef]
- Senanayake, U.; Das, S.; Vesely, P.; Alzoughbi, W.; Fröhlich, L.F.; Chowdhury, P.; Leuschner, I.; Hoefler, G.; Guertl, B. miR-192, miR-194, miR-215, miR-200c and miR-141 are downregulated and their common target ACVR2B is strongly expressed in renal childhood neoplasms. Carcinogenesis 2012, 33, 1014–1021. [Google Scholar] [CrossRef]
- Littooij, A.S.; Sebire, N.J.; Olsen, Ø.E. Whole-tumor apparent diffusion coefficient measurements in nephroblastoma: Can it identify blastemal predominance? J. Magn. Reson. Imaging 2017, 45, 1316–1324. [Google Scholar] [CrossRef]
- Hötker, A.M.; Lollert, A.; Mazaheri, Y.; Müller, S.; Schenk, J.P.; Mildenberger, P.C.; Akin, O.; Graf, N.; Staatz, G. Diffusion-weighted MRI in the assessment of nephroblastoma: Results of a multi-center trial. Abdom Radiol 2020, 45, 3202–3212. [Google Scholar] [CrossRef] [PubMed]
- Guertl, B.; Senanayake, U.; Nusshold, E.; Leuschner, I.; Mannweiler, S.; Ebner, B.; Hoefler, G. Lim1, an embryonal transcription factor, is absent in multicystic renal dysplasia, but reactivated in nephroblastomas. Pathobiology 2011, 78, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, J.; Jin, L.; Mi, T.; Zhang, Z.; Zhanghuang, C.; Li, M.; Wang, J.; Wu, X.; Wang, Z.; et al. ZSTK474 targeting PIK3R3 inhibits the Wilms’ tumor through G0 / G1 phase arrest. PLoS ONE 2024, 19, e0312178. [Google Scholar] [CrossRef] [PubMed]
- Stones, D.K.; Hadley, G.P.; Wainwright, R.D.; Stefan, D.C. The impact of ethnicity on Wilms tumor: Characteristics and outcome of a South African cohort. Int. J. Pediatr. 2015, 2015, 706058. [Google Scholar] [CrossRef]
- Ooms, A.H.A.G.; Vujanić, G.M.; D’Hooghe, E.; Collini, P.; L’Herminé-Coulomb, A.; Vokuhl, C.; Graf, N.; Heuvel-Eibrink, M.M.V.D.; de Krijger, R.R. Renal Tumors of Childhood-A Histopathologic Pattern-Based Diagnostic Approach. Cancers 2020, 12, 729. [Google Scholar] [CrossRef]
- Su, C.; Huang, R.; Yu, Z.; Zheng, J.; Liu, F.; Liang, H.; Mo, Z. Myelin and lymphocyte protein serves as a prognostic biomarker and is closely associated with the tumor microenvironment in the nephroblastoma. Cancer Med. 2022, 11, 1427–1438. [Google Scholar] [CrossRef]
- Benlhachemi, S.; Abouqal, R.; Coleman, N.; Murray, M.J.; Khattab, M.; El Fahime, E. Circulating microRNA profiles in Wilms tumour (WT): A systematic review and meta-analysis of diagnostic test accuracy. Noncoding RNA Res. 2023, 8, 413–425. [Google Scholar] [CrossRef]
- Zhang, J.; Hou, T.; Qi, X.; Wang, J.; Sun, X. SOX21-AS1 is associated with clinical stage and regulates cell proliferation in nephroblastoma. Biosci. Rep. 2019, 39, BSR20190602. [Google Scholar] [CrossRef]
- Lopes, R.I.; Lorenzo, A. Recent advances in the management of Wilms’ tumor. F1000Research 2017, 6, 670. [Google Scholar] [CrossRef]
- Wang, J.; Li, M.; Tang, D.; Gu, W.; Mao, J.; Shu, Q. Current treatment for Wilms tumor: COG and SIOP standards. World J. Pediatr. Surg. 2019, 2, e000038. [Google Scholar] [CrossRef]
- Benbrahim, F.Z.; Haddad, S.E.; Belkouchi, L.; Faraj, C.; Jellal, S.; Boutaleb, J.; Allali, N.; Chat, L. Acute pyelonephritis and subcapsular hematoma revealing a nephroblastoma. Radiol. Case Rep. 2024, 20, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.J.; Couitchere, L.; Atimere, Y.; Koné, D.; Azagoh-Kouadio, R.; Oulai, M.S.; Stefan, D.C. Childhood cancer in Côte d’Ivoire, 1995–2004: Challenges and hopes. South Afr. Med. J. 2012, 103, 113–115. [Google Scholar] [CrossRef]
- Lu, T.; Li, L.; Zhu, J.; Liu, J.; Lin, A.; Fu, W.; Liu, G.; Xia, H.; Zhang, T.; He, J. AURKA rs8173 G>C Polymorphism Decreases Wilms Tumor Risk in Chinese Children. J. Oncol. 2019, 2019, 9074908. [Google Scholar] [CrossRef] [PubMed]
- Blish, K.R.; Clausen, K.A.; Hawkins, G.A.; Garvin, A.J.; Willingham, M.C.; Turner, J.C.; Torti, F.M.; Torti, S.V. Loss of heterozygosity and SOSTDC1 in adult and pediatric renal tumors. J. Exp. Clin. Cancer Res. 2010, 29, 147. [Google Scholar] [CrossRef]
- Ahmad, E.; Qasim, B.; Hussein, A. Clinicopathological assessment of renal tumors in pediatric age group in a sample of Iraqi patients. Iraqi J. Cancer Med. Genet. 2023, 16, 72–86. [Google Scholar] [CrossRef]
- Li, G.; Jia, W.; Yin, Z.; Zhu, J.; Liu, G.; Xia, H.; He, J.; Fu, W. LMO1 Super-Enhancer rs2168101 G>T Polymorphism Reduces Wilms Tumor Risk. J. Cancer 2019, 10, 1808–1813. [Google Scholar] [CrossRef]
- Fu, W.; Zhuo, Z.J.; Jia, W.; Zhu, J.; Zhu, S.B.; Lin, Z.F.; Wang, F.H.; Xia, H.; He, J.; Liu, G.C. Association between TP53 gene Arg72Pro polymorphism and Wilms’ tumor risk in a Chinese population. Onco Targets Ther. 2017, 10, 1149–1154. [Google Scholar] [CrossRef]
- Lee, J.S.; Padilla, B.; DuBois, S.G.; Oates, A.; Boscardin, J.; Goldsby, R.E. Second malignant neoplasms among children, adolescents and young adults with Wilms tumor. Pediatr. Blood Cancer 2015, 62, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- Al-Hadidi, A.; Lapkus, M.; Novotny, N.M.; Gowans, L.K.; Chen, P.Y.; Stallion, A. Wilms Tumor with Pleural Metastasis. Glob. Pediatr. Health 2020, 7, 2333794X20952292. [Google Scholar] [CrossRef]
- Okbah, A.A.; Al-Shamahy, H.A. Nephroblastoma (Wilms’ Tumor): Sex and Age Distribution and Correlation Rate with Ages, Sex, and Kidney Side in Sana’a City, Yemen. J. Clin. Oncol. Case Rep. 2023, 2, 1–5. [Google Scholar]
- Nazemi, A.; Daneshmand, S.; Chang, A. Pediatric genitourinary tumors: Distribution, demographics, and outcomes. Pediatr. Investig. 2022, 6, 85–92. [Google Scholar] [CrossRef]
- Jones, E.A.; Stewart, A.; Stiller, C.; Douglas, F.; Bown, N. Wilms tumor incidence in children with 2q terminal deletions: A cohort study. Am. J. Med. Genet. A 2011, 155A, 2221–2223. [Google Scholar] [CrossRef] [PubMed]
- Chong, W.C.; Cain, J.E. Lessons learned from the developmental origins of childhood renal cancer. Anat. Rec. 2020, 303, 2561–2577. [Google Scholar] [CrossRef]
- Wesevich, A.; Mocha, G.; Kiwara, F.; Chao, C.; Shabani, I.; Igenge, J.Z.; Schroeder, K. Wilms tumor treatment protocol compliance and the influence on outcomes for children in Tanzania. Pediatr. Blood Cancer 2023, 70, e30704. [Google Scholar] [CrossRef] [PubMed]
- Libes, J.; Oruko, O.; Abdallah, F.; Githanga, J.; Ndung’U, J.; Musimbi, J.; Njuguna, F.; Patel, K.; White, J.; Axt, J.R.; et al. Risk factors for abandonment of Wilms tumor therapy in Kenya. Pediatr. Blood Cancer 2014, 62, 252–256. [Google Scholar] [CrossRef]
