A Multi-Omics Characterization of the Natural Product Potential of Tropical Filamentous Marine Cyanobacteria

 , , ,
, , ,  ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Results
2.1. Diverse Sampling of Environmental Cyanobacteria
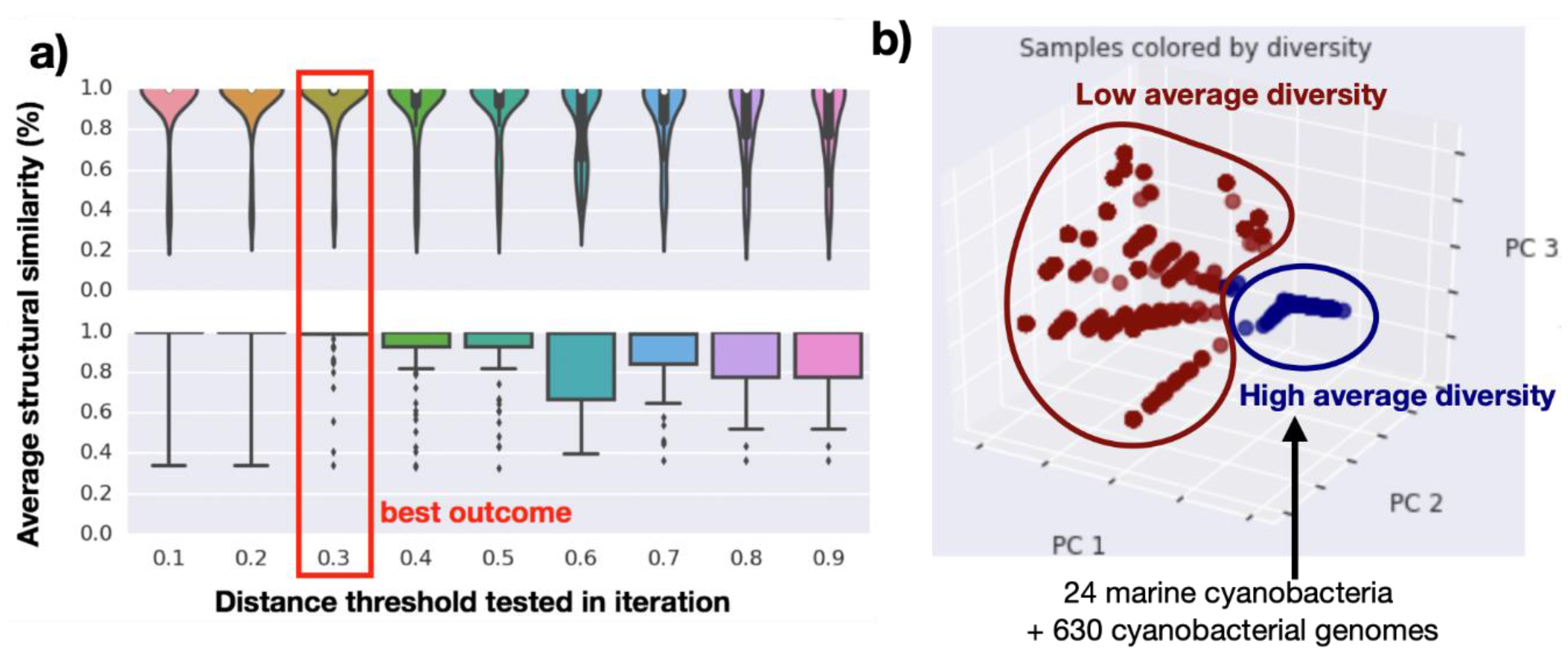
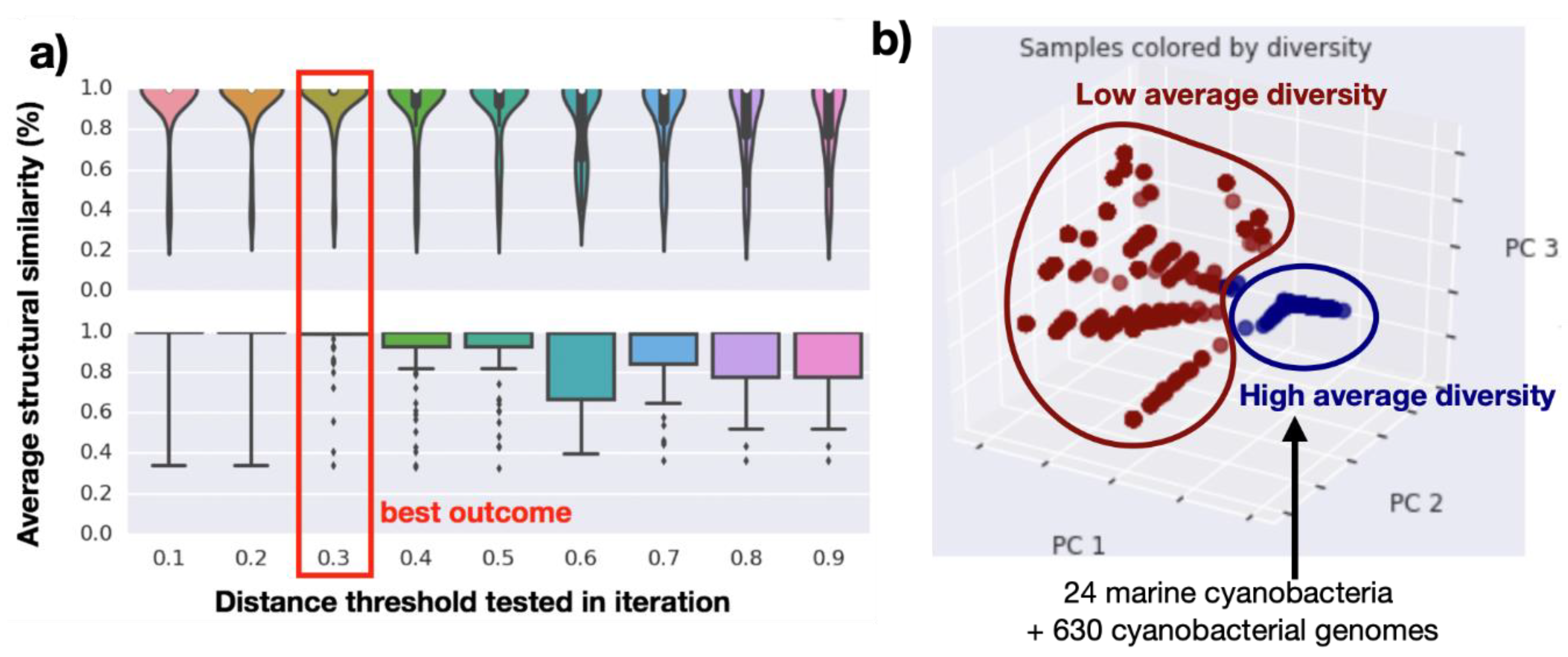
2.2. Genomics Assembly Pipeline and Phylogenomics
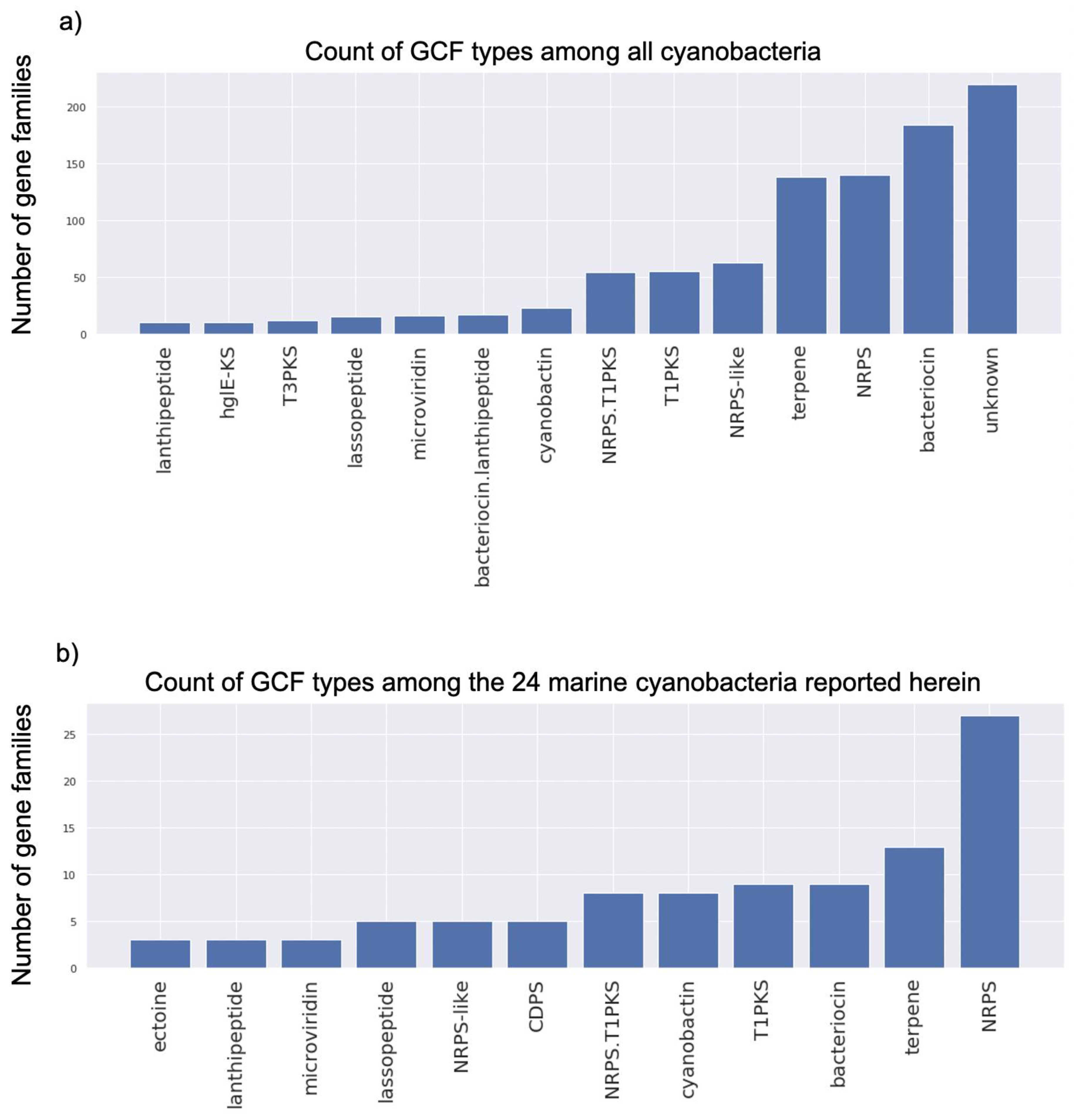
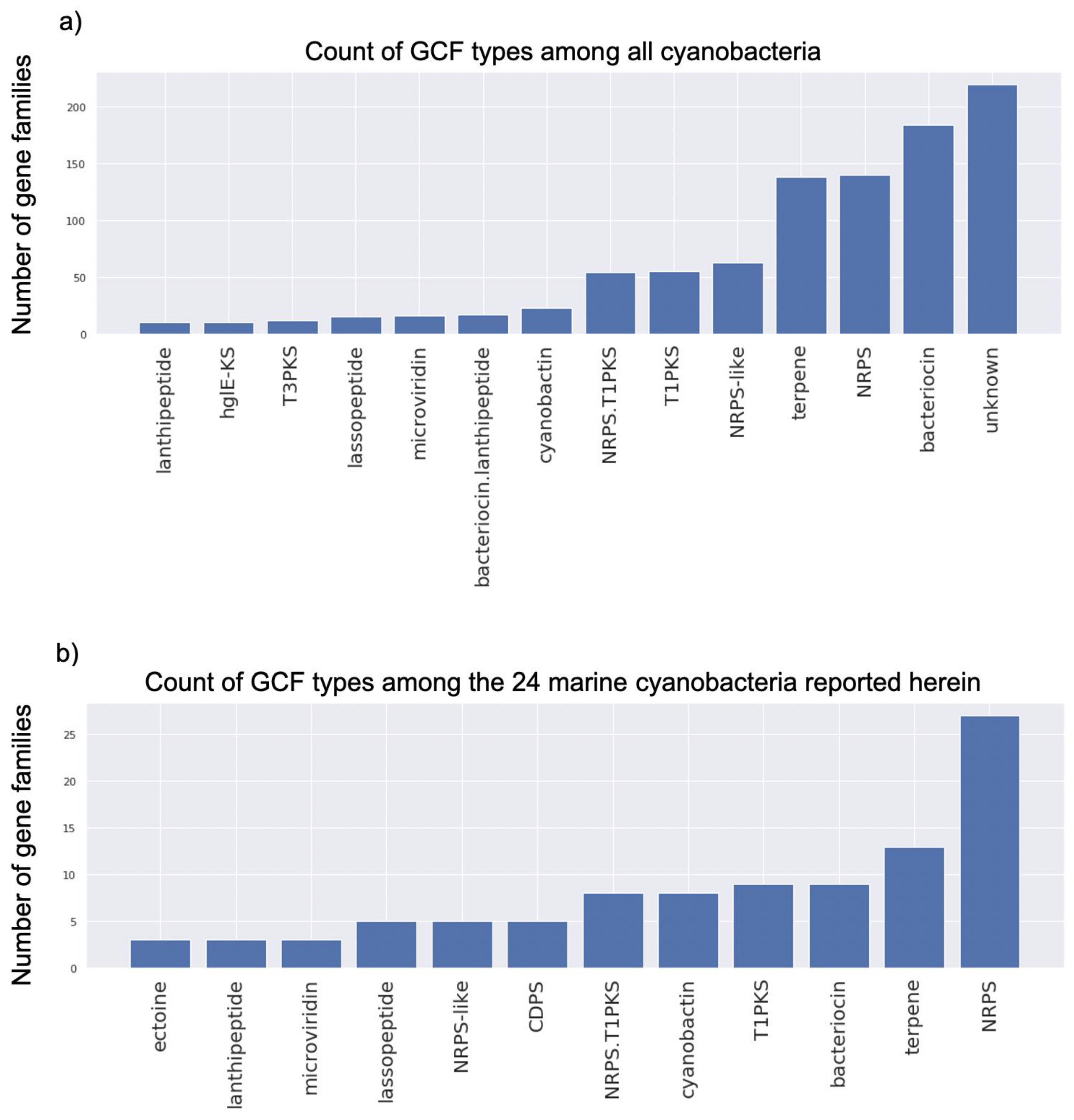
2.3. Genome Mining Pipeline
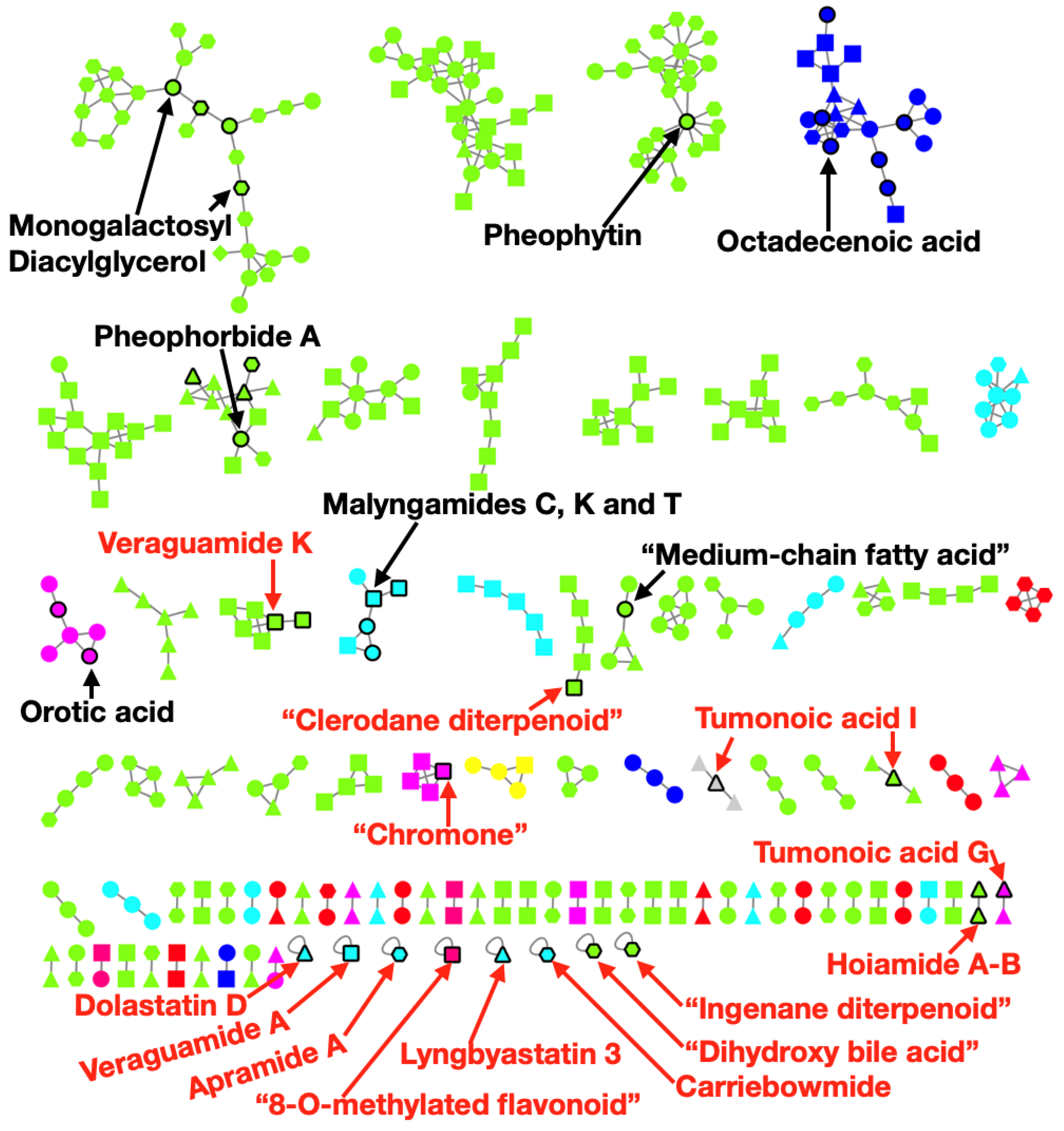
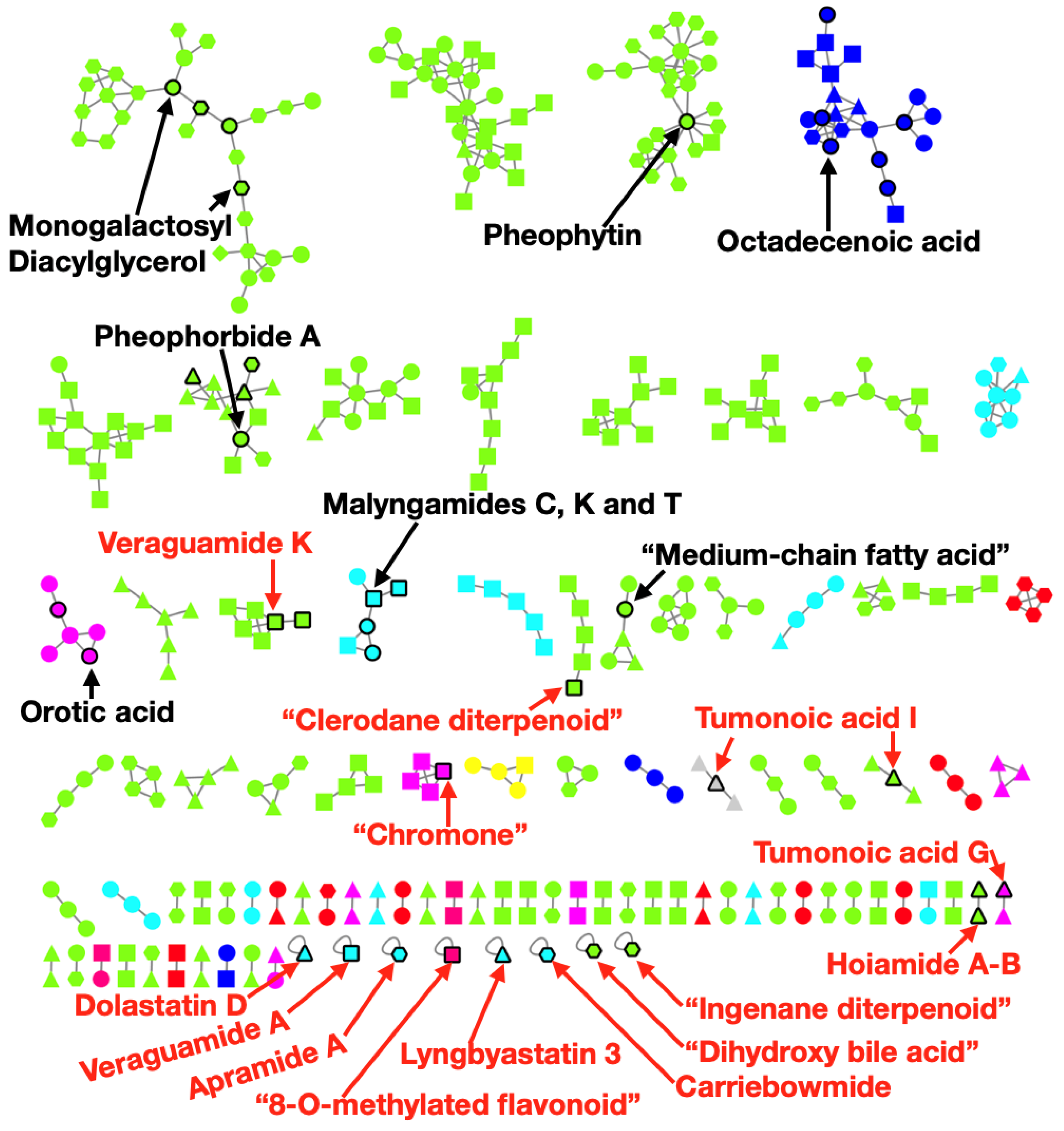
2.4. Metabolomics Pipeline
3. Discussion
4. Materials and Methods
4.1. Collection, DNA Extraction and Sequencing
4.2. Genome Assembly Pipeline
4.3. Phylogenomics
4.4. Gene Cluster Networking and Diversity Analysis
4.5. Extraction and UHPLC-MS/MS Analysis
4.6. GNPS Molecular Networking
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blin, K.; Kim, H.U.; Medema, M.H.; Weber, T. Recent development of antiSMASH and other computational approaches to mine secondary metabolite biosynthetic gene clusters. Brief. Bioinform. 2017, 1, 11. [Google Scholar] [CrossRef] [PubMed]
- Pye, C.R.; Bertin, M.J.; Lokey, R.S.; Gerwick, W.H.; Linington, R.G. Retrospective analysis of natural products provides insights for future discovery trends. Proc. Natl. Acad. Sci. USA 2017, 114, 5601–5606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komarek, J.; Kastovsky, J.; Mares, J.; Johansen, J.R. Taxonomic classification of cyanoprokaryotes (cyanobacterial genera) 2014, using a polyphasic approach. Preslia 2014, 86, 295–335. [Google Scholar]
- Keller, L.; Leão, T.; Gerwick, W.H. Chemical Biology of Marine Cyanobacteria. In Chemical Biology of Natural Products; Taylor & Francis Group: Oxfordshire, UK, 2017; pp. 43–87. ISBN 9781439841938. [Google Scholar]
- Flores, E.; López-lozano, A.; Herrero, A. Nitrogen Fixation in the Oxygenic (Cyanobacteria): The Fight Against Oxygen. Biol. Nitrogen Fixat. 2015, 2, 879–889. [Google Scholar] [CrossRef]