- Israëls, T.; Pidini, D.; Borgstein, E.; Bailey, S.; Tump, C.; Chagaluka, G.; Kamiza, S.; Molyneux, E. Survival of children with a Wilms tumor in Blantyre, Malawi. Pediatr. Hematol. Oncol. 2018, 35, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Chagaluka, G.; Paintsil, V.; Renner, L.; Weijers, J.; Chitsike, I.; Borgstein, E.; Kamiza, S.; Afungchwi, G.M.; Kouya, F.; Hesseling, P.; et al. Improvement of overall survival in the collaborative Wilms tumour Africa project. Pediatr. Blood Cancer 2020, 67, e28383. [Google Scholar] [CrossRef]
- Treger, T.D.; Chowdhury, T.; Pritchard-Jones, K.; Behjati, S. The genetic changes of Wilms tumor. Nat. Rev. Nephrol. 2019, 15, 240–251. [Google Scholar] [CrossRef]
- Zhu, S.; Zhou, R.; Tang, X.; Fu, W.; Jia, W. Hypoxia/inflammation-induced upregulation of HIF-1α and C/EBPβ promotes nephroblastoma cell EMT by improving HOXA11-AS transcription. Heliyon 2024, 10, e27654. [Google Scholar] [CrossRef]
- Sasso, G.; Greco, N.; Murino, P.; Sasso, F.S. Late toxicity in Wilms tumor patients treated with radiotherapy at 15 years of median follow-up. J. Pediatr. Hematol. Oncol. 2010, 32, e264-7. [Google Scholar] [CrossRef]
- Wellens, L.M.; Meulstee, J.; van de Ven, C.P.; Terwisscha van Scheltinga, C.E.J.; Littooij, A.S.; van den Heuvel-Eibrink, M.M.; Fiocco, M.; Rios, A.C.; Maal, T.; Wijnen, M.H.W.A. Comparison of 3-Dimensional and Augmented Reality Kidney Models With Conventional Imaging Data in the Preoperative Assessment of Children With Wilms Tumors. JAMA Netw. Open 2019, 2, e192633. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wu, Y.; Xu, W.; Liu, J.; Lv, Z. Teratoid Wilms Tumor and Classical Wilms Tumor: A Retrospective 10-Year Single-Center Study and Literature Review. Front. Surg. 2022, 8, 781060. [Google Scholar] [CrossRef]
- Geng, G.; Xu, Y.; Li, Q.; Li, Q.; Yuan, L.; Dong, M.; Ming, M. S100A16 cooperates with DEPDC1 to promote the progression and angiogenesis of nephroblastoma through PI3K/Akt/mTOR pathway. Pol. J. Pathol. 2023, 74, 182–193. [Google Scholar] [CrossRef]
- Davidoff, A.M.; Interiano, R.B.; Wynn, L.; Delos Santos, N.; Dome, J.S.; Green, D.M.; Brennan, R.C.; McCarville, M.B.; Krasin, M.J.; Kieran, K.; et al. Overall Survival and Renal Function of Patients With Synchronous Bilateral Wilms Tumor Undergoing Surgery at a Single Institution. Ann. Surg. 2015, 262, 570–576. [Google Scholar] [CrossRef]
- Sandoval, J.A.; Malkas, L.H.; Hickey, R.J. Clinical significance of serum biomarkers in pediatric solid mediastinal and abdominal tumors. Int. J. Mol. Sci. 2012, 13, 1126–1153. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, I.; Chicard, M.; Colmet-Daage, L.; Clément, N.; Danzon, A.; Lapouble, E.; Pierron, G.; Bohec, M.; Baulande, S.; Berrebi, D.; et al. Circulating tumor DNA analysis enables molecular characterization of pediatric renal tumors at diagnosis. Int. J. Cancer 2019, 144, 68–79. [Google Scholar] [CrossRef]
- Janeczko-Czarnecka, M.; Slezak, R.; Pietras, W.; Pstrusinska, K. Wilms tumor (nephroblastoma)—Clinical and genetic aspects. Nowotw. J. Oncol. 2022, 72, 259–264. [Google Scholar] [CrossRef]
- Zina, D.; Rosita, K.; Kristina, Z.; Giedre, R.; Jurate, M. Case Report: Autosomal dominant polycystic kidney disease and Wilms’ tumor in infancy and chilhood. Front. Pediatr. 2024, 12, 1322142. [Google Scholar] [CrossRef] [PubMed]
- Seminara, C.; Planells, M.C.; Pogonza, R.E.; Morales, M.; Tissera, R. Wilms tumor: 15 years of experience at a children’s hospital in Córdoba, Argentina. Arch. Argent. Pediatr. 2019, 117, 263–270. [Google Scholar]
- Solomon, Z.; Withers, A.; Govender, T.; Je, P.; Wainwright, R.; Candy, G.; Loveland, J. Bilateral wilms’ tumor: A ten-year experience of two academic centers in Johannesburg. South. Afr. J. Child. Health 2021, 15, 8. [Google Scholar] [CrossRef]
- Ma, X.H.; Yang, J.; Jia, X.; Zhou, H.C.; Liang, J.W.; Ding, Y.S.; Shu, Q.; Niu, T. Preoperative radiomic signature based on CT images for noninvasive evaluation of localized nephroblastoma in pediatric patients. Front. Oncol. 2023, 13, 1122210. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.K.; Goldin, A.B.; Ehrlich, P.F.; Beierle, E.A.; Doski, J.J.; Goldfarb, M.; Langer, M.; Nuchtern, J.G.; Vasudevan, S.; Gow, K.W. Partial Nephrectomy for Nephroblastoma: A National Cancer Data Base Review. Am. Surg. 2018, 84, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Nirgude, S.; Naveh, N.S.S.; Kavari, S.L.; Traxler, E.M.; Kalish, J.M. Cancer predisposition signaling in Beckwith-Wiedemann Syndrome drives Wilms tumor development. Br. J. Cancer 2024, 130, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Imrani, K.; Chat, L. Bilateral nephroblastoma: A case report. Int. J. Case Rep. Images 2020, 11, 1. [Google Scholar] [CrossRef]
- Raghunath, B.; Jadhav, V.; Shankar, G.; Narendrababu, M.; Ramesh, S. Management of Bilateral Wilms’ Tumor: Our Experience. Indian. J. Surg. Oncol. 2017, 8, 4–8. [Google Scholar] [CrossRef]
- Furtwängler, R.; Schmolze, M.; Gräber, S.; Leuschner, I.; Amann, G.; Schenk, J.P.; Niggli, F.; Kager, L.; von Schweinitz, D.; Graf, N. Pretreatment for bilateral nephroblastomatosis is an independent risk factor for progressive disease in patients with stage V nephroblastoma. Klin. Padiatr. 2014, 226, 175–181. [Google Scholar] [CrossRef]
- Xu, S.; Sun, N.; Zhang, W.P.; Song, H.C.; Huang, C.R. Management of Wilms tumor with intravenous thrombus in children: A single center experience. World J. Pediatr. 2019, 15, 476–482. [Google Scholar] [CrossRef]
- Xu, G.; Hu, J.; Wu, Y.; Xiao, Y.; Xu, M. Botryoid Wilms’ tumor: A case report and review of the literature. World J. Surg. Oncol. 2013, 11, 102. [Google Scholar] [CrossRef]
- Sakthivel, V.; Adeeb, I.Z.; Vijayabalan, D. Recent Improvements in Adult Wilms Tumor Diagnosis and Management: Review of Literature. J. Kidney Cancer VHL 2023, 10, 32–36. [Google Scholar] [CrossRef]
- Sethasathien, S.; Choed-Amphai, C.; Saengsin, K.; Sathitsamitphong, L.; Charoenkwan, P.; Tepmalai, K.; Silvilairat, S. Wilms tumor with dilated cardiomyopathy: A case report. World J. Clin. Oncol. 2019, 10, 293–299. [Google Scholar] [CrossRef]
- Miura, H.; Kawana, S.; Sugino, S.; Kikuchi, C.; Yamauchi, M. Successful management of an infant with hypertensive heart failure associated with Wilms’ tumor: A case report. JA Clin. Rep. 2020, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Honda, A.; Shima, M.; Onoe, S.; Hanada, M.; Nagai, T.; Nakajima, S.; Okada, S. Botryoid Wilms tumor: Case report and review of literature. Pediatr. Nephrol. 2000, 14, 59–61. [Google Scholar] [CrossRef]