- Leao, T.; Castelão, G.; Korobeynikov, A.; Monroe, E.A.; Podell, S.; Glukhov, E.; Allen, E.E.; Gerwick, W.H.; Gerwick, L. Comparative genomics uncovers the prolific and distinctive metabolic potential of the cyanobacterial genus Moorea. Proc. Natl. Acad. Sci. USA 2017, 114, 3198–3203. [Google Scholar] [CrossRef] [Green Version]
- Alvarenga, D.O.; Fiore, M.F.; Varani, A.M. A Metagenomic Approach to Cyanobacterial Genomics. Front. Microbiol. 2017, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Tronholm, A.; Engene, N. Moorena gen. nov., a valid name for “Moorea Engene & al.” nom. inval. Oscillatoriaceae Cyanobacteria 2019, 122, 1–2. [Google Scholar]
- Dittmann, E.; Gugger, M.; Sivonen, K.; Fewer, D.P. Natural Product Biosynthetic Diversity and Comparative Genomics of the Cyanobacteria. Trends Microbiol. 2015, 23, 642–652. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P. metaSPAdes: A new versatile de novo metagenomics assembler. Quant. Biol. Genom. 2016, 27, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Podell, S.; Gaasterland, T. DarkHorse: A method for genome-wide prediction of horizontal gene transfer. Genome Biol. 2007, 8, R16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, N.A.; Seiler, G.; Leão, T.F.; Castro-Falcón, G.; Gerwick, L.; Hughes, C.C.; Gerwick, W.H. Nature’s Combinatorial Biosynthesis Produces Vatiamides A-F. Angew. Chem. Int. Ed. 2019, 58, 9027–9031. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, N.A.; Leão, T.; Rankin, M.R.; McCullough, T.M.; Qu, P.; Korobeynikov, A.; Smith, J.L.; Gerwick, L.; Gerwick, W.H. Ketoreductase Domain Dysfunction Expands Ketoreductase Domain Dysfunction Expands Chemodiversity: Malyngamide Biosynthesis in the Cyanobacterium Okeania hirsuta. ACS Chem. Biol. 2018, 13, 3385–3395. [Google Scholar] [CrossRef] [PubMed]
- Calteau, A.; Fewer, D.P.; Latifi, A.; Coursin, T.; Laurent, T.; Jokela, J.; Kerfeld, C.A.; Sivonen, K.; Piel, J.; Gugger, M. Phylum-wide comparative genomics unravel the diversity of secondary metabolism in Cyanobacteria. BMC Genom. 2014, 15, 977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. AntiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [Green Version]
- Navarro-muñoz, J.C.; Selem-mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; Santos, E.L.C.D.L.; Yeong, M.; Cruz-morales, P.; Abubucker, S.; et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol. 2019, 16, 60–68. [Google Scholar] [CrossRef]
- Medema, M.H.; Kottmann, R.; Yilmaz, P.; Cummings, M.; Biggins, J.B.; Blin, K.; De Bruijn, I.; Chooi, Y.H.; Claesen, J.; Coates, R.C.; et al. The Minimum Information about a Biosynthetic Gene cluster (MIBiG) specification. Nat. Chem. Biol. 2015, 11, 625–631. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [Green Version]
- Ernst, M.; Kang, K.B.; Caraballo-Rodríguez, A.M.; Nothias, L.F.; Wandy, J.; Chen, C.; Wang, M.; Rogers, S.; Medema, M.H.; Dorrestein, P.C.; et al. Molnetenhancer: Enhanced molecular networks by integrating metabolome mining and annotation tools. Metabolites 2019, 9, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- da Silva, R.R.; Wang, M.; Nothias, L.F.; van der Hooft, J.J.J.; Caraballo-Rodríguez, A.M.; Fox, E.; Balunas, M.J.; Klassen, J.L.; Lopes, N.P.; Dorrestein, P.C. Propagating annotations of molecular networks using in silico fragmentation. PLoS Comput. Biol. 2018, 14, e1006089. [Google Scholar] [CrossRef] [PubMed]
- Mohimani, H.; Gurevich, A.; Shlemov, A.; Mikheenko, A.; Korobeynikov, A.; Cao, L.; Shcherbin, E.; Nothias, L.F.; Dorrestein, P.C.; Pevzner, P.A. Dereplication of microbial metabolites through database search of mass spectra. Nat. Commun. 2018, 9, 4035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djoumbou Feunang, Y.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated chemical classification with a comprehensive, computable taxonomy. J. Cheminform. 