- Collins, A.; Molina Kuna, E.; Anderson-Mellies, A.; Cost, C.; Green, A.L. Investigating the Impact of Tumor Biology and Social Determinants on Time to Diagnosis and Stage at Presentation of Wilms Tumor. J. Pediatr. Hematol. Oncol. 2024, 46, 147–153. [Google Scholar] [CrossRef]
- Chandrasekaran, A. Neonatal solid tumors. Pediatr. Neonatol. 2018, 59, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Hu, J.; Cui, Y.; Liang, S.; Gao, X.; Zhang, J.; Jia, W. Knockdown of SENP1 inhibits HIF-1α SUMOylation and suppresses oncogenic CCNE1 in Wilms tumor. Mol. Ther. Oncolytics 2021, 23, 355–366. [Google Scholar] [CrossRef]
- Jeevarathi, T.; Vadivelu, G. Long-term outcome of Wilms’ tumor in tertiary care hospital. Int. Surg. J. 2020, 7, 3234. [Google Scholar]
- Song, J.S.; Kim, I.K.; Kim, Y.M.; Khang, S.K.; Kim, K.R.; Lee, Y. Extrarenal teratoid Wilms’ tumor: Two cases in unusual locations, one associated with elevated serum AFP. Pathol. Int. 2010, 60, 35–41. [Google Scholar] [CrossRef]
- Green, D.M.; Cotton, C.A.; Malogolowkin, M.; Breslow, N.E.; Perlman, E.; Miser, J.; Ritchey, M.L.; Thomas, P.R.; Grundy, P.E.; D’Angio, G.J.; et al. Treatment of Wilms tumor relapsing after initial treatment with vincristine and actinomycin D: A report from the National Wilms Tumor Study Group. Pediatr. Blood Cancer 2007, 48, 493–499. [Google Scholar] [CrossRef]
- Hohenstein, P.; Pritchard-Jones, K.; Charlton, J. The yin and yang of kidney development and Wilms’ tumors. Genes. Dev. 2015, 29, 467–482. [Google Scholar] [CrossRef]
- Popov, S.D.; Sebire, N.J.; Vujanic, G.M. Wilms’ Tumour—Histology and Differential Diagnosis. In Wilms Tumor; Van den Heuvel-Eibrink, M.M., Ed.; Codon Publications: Brisbane, Australia, 2016; Chapter 1. [Google Scholar]
- Turner, J.T.; Brzezinski, J.; Dome, J.S. Wilms Tumor Predisposition. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2003. [Google Scholar]
- Murphy, A.J.; Cheng, C.; Williams, J.; Shaw, T.I.; Pinto, E.M.; Dieseldorff-Jones, K.; Brzezinski, J.; Renfro, L.A.; Tornwall, B.; Huff, V.; et al. Genetic and epigenetic features of bilateral Wilms tumor predisposition in patients from the Children’s Oncology Group AREN18B5-Q. Nat. Commun. 2023, 14, 8006. [Google Scholar] [CrossRef]
- Charlton, J.; Irtan, S.; Bergeron, C.; Pritchard-Jones, K. Bilateral Wilms tumour: A review of clinical and molecular features. Expert. Rev. Mol. Med. 2017, 19, e8. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.T.; Loh, A.H.P. Inter-Ethnic Variations in the Clinical, Pathological, and Molecular Characteristics of Wilms Tumor. Cancers 2024, 16, 3051. [Google Scholar] [CrossRef] [PubMed]
- Waehle, V.; Ungricht, R.; Hoppe, P.S.; Betschinger, J. The tumor suppressor WT1 drives progenitor cell progression and epithelialization to prevent Wilms tumorigenesis in human kidney organoids. Stem Cell Rep. 2021, 16, 2107–2117. [Google Scholar] [CrossRef] [PubMed]
- Sangkhathat, S.; Kanngurn, S.; Chaiyapan, W.; Gridist, P.; Maneechay, W. Wilms’ tumor 1 gene (WT1) is overexpressed and provides an oncogenic function in pediatric nephroblastomas harboring the wild-type WT1. Oncol. Lett. 2010, 1, 615–619. [Google Scholar] [CrossRef]
- Li, Y.; Lei, C.; Xiang, B.; Li, F.; Wang, C.; Wang, Q.; Chen, S.; Ji, Y. Extrarenal teratoma with nephroblastoma in the retroperitoneum: Case report and literature review. Medicine 2017, 96, e8670. [Google Scholar] [CrossRef]
- Hu, J.; Jin, L.U.; He, T.; Li, Y.; Zhao, Y.; Ding, Y.U.; Li, X.; Liu, Y.; Gui, Y.; Mao, X.; et al. Wilms’ tumor in a 51-year-old patient: An extremely rare case and review of the literature. Mol. Clin. Oncol. 2016, 4, 1013–1016. [Google Scholar] [CrossRef]
- Ko, S. A Rare Antitiy Extrarenal Wilms Tumor: A Case Report. Med. J. Clin. Trials Case Stud. 2017, 1, 1–5. [Google Scholar] [CrossRef]
- Sinha, A.; Phukan, J.P.; Bandyopadhyay, G.; Mukherjee, S. Teratoid Wilms’ tumor in a child: A report of a rare case. Int. J. Appl. Basic. Med. Res. 2013, 3, 72–74. [Google Scholar] [CrossRef]
- Shukrun, R.; Pode-Shakked, N.; Pleniceanu, O.; Omer, D.; Vax, E.; Peer, E.; Pri-Chen, S.; Jacob, J.; Hu, Q.; Harari-Steinberg, O.; et al. Wilms’ tumor blastemal stem cells dedifferentiate to propagate the tumor bulk. Stem Cell Rep. 2014, 3, 24–33. [Google Scholar] [CrossRef]
- Li, H.; Hohenstein, P.; Kuure, S. Embryonic Kidney Development, Stem Cells and the Origin of Wilms Tumor. Genes 2021, 12, 318. [Google Scholar] [CrossRef]
- Ferreira Junior, J.A.; Rissi, D.R.; Elias, M.A.; Leonardo, A.S.; Nascimento, K.A.; Macêdo, J.T.S.A.; Pedroso, P. Nephroblastoma in a black-tufted marmoset (Callithrix penicillata). Pesqui. Veterinária Bras. 2018, 38, 2155–2158. [Google Scholar] [CrossRef]
- Gadd, S.; Huff, V.; Walz, A.L.; Ooms, A.H.A.G.; Armstrong, A.E.; Gerhard, D.S.; Smith, M.A.; Auvil, J.M.G.; Meerzaman, D.; Chen, Q.R.; et al. A Children’s Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat. Genet. 2017, 49, 1487–1494. [Google Scholar] [CrossRef]
- Numakura, Y.; Konishi, S.; Kumabe, S.; Kotera, T.; Ueda, M. A case of spontaneous nephroblastoma characterized by two distinct morphologies in a Slc:CD(SD)IGS rat. J. Toxicol. Pathol. 2020, 33, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chu, X.; Song, L.; Tang, W. A novel model incorporating chromatin regulatory factors for risk stratification, prognosis prediction, and characterization of the microenvironment in Wilms tumor. J. Gene Med. 2024, 26, e3574. [Google Scholar] [CrossRef]
- Vujanić, G.M.; Sandstedt, B.; Harms, D.; Kelsey, A.; Leuschner, I.; de Kraker, J.; SIOP Nephroblastoma Scientific Committee. Revised International Society of Paediatric Oncology (SIOP) working classification of renal tumors of childhood. Med. Pediatr. Oncol. 2002, 38, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.; van Peer, S.E.; de Witte, M.M.; Tytgat, G.A.M.; Karim-Kos, H.E.; van Grotel, M.; van de Ven, C.P.; Mavinkurve-Groothuis, A.M.C.; Merks, J.H.M.; Kuiper, R.P.; et al. Characteristics and outcome of children with renal tumors in the Netherlands: The first five-years’ experience of national centralization. PLoS ONE 2022, 17, e0261729. [Google Scholar] [CrossRef]
- Hu, Q.; Gao, F.; Tian, W.; Ruteshouser, E.C.; Wang, Y.; Lazar, A.; Stewart, J.; Strong, L.C.; Behringer, R.R.; Huff, V. Wt1 ablation and Igf2 upregulation in mice result in Wilms tumors with elevated ERK1/2 phosphorylation. J. Clin. Invest. 2011, 121, 174–183. [Google Scholar] [CrossRef]
- Rakheja, D.; Khokhar, S.; Mitui, M.; Cost, N.G. Immunohistochemical expression of GLUT1 and its correlation with unfavorable histology and TP53 codon 72 polymorphism in Wilms tumors. Pediatr. Dev. Pathol. 2012, 15, 286–292. [Google Scholar] [CrossRef]
- Popov, S.D.; Sebire, N.J.; Pritchard-Jones, K.; Vujanić, G.M. Renal tumors in children aged 10-16 Years: A report from the United Kingdom Children’s Cancer and Leukaemia Group. Pediatr. Dev. Pathol. 2011, 14, 189–193. [Google Scholar] [CrossRef]
- van den Hoek, J.; de Krijger, R.; van de Ven, K.; Lequin, M.; van den Heuvel-Eibrink, M.M. Cystic nephroma, cystic partially differentiated nephroblastoma and cystic Wilms’ tumor in children: A spectrum with therapeutic dilemmas. Urol. Int. 2009, 82, 65–70. [Google Scholar] [CrossRef]
- Davidoff, A.M.; Giel, D.W.; Jones, D.P.; Jenkins, J.J.; Krasin, M.J.; Hoffer, F.A.; Williams, M.A.; Dome, J.S. The feasibility and outcome of nephron-sparing surgery for children with bilateral Wilms tumor: The St Jude Children’s Research Hospital experience: 1999–2006. Cancer 2008, 112, 2060–2070. [Google Scholar] [CrossRef] [PubMed]
- Venkatramani, R.; Chi, Y.-Y.; Coppes, M.J.; Malogolowkin, M.; Kalapurakal, J.A.; Tian, J.; Dome, J.S. Outcome of patients with intracranial relapse enrolled on national Wilms tumor study group clinical trials. Pediatr. Blood Cancer 2017, 64, e26406. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, J.; Zhu, J.; Chen, S.; Fu, W.; Tian, X.; Lou, S.; Ruan, J.; He, J.; Zhou, H. MYCN gene polymorphisms and Wilms tumor susceptibility in Chinese children. J. Clin. Lab. Anal. 2019, 33, e22988. [Google Scholar] [CrossRef]
- Krishna, O.; Kayla, G.; Aleem, M.; Malleboyina, R.; Kota, R. Immunohistochemical expression of Ki67 and p53 in Wilms tumor and its relationship with tumor histology and stage at presentation. Pathol. Res. Int. 2016, 2016, 6123951. [Google Scholar] [CrossRef]
- Thevendran, G. Wilms’ tumor in a 37 years old. J. Clin. Med. Res. 2010, 2, 194–197. [Google Scholar] [CrossRef]
- Chan, C.C.; To, K.F.; Yuen, H.L.; Chiang, A.K.S.; Ling, S.C.; Li, C.H.; Cheuk, D.K.L.; Li, C.K.; Shing, M.M.K. A 20-year prospective study of Wilms tumor and other kidney tumors. J. Pediatr. Hematol. /Oncol. 2014, 36, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Kieran, K.; Anderson, J.R.; Dome, J.S.; Ehrlich, P.F.; Ritchey, M.L.; Shamberger, R.C.; Perlman, E.J.; Green, D.M.; Davidoff, A.M. Is adrenalectomy necessary during unilateral nephrectomy for wilms tumor? A report from the children’s oncology group. J. Pediatr. Surg. 2013, 48, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- Trink, A.; Kanter, I.; Pode-Shakked, N.; Urbach, A.; Dekel, B.; Kalisky, T. Geometry of gene expression space of Wilms’ tumors from human patients. Neoplasia 2018, 20, 871–881. [Google Scholar] [CrossRef]
- Yao, W.; Weng, S.; Li, K.; Shen, J.; Dong, R.; Dong, K. Bilateral Wilms tumor: 10-year experience from a single center in China. Transl. Cancer Res. 2024, 13, 879–887. [Google Scholar] [CrossRef]
- Fang, Y.W.; Song, H.C.; Sun, N.; Zhang, W.P. Non-Wilms’ renal tumors in children: Experience with 139 cases treated at a single center. BMC Urol. 2022, 22, 89. [Google Scholar] [CrossRef]
- Bozlu, G.; Çıtak, E.Ç. Evaluation of renal tumors in children. Turk. J. Urol. 2018, 44, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Dorwal, P.; Abou-Seif, C.; Ng, J.; Super, L.; Chan, Y.; Rathi, V. Clear Cell Sarcoma of the Kidney (CCSK) With BCOR-CCNB3 Fusion: A Rare Case Report With a Brief Review of the Literature. Pediatr. Dev. Pathol. 2023, 26, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Raffaele, A.; Gazzaneo, M.; Busel, A.; Vatta, F.; Belgiovine, C.; Parigi, G.B.; Riccipetitoni, G. Meta-Analysis on Long-Term Outcomes of Pediatric Renal Cancer Survivors Following COG and SIOP Protocols. Eur. J. Pediatr. Surg. 2023, 33, 17–25. [Google Scholar] [CrossRef]
- Mahamdallie, S.; Yost, S.; Poyastro-Pearson, E.; Holt, E.; Zachariou, A.; Seal, S.; Elliott, A.; Clarke, M.; Warren-Perry, M.; Hanks, S.; et al. Identification of new Wilms tumour predisposition genes: An exome sequencing study. Lancet Child. Adolesc. Health 2019, 3, 322–331. [Google Scholar] [CrossRef]
- Liu, P.; Zhuo, Z.; Li, W.; Cheng, J.; Zhou, H.; He, J.; Zhang, J.; Wang, J. TP53 rs1042522 C>G polymorphism and Wilms tumor susceptibility in Chinese children: A four-center case-control study. Biosci. Rep. 2019, 39, BSR20181891. [Google Scholar] [CrossRef]
- Liu, G.C.; Zhuo, Z.J.; Zhu, S.B.; Zhu, J.; Jia, W.; Zhao, Z.; Hu, J.H.; He, J.; Wang, F.H.; Fu, W. Associations between LMO1 gene polymorphisms and Wilms’ tumor susceptibility. Oncotarget 2017, 8, 50665–50672. [Google Scholar] [CrossRef]
- Zhu, J.; Jia, W.; Wu, C.; Fu, W.; Xia, H.; Liu, G.; He, J. Base Excision Repair Gene Polymorphisms and Wilms Tumor Susceptibility. EBioMedicine 2018, 33, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Chu, A.; Heck, J.E.; Ribeiro, K.B.; Brennan, P.; Boffetta, P.; Buffler, P.; Hung, R.J. Wilms’ tumour: A systematic review of risk factors and meta-analysis. Paediatr. Perinat. Epidemiol. 2010, 24, 449–469. [Google Scholar] [CrossRef]
- Politis, G.; Wagenpfeil, S.; Welter, N.; Mergen, M.; Furtwängler, R.; Graf, N. An Observational Case-Control Study on Parental Age and Childhood Renal Tumors. Cancers 2023, 15, 5144. [Google Scholar] [CrossRef]
- Contreras, Z.A.; Hansen, J.; Ritz, B.; Olsen, J.; Yu, F.; Heck, J.E. Parental age and childhood cancer risk: A Danish population-based registry study. Cancer Epidemiol. 2017, 49, 202–215. [Google Scholar] [CrossRef]
- Ayaz, E.; Ozcan, H.N.; Oguz, B.; Haliloglu, M. Beyond Wilms tumor: Imaging findings and outcomes of rare renal tumors in children. Pediatr. Radiol. 2022, 52, 2557–2567. [Google Scholar] [CrossRef] [PubMed]
- Ota, Y.; Naiki, T.; Nakagawa, M.; Naiki-Ito, A.; Hamamoto, S.; Etani, T.; Iida, K.; Nozaki, S.; Ando, R.; Kawai, N.; et al. Laparoscopic radical surgery produces a good prognosis in an elderly patient with small Wilms’ tumor. IJU Case Rep. 2019, 2, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Jurić, I.; Pogorelić, Z.; Kuzmić-Prusac, I.; Biocić, M.; Jakovljević, G.; Stepan, J.; Zupancić, B.; Culić, S.; Kruslin, B. Expression and prognostic value of the Ki-67 in Wilms’ tumor: Experience with 48 cases. Pediatr. Surg. Int. 2010, 26, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Zhang, J.; Gao, X.; Tang, X.; Cui, Y.; Li, D.; Jia, W. Silencing of long noncoding RNA MYLK-AS1 suppresses nephroblastoma via down-regulation of CCNE1 through transcription factor TCF7L2. J. Cell Physiol. 2021, 236, 5757–5770. [Google Scholar] [CrossRef]