2016, 8, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Mevers, E.; Liu, W.T.; Engene, N.; Mohimani, H.; Byrum, T.; Pevzner, P.A.; Dorrestein, P.C.; Spadafora, C.; Gerwick, W.H. Cytotoxic veraguamides, alkynyl bromide-containing cyclic depsipeptides from the marine cyanobacterium cf. Oscillatoria margaritifera. J. Nat. Prod. 2011, 74, 928–936. [Google Scholar] [CrossRef] [Green Version]
- Clark, B.R.; Engene, N.; Teasdale, M.E.; Rowley, D.C.; Matainaho, T.; Valeriote, F.A.; Gerwick, W.H. Natural products chemistry and taxonomy of the marine cyanobacterium Blennothrix cantharidosmum. J. Nat. Prod. 2008, 71, 1530–1537. [Google Scholar] [CrossRef] [Green Version]
- Williams, P.G.; Moore, R.E.; Paul, V.J. Isolation and Structure Determination of Lyngbyastatin 3, a Lyngbyastatin 1 Homologue from the Marine Cyanobacterium Lyngbya majuscula. Determination of the Configuration of the 4-Amino-2,2-dimethyl-3-oxopentanoic Acid Unit in Majusculamide C., Dolastatin. J. Nat. Prod. 2003. [Google Scholar] [CrossRef]
- Mitchell, S.S.; Faulkner, D.J.; Rubins, K.; Bushman, F.D. Dolastatin 3 and two novel cyclic peptides from a palauan collection of Lyngbya majuscula. J. Nat. Prod. 2000, 63, 279–282. [Google Scholar] [CrossRef] [Green Version]
- Pereira, A.; Cao, Z.; Murray, T.F.; Gerwick, W.H. Hoiamide A, a Sodium Channel Activator of Unusual Architecture from a Consortium of Two Papua New Guinea Cyanobacteria. Chem. Biol. 2009, 16, 1208. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.; Pereira, A.R.; Cao, Z.; Shuman, C.F.; Engene, N.; Byrum, T.; Matainaho, T.; Murray, T.F.; Mangoni, A.; Gerwick, W.H. The hoiamides, structurally intriguing neurotoxic Lipopeptides from Papua New Guinea marine cyanobacteria. J. Nat. Prod. 2010, 73, 1411–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luesch, H.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Apramides A-G, novel lipopeptides from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2000, 63, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Gunasekera, S.P.; Ritson-Williams, R.; Paul, V.J. Carriebowmide, a new cyclodepsipeptide from the marine cyanobacterium Lyngbya polychroa. J. Nat. Prod. 2008, 71, 2060–2063. [Google Scholar] [CrossRef]