- Taros, T.; Chabot, M.; Sokoloff, M.; Wollin, M. Report of a rare testicular teratoid wilms tumor in an adult patient. Urol. Case Rep. 2022, 40, 101894. [Google Scholar] [CrossRef]
- Liu, H.; Tang, C.; Yang, Y. Identification of Nephrogenic Therapeutic Biomarkers of Wilms Tumor Using Machine Learning. J. Oncol. 2021, 2021, 6471169. [Google Scholar] [CrossRef]
- Perrotta, G.; Castellani, D. Wilms Tumor: Updates about Pathogenesis and New Possible Clinical Treatments of the Most Frequent Pediatric Urogenital Cancer: A Narrative Review. Surgeries 2023, 4, 678–697. [Google Scholar] [CrossRef]
- Stanhope-Baker, P.; Kessler, P.M.; Li, W.; Agarwal, M.L.; Williams, B.R. The Wilms tumor suppressor-1 target gene podocalyxin is transcriptionally repressed by p53. J. Biol. Chem. 2004, 279, 33575–33585. [Google Scholar] [CrossRef]
- Liu, K.; He, B.; Xu, J.; Li, Y.; Guo, C.; Cai, Q.; Wang, S. miR-483-5p Targets MKNK1 to Suppress Wilms’ Tumor Cell Proliferation and Apoptosis In Vitro and In Vivo. Med. Sci. Monit. 2019, 25, 1459–1468. [Google Scholar] [CrossRef]
- Royer-Pokora, B.; Wruck, W.; Adjaye, J.; Beier, M. Gene expression studies of WT1 mutant Wilms tumor cell lines in the frame work of published kidney development data reveals their early kidney stem cell origin. PLoS ONE 2023, 18, e0270380. [Google Scholar] [CrossRef]
- Ehrlich, D.; Bruder, E.; Thome, M.A.; Gutt, C.N.; von Knebel Doeberitz, M.; Niggli, F.; Perantoni, A.O.; Koesters, R. Nuclear accumulation of beta-catenin protein indicates activation of wnt signaling in chemically induced rat nephroblastomas. Pediatr. Dev. Pathol. 2010, 13, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Li, Y.; Wu, J.; Zhang, B.; Xie, S.; Zheng, X.; Jiang, Z. An m6A/m5C/m1A/m7G-Related Long Non-coding RNA Signature to Predict Prognosis and Immune Features of Glioma. Front. Genet. 2022, 13, 903117. [Google Scholar] [CrossRef]
- Luo, B.; Feng, S.; Li, T.; Wang, J.; Qi, Z.; Zhao, Y.; Hu, B. Transcription factor HOXB2 upregulates NUSAP1 to promote the proliferation, invasion and migration of nephroblastoma cells via the PI3K/Akt signaling pathway. Mol. Med. Rep. 2022, 25, 205. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Liu, Q. FOXO3a inhibits nephroblastoma cell proliferation, migration and invasion, and induces apoptosis through downregulating the Wnt/β-catenin signaling pathway. Mol. Med. Rep. 2021, 24, 796. [Google Scholar] [CrossRef] [PubMed]
- Geng, G.; Li, Q.; Guo, X.; Ni, Q.; Xu, Y.; Ma, Z.; Wang, Y.; Ming, M. FOXO3a-modulated DEPDC1 promotes malignant progression of nephroblastoma via the Wnt/β-catenin signaling pathway. Mol. Med. Rep. 2022, 26, 272. [Google Scholar] [CrossRef]
- Che, G.; Gao, H.; Tian, J.; Hu, Q.; Xie, H.; Zhang, Y. MicroRNA-483-3p Promotes Proliferation, Migration, and Invasion and Induces Chemoresistance of Wilms’ Tumor Cells. Pediatr. Dev. Pathol. 2020, 23, 144–151. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Shiina, M.; Dasgupta, P.; Kulkarni, P.; Kato, T.; Wong, R.K.; Tanaka, Y.; Shahryari, V.; Maekawa, S.; Yamamura, S.; et al. Upregulation of miR-130b Contributes to Risk of Poor Prognosis and Racial Disparity in African-American Prostate Cancer. Cancer Prev. Res. 2019, 12, 585–598. [Google Scholar] [CrossRef]
- Ludwig, N.; Werner, T.V.; Backes, C.; Trampert, P.; Gessler, M.; Keller, A.; Lenhof, H.P.; Graf, N.; Meese, E. Combining miRNA and mRNA Expression Profiles in Wilms Tumor Subtypes. Int. J. Mol. Sci. 2016, 17, 475. [Google Scholar] [CrossRef]
- Zheng, H.; Liu, J.; Pan, X.; Cui, X. Biomarkers for patients with Wilms tumor: A review. Front. Oncol. 2023, 13, 1137346. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, J.; Dong, R.; Yang, S.; Zheng, S. Identification of novel serum biomarkers in child nephroblastoma using proteomics technology. Mol. Biol. Rep. 2011, 38, 631–638. [Google Scholar] [CrossRef]
- Percicote, A.; Leme, F.; Almeida, T.; Freitas, A.; Gugelmin, E.; Noronha, L. Immunohistochemical expression of p53, bcl-2, bax and vegfr1 proteins in nephroblastomas. J. Bras. De Patol. E Med. Lab. 2013, 49, 50–56. [Google Scholar] [CrossRef]
- Chan, E.S.; Pawel, B.R.; Corao, D.A.; Venneti, S.; Russo, P.; Santi, M.; Sullivan, L.M. Immunohistochemical expression of glypican-3 in pediatric tumors: An analysis of 414 cases. Pediatr. Dev. Pathol. 2013, 16, 272–277. [Google Scholar] [CrossRef]
- Walz, A.L.; Maschietto, M.; Crompton, B.; Evageliou, N.; Dix, D.; Tytgat, G.; Gessler, M.; Gisselsson, D.; Daw, N.C.; Wegert, J. Tumor biology, biomarkers, and liquid biopsy in pediatric renal tumors. Pediatr. Blood Cancer 2023, 70 (Suppl. S2), e30130. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Han, L.; Shahen, M.; Hu, C.; Li, F. Multi-Omics Integration Reveals a Competitive Endogenous RNAs Network for the Identification of Progression Biomarkers and the Stratification of Patients Diagnosed With Nephroblastoma. Front. Oncol. 2020, 10, 444. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.; Keller, A.; Nourkami-Tutdibi, N.; Heisel, S.; Habel, N.; Leidinger, P.; Ludwig, N.; Gessler, M.; Graf, N.; Berthold, F.; et al. Autoantibody signature differentiates Wilms tumor patients from neuroblastoma patients. PLoS ONE 2011, 6, e28951. [Google Scholar] [CrossRef]
- El-Hawary, A.; Zalata, K.; El-Baz, M.; Refaei, H.; Enein, H. Adult teratoid Wilms tumor: A case report of rare variant with predominant neuroepithelium. Case Rep. Clin. Pathol. 2014, 1, 42–46. [Google Scholar] [CrossRef]
- Khanna, G.; Rosen, N.; Anderson, J.R.; Ehrlich, P.F.; Dome, J.S.; Gow, K.W.; Perlman, E.; Barnhart, D.; Karolczuk, K.; Grundy, P. Evaluation of diagnostic performance of CT for detection of tumor thrombus in children with Wilms tumor: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2012, 58, 551–555. [Google Scholar] [CrossRef]
- Meni, D. Wilms tumor and associated predisposing syndromes and conditions. JAAPA 2025, 38, 27–33. [Google Scholar] [CrossRef]
- Bach, A.; Mi, J.; Hunter, M.; Halliday, B.J.; García-Miñaúr, S.; Sperotto, F.; Trevisson, E.; Markie, D.; Morison, I.M.; Shinawi, M.; et al. Wilms tumor in patients with osteopathia striata with cranial sclerosis. Eur. J. Hum. Genet. 2021, 29, 396–401. [Google Scholar] [CrossRef]
- Gariépy-Assal, L.; Gilbert, R.D.; Žiaugra, A.; Foster, B.J. Management of Denys-Drash syndrome: A case series based on an international survey. Clin. Nephrol. Case Stud. 2018, 6, 36–44. [Google Scholar] [CrossRef]
- Demirel, G.; Oguz, S.S.; Celik, I.H.; Uras, N.; Erdeve, O.; Dilmen, U. Rare clinical entity Perlman syndrome: Is cholestasis a new finding? Congenit. Anom. 2011, 51, 43–45. [Google Scholar] [CrossRef]
- Diets, I.J.; Hoyer, J.; Ekici, A.B.; Popp, B.; Hoogerbrugge, N.; van Reijmersdal, S.V.; Bhaskaran, R.; Hadjihannas, M.; Vasileiou, G.; Thiel, C.T.; et al. TRIM28 haploinsufficiency predisposes to Wilms tumor. Int. J. Cancer 2019, 145, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Zhuo, Z.; Hua, R.X.; Fu, K.; Jia, W.; Zhu, J.; Zhang, J.; Cheng, J.; Zhou, H.; Xia, H.; et al. Association of KRAS and NRAS gene polymorphisms with Wilms tumor risk: A four-center case-control study. Aging 2019, 11, 1551–1563. [Google Scholar] [CrossRef] [PubMed]
- Avellano, P.; Abellera, J.M.; Alegarbes, R.; Isabedra, N. Management of Wilms’ Tumor in a Horseshoe Kidney at the Time of COVID-19 Pandemic: A Case Report. Philipp. J. Surg. Spec. 2021, 76, 73–78. [Google Scholar] [CrossRef]
- Holmes, D.M.; Matatiyo, A.; Mpasa, A.; Huibers, M.H.W.; Manda, G.; Tomoka, T.; Mulenga, M.; Namazzi, R.; Mehta, P.; Zobeck, M.; et al. Outcomes of Wilms tumor therapy in Lilongwe, Malawi, 2016–2021: Successes and ongoing research priorities. Pediatr. Blood Cancer 2023, 70, e30242. [Google Scholar] [CrossRef]
- Palmisani, F.; Kovar, H.; Kager, L.; Amann, G.; Metzelder, M.; Bergmann, M. Systematic review of the immunological landscape of Wilms tumors. Mol. Ther. Oncolytics 2021, 22, 454–467. [Google Scholar] [CrossRef]
- Abdelhafeez, A.H.; Reljic, T.; Kumar, A.; Banu, T.; Cox, S.; Davidoff, A.M.; Elgendy, A.; Ghandour, K.; Gerstle, J.T.; Karpelowsky, J.; et al. Evidence-based surgical guidelines for treating children with Wilms tumor in low-resource settings. Pediatr. Blood Cancer 2022, 69, e29906. [Google Scholar] [CrossRef] [PubMed]
- Huszno, J.; Starzyczny–Słota, D.; Jaworska, M.; Nowara, E. Adult Wilms’ Tumor—Diagnosis and Current Therapy. Cent. Eur. J. Urol. 2013, 66, 39. [Google Scholar] [CrossRef]
- Luo, J.-Y.; Yan, S.-B.; Chen, G.; Chen, P.; Liang, S.-W.; Xu, Q.-Q.; Gu, J.-H.; Huang, Z.-G.; Qin, L.-T.; Lu, H.-P.; et al. RNA-Sequencing, Connectivity Mapping, and Molecular Docking to Investigate Ligand-Protein Binding for Potential Drug Candidates for the Treatment of Wilms Tumor. Med. Sci. Monit. 2020, 26, e920725. [Google Scholar] [CrossRef]
- Lee, S.H.; Bae, M.H.; Choi, S.H.; Lee, J.S.; Cho, Y.S.; Joo, K.J.; Kwon, C.H.; Park, H.J. Wilms’ Tumor in a Horseshoe Kidney. Korean J. Urol. 2012, 53, 577. [Google Scholar] [CrossRef]
- Zhao, Q.; Xiong, Q.; Song, Q. Metastatic Adult Wilms’ Tumor Managed by Chemotherapy, Immunotherapy and Targeted Therapy: A Case Report. Future Sci. OA 2024, 10, FSO915. [Google Scholar] [CrossRef] [PubMed]
- Venkatramani, R.; Malogolowkin, M.H.; Mascarenhas, L. Treatment of Multiply Relapsed Wilms Tumor with Vincristine, Irinotecan, Temozolomide and Bevacizumab. Pediatr. Blood Cancer 2014, 61, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guo, F.; Wang, L.; Zhao, W.; Zhang, D.; Yang, H.; Yu, J.; Niu, L.; Yang, F.; Zheng, S.; et al. Identification of Apolipoprotein C-I as a Potential Wilms’ Tumor Marker After Excluding Inflammatory Factors. Int. J. Mol. Sci. 2014, 15, 16186–16195. [Google Scholar] [CrossRef]
- Chalfant, V.; Riveros, C.; Stec, A.A. Effect of social disparities on 10 year survival in pediatric patients with Wilms’ tumor. Cancer Med. 2023, 12, 3452–3459. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lai, Y.; Zhu, L.; Lu, Z.; Hu, C.; Zhou, H.; Lu, Z.; Tang, Z.; He, Z.; Tang, F. A Novel Inflammation-Related Gene Signature for Overall Survival Prediction and Comprehensive Analysis in Pediatric Patients with Wilms Tumor. Dis. Markers 2022, 2022, 2651105. [Google Scholar] [CrossRef]
- Dome, J.S.; Fernandez, C.V.; Mullen, E.A.; Kalapurakal, J.A.; Geller, J.I.; Huff, V.; Gratias, E.J.; Dix, D.B.; Ehrlich, P.F.; Khanna, G.; et al. COG Renal Tumors Committee. Children’s Oncology Group’s 2013 blueprint for research: Renal tumors. Pediatr. Blood Cancer 2013, 60, 994–1000. [Google Scholar] [CrossRef]
- Dix, D.B.; Seibel, N.L.; Chi, Y.Y.; Khanna, G.; Gratias, E.; Anderson, J.R.; Mullen, E.A.; Geller, J.I.; Kalapurakal, J.A.; Paulino, A.C.; et al. Treatment of Stage IV Favorable Histology Wilms Tumor With Lung Metastases: A Report From the Children’s Oncology Group AREN0533 Study. J. Clin. Oncol. 2018, 36, 1564–1570. [Google Scholar] [CrossRef]
- Brok, J.; Treger, T.D.; Gooskens, S.L.; van den Heuvel-Eibrink, M.M.; Pritchard-Jones, K. Biology and treatment of renal tumours in childhood. Eur. J. Cancer 2016, 68, 179–195. [Google Scholar] [CrossRef]
- Aldrink, J.H.; Heaton, T.E.; Dasgupta, R.; Lautz, T.B.; Malek, M.M.; Abdessalam, S.F.; Weil, B.R.; Rhee, D.S.; Baertschiger, R.; Ehrlich, P.F.; et al. American Pediatric Surgical Association Cancer Committee. Update on Wilms tumor. J. Pediatr. Surg. 2019, 54, 390–397. [Google Scholar] [CrossRef]
- Huang, X.; Zhao, J.; Fu, W.; Zhu, J.; Lou, S.; Tian, X.; Chen, S.; Ruan, J.; He, J.; Zhou, H. The association of RAN and RANBP2 gene polymerphisms with Wilms tumor risk in Chinese children. J. Cancer 2020, 11, 804–809. [Google Scholar] [CrossRef]
- Woods, A.D.; Berlow, N.E.; Ortiz, M.V.; Dela Cruz, F.; Siddiquee, A.; Coutinho, D.F.; Purohit, R.; Freier, K.E.T.; Michalek, J.E.; Lathara, M.; et al. Bromodomain 4 inhibition leads to MYCN downregulation in Wilms tumor. Pediatr. Blood Cancer 2022, 69, e29401. [Google Scholar] [CrossRef] [PubMed]
- Njuguna, F.; Martijn, H.A.; Kuremu, R.T.; Saula, P.; Kirtika, P.; Olbara, G.; Langat, S.; Martin, S.; Skiles, J.; Vik, T.; et al. Wilms Tumor Treatment Outcomes: Perspectives From a Low-Income Setting. J. Glob. Oncol. 2016, 3, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Israels, T.; Borgstein, E.; Pidini, D.; Chagaluka, G.; de Kraker, J.; Kamiza, S.; Molyneux, E.M. Management of children with a Wilms tumor in Malawi, sub-Saharan Africa. J. Pediatr. Hematol. Oncol. 2012, 34, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Al-Hadidi, A.; Rinehardt, H.N.; Sutthatarn, P.; Talbot, L.J.; Murphy, A.J.; Whitlock, R.; Condon, S.; Naik-Mathuria, B.; Utria, A.F.; Rothstein, D.H.; et al. Incidence and management of pleural effusions in patients with Wilms tumor: A Pediatric Surgical Oncology Research Collaborative study. Int. J. Cancer 2022, 151, 1696–1702. [Google Scholar] [CrossRef]