- Orjala, J.; Gerwick, W.H. Barbamide, a chlorinated metabolite with molluscicidal activity from the Caribbean cyanobacterium Lyngbya majuscula. J. Nat. Prod. 1996, 59, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Naman, C.B.; Alexander, K.L.; Guan, H.; Gerwick, W.H. The Chemistry, Biochemistry, and Pharmacology of Marine Natural Products from Leptolyngbya, a Chemically Endowed Genus of Cyanobacteria. Mar. Drugs 2020, 18, 508. [Google Scholar] [CrossRef]
- Pettit, G.R.; Kamano, Y.; Kizu, H.; Dufresne, C.; Herald, C.L.; Bontems, R.J.; Schmidt, J.M.; Boettner, F.E.; Nieman, R.A. Isolation and structure of the cell growth inhibitory depsipeptides dolastatins 11 and 12. Heterocycles 1989, 28, 553–558. [Google Scholar] [CrossRef]
- Pereira, A.R.; Cao, Z.; Engene, N.; Soria-Mercado, I.E.; Murray, T.F.; Gerwick, W.H. Palmyrolide A, an unusually stabilized neuroactive macrolide from palmyra atoll cyanobacteria. Org. Lett. 2010, 12, 4490–4493. [Google Scholar] [CrossRef] [Green Version]
- Williamson, R.T.; Boulanger, A.; Vulpanovici, A.; Roberts, M.A.; Gerwick, W.H. Structure and Absolute Stereochemistry of Phormidolide, a New Toxic Metabolite from the Marine Cyanobacterium Phormidium sp. J. Org. Chem. 2003, 67, 7927–7936. [Google Scholar] [CrossRef]
- Rastogi, R.P.; Incharoensakdi, A. Characterization of UV-screening compounds, mycosporine-like amino acids, and scytonemin in the cyanobacterium Lyngbya sp. CU2555. FEMS Microbiol. Ecol. 2014, 87, 244–256. [Google Scholar] [CrossRef] [Green Version]
- Joshi, D.; Mohandass, C.; Dhale, M. Effect of UV-B Radiation and Desiccation Stress on Photoprotective Compounds Accumulation in Marine Leptolyngbya sp. Appl. Biochem. Biotechnol. 2018, 184, 35–47. [Google Scholar] [CrossRef]