- Dome, J.S.; Graf, N.; Geller, J.I.; Fernandez, C.V.; Mullen, E.A.; Spreafico, F.; Van den Heuvel-Eibrink, M.; Pritchard-Jones, K. Advances in Wilms Tumor Treatment and Biology: Progress Through International Collaboration. J. Clin. Oncol. 2015, 33, 2999–3007. [Google Scholar] [CrossRef]
- Chen, X.; Bao, Y.; Sun, G.; Wang, X.; Zhu, J. UNC13B Regulates the Sensitivity of Wilms’ Tumor Cells to Doxorubicin by Modulating Lysosomes. Oncol. Lett. 2024, 28, 446. [Google Scholar] [CrossRef]
- Zhang, J.; Quan, Y.; Su, X.; Qiu, B.; Dong, Q. Circ-PRMT5 Stimulates the Proliferative Ability in Wilms’ Tumor Through the miR-7-5p/KLF4 Axis. Cell. Mol. Biol. 2023, 69, 232–236. [Google Scholar] [CrossRef]
- Sun, F.; Li, W.; Wang, L.; Jiao, C. Expression of Phosphatase of Regenerating Liver-3 Is Associated with Prognosis of Wilms’ Tumor. OncoTargets Ther. 2017, 10, 311–317. [Google Scholar] [CrossRef]
- Li, W.; Li, L.; Xi-Qiang, D.; Liu, J.; Tan, X.; Huang, Q.; Li, H. MicroRNA-215-5p Inhibits the Proliferation and Migration of Wilms’ Tumor Cells by Targeting CRK. Technol. Cancer Res. Treat. 2021, 20, 15330338211036523. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, Y. LncRNA LINC01339 Hinders the Development of Wilms’ Tumor via the miR-135b-3p/ADH1C Axis. Horm. Metab. Res. 2023, 56, 244–254. [Google Scholar] [CrossRef]
- Kapur, P.; Rakheja, D. Immunohistochemical Expression of Neural Cell Adhesion Molecule in Wilms Tumors, Nephrogenic Rests, and Fetal and Postnatal Renal Cortices. Pediatr. Dev. Pathol. 2011, 14, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Kim, S.; Seong, S.; Cho, W.; Yu, H. Stage IV Wilms Tumor Treated by Korean Medicine, Hyperthermia, and Thymosin-α1: A Case Report. Case Rep. Oncol. 2016, 9, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Wang, W.; Hou, T.; Kuo, C.; Lai, Y. Identification of Molecular Biomarkers in Taiwanese Patients with Wilms Tumor Using a Methylation-Specific Multiplex Ligation-Dependent Probe Amplification (MS-MLPA)-Based Approach. Preprints 2020. [Google Scholar] [CrossRef]
- Hont, A.B.; Cruz, C.R.; Ulrey, R.; O’brien, B.; Stanojevic, M.; Datar, A.; Albihani, S.; Saunders, D.; Hanajiri, R.; Panchapakesan, K.; et al. Immunotherapy of Relapsed and Refractory Solid Tumors with ex vivo Expanded Multi-Tumor Associated Antigen Specific Cytotoxic T Lymphocytes: A Phase I Study. J. Clin. Oncol. 2019, 37, 2349–2359. [Google Scholar] [CrossRef]
- Treetipsatit, J.; Raveesunthornkiet, M.; Ruangtrakool, R.; Sanpakit, K.; Thorner, P. Teratoid Wilms’ Tumor: Case Report of a Rare Variant That Can Mimic Aggressive Biology During Chemotherapy. J. Pediatr. Surg. 2011, 46, e1–e6. [Google Scholar] [CrossRef]

{kind=link}
| Classification Category | Subtype/Feature | Description | Clinical Significance/ Prognostic Value |
|---|---|---|---|
| Histological Subtypes | Favorable Histology (FH) | Characterized by well-differentiated blastemal, epithelial, and stromal components without anaplasia. | Generally associated with a good prognosis. |
| Anaplastic Histology (AH) | Presence of large, hyperchromatic nuclei with multipolar mitoses. It can be focal (localized) or diffuse (widespread). | Associated with a less favorable prognosis, especially in diffuse anaplasia. Requires more aggressive treatment. | |
| Clear Cell Sarcoma of Kidney (CCSK) | A distinct histological subtype characterized by cells with clear cytoplasm and a prominent vascular network. | Generally more aggressive than an FH Wilms tumor. Requires specific treatment protocols, including anthracycline-based chemotherapy. | |
| Rhabdoid Tumor of Kidney (RTK) | Highly aggressive tumors are composed of cells with eccentric nuclei, prominent nucleoli, and eosinophilic cytoplasm, and they often display a loss of INI1/SMARCB1 expression. | Very poor prognosis. Often presents at advanced stages and is associated with central nervous system involvement. Requires intensive multi-modal therapy. | |
| Congenital Mesoblastic Nephroma (CMN) | Typically diagnosed in infants < 1 year. Usually presents as a large, solid renal mass. Characterized by spindle cells. | Usually favorable prognosis, especially when completely resected. Cellular variants may be more aggressive. | |
| Wilms Tumor with Regressive Features | Tumors showing regressive changes like necrosis, hyalinization in the stroma, and/or cystic changes. | Associated with good prognosis. | |
| Genetic Mutations | WT1 mutations | Mutations in the WT1 gene (Wilms Tumor 1) on chromosome 11p13. | Associated with certain syndromes (e.g., WAGR, Denys-Drash, Frasier) and increased risk of Wilms tumor. It can influence treatment response. |
| CTNNB1 mutations | Mutations in the CTNNB1 gene (beta-catenin) are found in a subset of Wilms tumors, particularly those with epithelial predominance. | It may be associated with specific histological features and potentially influence prognosis. | |
| TP53 mutations | Mutations in the TP53 gene (tumor protein p53) are often associated with anaplastic histology. | Generally associated with a poorer prognosis and increased risk of relapse. | |
| MYCN amplification | Amplification of the MYCN oncogene. | Associated with aggressive tumor behavior and poorer outcomes. More commonly seen in relapsed Wilms tumors. | |
| SIX2 and SIX1 | Overexpression of SIX2 and SIX1 genes. | Involved in early kidney development. Overexpression leads to increased blastemal proliferation. | |
| Loss of heterozygosity (LOH) at 1p and 16q | Loss of genetic material on chromosomes 1p and 16q. | Associated with increased risk of relapse or adverse outcomes. | |
| Molecular Features | Loss of Imprinting (LOI) at 11p15 | Loss of imprinting at the 11p15 region affects genes like IGF2 and H19. | Common in Wilms tumors and can contribute to tumor development. |
| MicroRNA (miRNA) expression profiles | Specific miRNA expression patterns can differentiate between FH and AH tumors and predict prognosis. | Potential biomarkers for risk stratification and targeted therapy. | |
| Aberrant Wnt signaling | Activation of the Wnt signaling pathway, often through CTNNB1 mutations. | Drives cell proliferation and survival. Potential target for therapeutic intervention. |
| Stage | Description | Key Features | Approximate 5-Year Overall Survival |
|---|---|---|---|
| I | The tumor is limited to the kidney and completely resected. The tumor is confined within the renal capsule and has not spread to surrounding tissues or lymph nodes. The renal capsule is intact, and there is no rupture of the tumor before or during surgery. | Complete surgical resection; tumor confined to the kidney; no involvement of renal sinus vessels; no spillage of tumor during surgery. | >95% |
| II | The tumor extends beyond the kidney but is completely resected. The tumor has grown beyond the kidney but has been completely removed surgically. This may include penetration through the renal capsule into the perirenal tissues or involvement of the renal sinus blood vessels. There is no residual tumor left behind after surgery. | The tumor extends beyond the kidney but is completely resected; penetration of the renal capsule involvement of the renal sinus vessels; no residual tumor after surgery. Lymph nodes may or may not be involved, but if they are, the surgeon must completely remove them. | >90% |
| III | The tumor extends beyond the kidney and is not completely resected. The tumor has spread beyond the kidney and cannot be completely removed surgically. This may be due to tumor spillage during surgery, the involvement of regional lymph nodes that cannot be completely resected, or tumor invasion into adjacent organs or structures. | Incomplete surgical resection; tumor spillage during surgery; involvement of regional lymph nodes that cannot be completely resected; tumor invasion into adjacent organs (e.g., bowel, muscles). | 80–90% |
| IV | Distant metastases are present. The tumor has spread to distant organs, such as the lungs, liver, bone, or brain. | Distant metastases to the lungs, liver, bone, brain, or other organs. | 70–80% |
| V | Bilateral Wilms’ tumor: the tumor is present in both kidneys at the time of diagnosis. Staging is assigned to each kidney individually after biopsy and/or resection. | Tumors are present in both kidneys. Initial therapy often involves chemotherapy to shrink the tumors, followed by bilateral partial nephrectomies to preserve renal function. | >70% (dependent on individual staging of each kidney) |
| NWTSG | SIOP | PPGGL | |
|---|---|---|---|
| 1. Initial Treatment Approach | Recommends initial surgical resection of the primary tumor before chemotherapy. This approach is aimed at effective local control and reducing the tumor burden before systemic treatment. | Advocates for administering chemotherapy for 4 weeks before surgery, called preoperative chemotherapy. This strategy is intended to shrink the tumor and reduce the risk of tumor rupture during surgery. | Aligns more closely with SIOP protocols, generally favoring preoperative chemotherapy for similar reasons, while adapting treatment based on individual patient circumstances and outcomes. |
| 2. Chemotherapy Regimens | Employs multi-agent chemotherapy consisting of drugs like Actinomycin D (ACT), Vincristine (VCR), and Doxorubicin (ADM). The duration and intensity of treatment depend on the stage of the disease, typically involving a longer regimen for higher-stage tumors. | Utilizes a similar array of chemotherapeutics as NWTSG but allows for some variation in treatment duration and dosing. For lower-stage tumors, less aggressive combinations may be used, whereas higher stages receive more intensive regimens. The SIOP protocols also dictate the timing of postoperative chemotherapy based on initial staging and response. | Treatment regimens are tailored to patient needs, often referencing SIOP guidelines. The chemotherapeutic agents and their dosages are adapted according to tumor histology and response to initial treatment, keeping in mind the higher toxicity levels seen in adult patients compared to children. |
| 3. Radiotherapy Recommendations | Radiotherapy is primarily recommended for advanced stages (III and IV) or for cases with unfavorable histology. Standard doses range from 10 to 20 grays (Gy) depending on the tumor stage and characteristics. | Similar to NWTSG, SIOP recommends radiotherapy mainly for advanced cases and some earlier stages, depending on the tumor’s histological features. The dosage is adjustable, with frequent doses seen between 10 and 15 Gy or more for specific cases. | Follows principles similar to SIOP but may have specific adaptations based on the outcomes of the Polish cohort. The use of radiotherapy is selectively based on individual patient circumstances, often after the assessment of chemotherapy responses. |
| 4. Prognostic Factors and Follow-Up | Uses a detailed classification system based on histological features for risk stratification, impacting treatment decisions and follow-up protocols. | Additionally, it emphasizes prognostic factors like response to preoperative chemotherapy, influencing not just treatment plans but also ongoing monitoring and potential adjustments. | Establishes follow-up protocols that consider both clinical and biological prognostic factors, focusing on long-term outcomes and quality of life for adult patients transitioning from pediatric protocols. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Lithuanian University of Health Sciences. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neagu, M.C.; David, V.L.; Iacob, E.R.; Chiriac, S.D.; Muntean, F.L.; Boia, E.S. Wilms’ Tumor: A Review of Clinical Characteristics, Treatment Advances, and Research Opportunities. Medicina 2025, 61, 491. https://doi.org/10.3390/medicina61030491
Neagu MC, David VL, Iacob ER, Chiriac SD, Muntean FL, Boia ES. Wilms’ Tumor: A Review of Clinical Characteristics, Treatment Advances, and Research Opportunities. Medicina. 2025; 61(3):491. https://doi.org/10.3390/medicina61030491
Chicago/Turabian StyleNeagu, Mihai Cristian, Vlad Laurenţiu David, Emil Radu Iacob, Sorin Dan Chiriac, Florin Lucian Muntean, and Eugen Sorin Boia. 2025. "Wilms’ Tumor: A Review of Clinical Characteristics, Treatment Advances, and Research Opportunities" Medicina 61, no. 3: 491. https://doi.org/10.3390/medicina61030491
APA StyleNeagu, M. C., David, V. L., Iacob, E. R., Chiriac, S. D., Muntean, F. L., & Boia, E. S. (2025). Wilms’ Tumor: A Review of Clinical Characteristics, Treatment Advances, and Research Opportunities. Medicina, 61(3), 491. https://doi.org/10.3390/medicina61030491