- Inayat-Hussain, S.H.; Thomas, N.F. Recent advances in the discovery and development of stilbenes and lactones in anticancer therapy. Expert Opin. Ther. Pat. 2004, 14, 819–835. [Google Scholar] [CrossRef]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [Green Version]
- Bofinger, M.R.; De Sousa, L.S.; Fontes, J.E.N.; Marsaioli, A.J. Diketopiperazines as Cross-Communication Quorum-Sensing Signals between Cronobacter sakazakii and Bacillus cereus. ACS Omega 2017, 2, 1003–1008. [Google Scholar] [CrossRef] [Green Version]
- Taton, A.; Ecker, A.; Diaz, B.; Moss, N.A.; Anderson, B.; Leão, T.F.; Simkovsky, R.; Dorrestein, P.C.; Gerwick, L.; William, H. Heterologous expression of cryptomaldamide in a cyanobacterial host. ACS Synth. Biol. 2020, 9, 33643376. [Google Scholar] [CrossRef]
- Moss, N.; Leao, T.; Glukhov, E.; Gerwick, L.; Gerwick, W.H. Collection, Culturing, and Genome Analyses of Tropical Marine Filamentous Benthic Cyanobacteria. Methods Enzymol. 2018, 604, 3–43. [Google Scholar]
- Glenn, T.C.; Nilsen, R.A.; Kieran, T.J.; Sanders, J.G.; Bayona-Vásquez, N.J.; Finger, J.W.; Pierson, T.W.; Bentley, K.E.; Hoffberg, S.L.; Louha, S.; et al. Adapterama I: Universal stubs and primers for 384 unique dual-indexed or 147,456 combinatorially-indexed Illumina libraries (iTru & iNext). bioRxiv 2019, 049114. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Podell, S.; Gaasterland, T.; Allen, E.E. A database of phylogenetically atypical genes in archaeal and bacterial genomes, identified using the DarkHorse algorithm. BMC Bioinform. 2008, 9, 419. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Bosi, E.; Donati, B.; Galardini, M.; Brunetti, S.; Sagot, M.F.; Lió, P.; Crescenzi, P.; Fani, R.; Fondi, M. MeDuSa: A multi-draft based scaffolder. Bioinformatics 2015, 31, 2443–2451. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive κ-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Muller, R.; Wohlleben, W.; et al. antiSMASH 3.0--a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Luzzatto-Knaan, T.; Garg, N.; Wang, M.; Glukhov, E.; Peng, Y.; Ackermann, G.; Amir, A.; Duggan, B.M.; Ryazanov, S.; Gerwick, L.; et al. Digitizing mass spectrometry data to explore the chemical diversity and distribution of marine cyanobacteria and algae. Elife 2017, 6, 1686–1699. [Google Scholar] [CrossRef]
- Garg, N.; Kapono, C.A.; Lim, Y.W.; Koyama, N.; Vermeij, M.J.A.; Conrad, D.; Rohwer, F.; Dorrestein, P.C. Mass spectral similarity for untargeted metabolomics data analysis of complex mixtures. Int. J. Mass Spectrom. 2015, 377, 719–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Collection Code | Genome ID | # of Scaffolds | Frag./Comp. BGC | Taxonomic ID | NCBI Accession | |
|---|---|---|---|---|---|---|
| 1 | ISB3NOV948BCUL | SIOISBB | 3 | 0/11 | Leptolyngbya | JAAHII01 |
| 2 | ASI16JUL142CUL | SIOASIH | 25 | 5/41 | Moorena | JAAHIH01 |
| 3 | NAC09DEC082ENV | SIO1I7 | 110 | 5/12 | Okeania | JAAHGF01 |
| 4 | SPB31JAN131ENV | SIO3A2 | 132 | 23/8 | Moorena | JAAHHC01 |
| 5 | NAC18DEC082ENV | SIO3A5 | 164 | 17/11 | Moorena | JAAHHD01 |
| 6 | ASI16JUL149ENV | SIO1A7 | 183 | 6/8 | Oscillatoria | JAAHFN01 |
| 7 | PAB18MAY117ENV | SIO2C2 | 184 | 8/6 | Okeania | JAAHGM01 |
| 8 | PAB05APR064ENV | SIO1H4 | 192 | 11/3 | Okeania | JAAHGB01 |
| 9 | PRL23MAR111ENV | SIO1F9 | 198 | 5/6 | Okeania | JAAHFW01 |
| 10 | PRM25MAR112ENV | SIO1G6 | 205 | 20/12 | Moorena | JAAHFZ01 |
| 11 | ASY22JUL141ENV | SIO1C2 | 246 | 16/11 | Symploca | JAAHFP01 |
| 12 | PAB17MAY117ENV | SIO2B3 | 250 | 14/2 | Okeania | JAAHGH01 |
| 13 | PAB07APR054ENV | SIO1H5 | 263 | 13/3 | Okeania | JAAHGC01 |
| 14 | PAB03APR065ENV | SIO1H2 | 279 | 11/2 | Okeania | JAAHGA01 |
| 15 | PAP14JUN083ENV | SIO3C6 | 290 | 6/11 | Symploca | JAAHHJ01 |
| 16 | PAC17FEB109ENV | SIO2C9 | 318 | 14/8 | Okeania | JAAHGO01 |
| 17 | PAL11AUG091ENV | SIO2F5 | 334 | 18/1 | Okeania | JAAHGU01 |
| 18 | PAL01AUG091ENV | SIO2G4 | 339 | 16/3 | Okeania | JAAHGW01 |
| 19 | PNG21MAY053ENV | SIO2E9 | 384 | 16/5 | Symploca | JAAHGS01 |
| 20 | PLP20MAR122ENV | SIO3E8 | 387 | 23/7 | Moorena | JAAHHM01 |
| 21 | PNG22APR061CUL | SIO3F7 | 399 | 22/7 | Moorena | JAAHHP01 |
| 22 | ASG15JUL146CUL | SIO3F2 | 454 | 10/0 | Spirulina | JAAHHN01 |
| 23 | PAB17MAY115ENV | SIO2C1 | 471 | 25/4 | Symploca | JAAHGL01 |
| 24 | ASX22JUL142CUL | SIO1E4_02 | 12 | 3/17 | Leptolyngbya | JAAHFU01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leão, T.; Wang, M.; Moss, N.; da Silva, R.; Sanders, J.; Nurk, S.; Gurevich, A.; Humphrey, G.; Reher, R.; Zhu, Q.; et al. A Multi-Omics Characterization of the Natural Product Potential of Tropical Filamentous Marine Cyanobacteria. Mar. Drugs 2021, 19, 20. https://doi.org/10.3390/md19010020
Leão T, Wang M, Moss N, da Silva R, Sanders J, Nurk S, Gurevich A, Humphrey G, Reher R, Zhu Q, et al. A Multi-Omics Characterization of the Natural Product Potential of Tropical Filamentous Marine Cyanobacteria. Marine Drugs. 2021; 19(1):20. https://doi.org/10.3390/md19010020
Chicago/Turabian StyleLeão, Tiago, Mingxun Wang, Nathan Moss, Ricardo da Silva, Jon Sanders, Sergey Nurk, Alexey Gurevich, Gregory Humphrey, Raphael Reher, Qiyun Zhu, and et al. 2021. "A Multi-Omics Characterization of the Natural Product Potential of Tropical Filamentous Marine Cyanobacteria" Marine Drugs 19, no. 1: 20. https://doi.org/10.3390/md19010020
APA StyleLeão, T., Wang, M., Moss, N., da Silva, R., Sanders, J., Nurk, S., Gurevich, A., Humphrey, G., Reher, R., Zhu, Q., Belda-Ferre, P., Glukhov, E., Whitner, S., Alexander, K. L., Rex, R., Pevzner, P., Dorrestein, P. C., Knight, R., Bandeira, N., ... Gerwick, L. (2021). A Multi-Omics Characterization of the Natural Product Potential of Tropical Filamentous Marine Cyanobacteria. Marine Drugs, 19(1), 20. https://doi.org/10.3390/md19010020