
Pan-Genomic and Transcriptomic Analyses of Marine Pseudoalteromonas agarivorans Hao 2018 Revealed Its Genomic and Metabolic Features

 ,
,
Abstract
:1. Introduction
2. Results
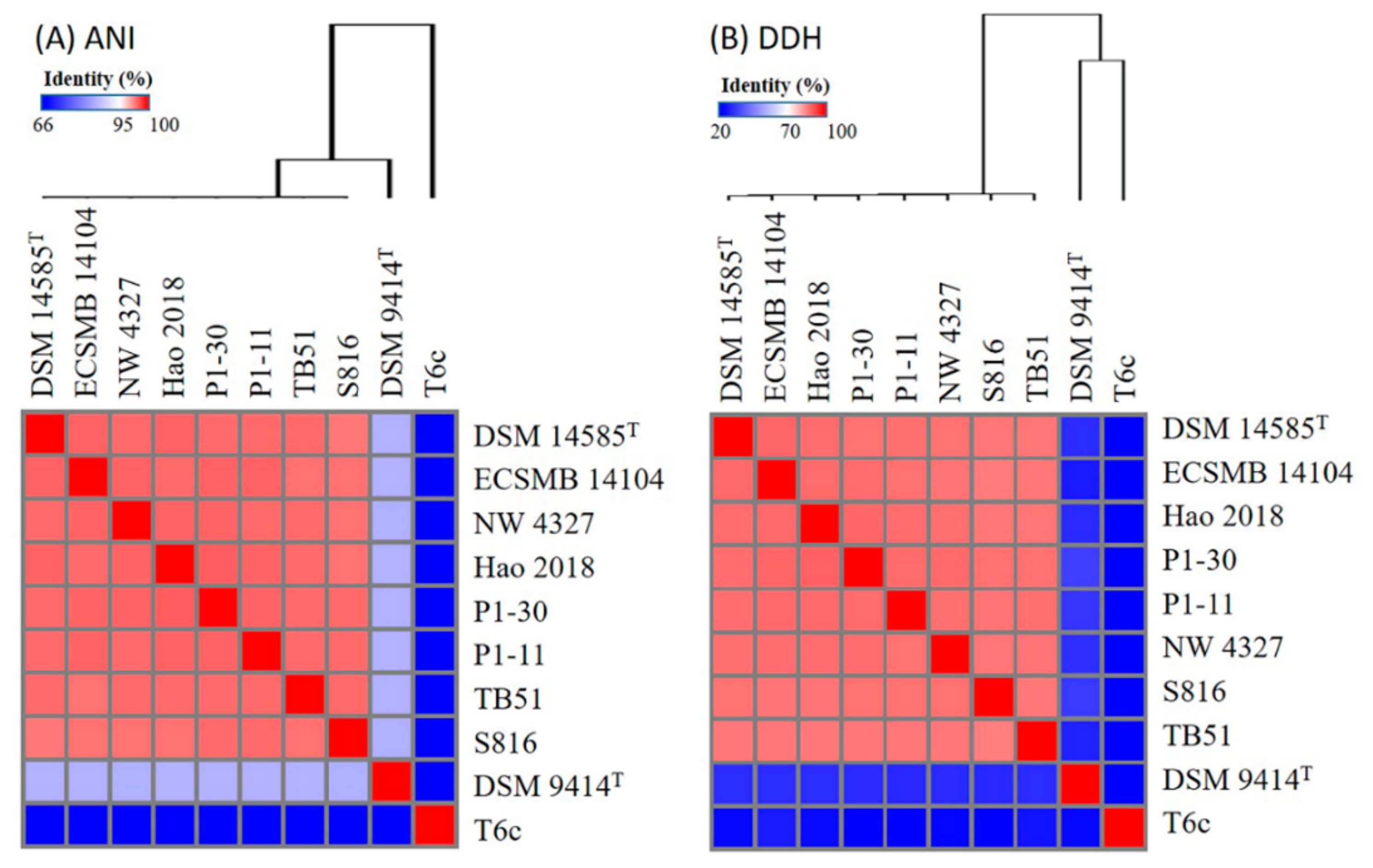
2.1. Genome Collection of the Pseudoalteromonas Strains
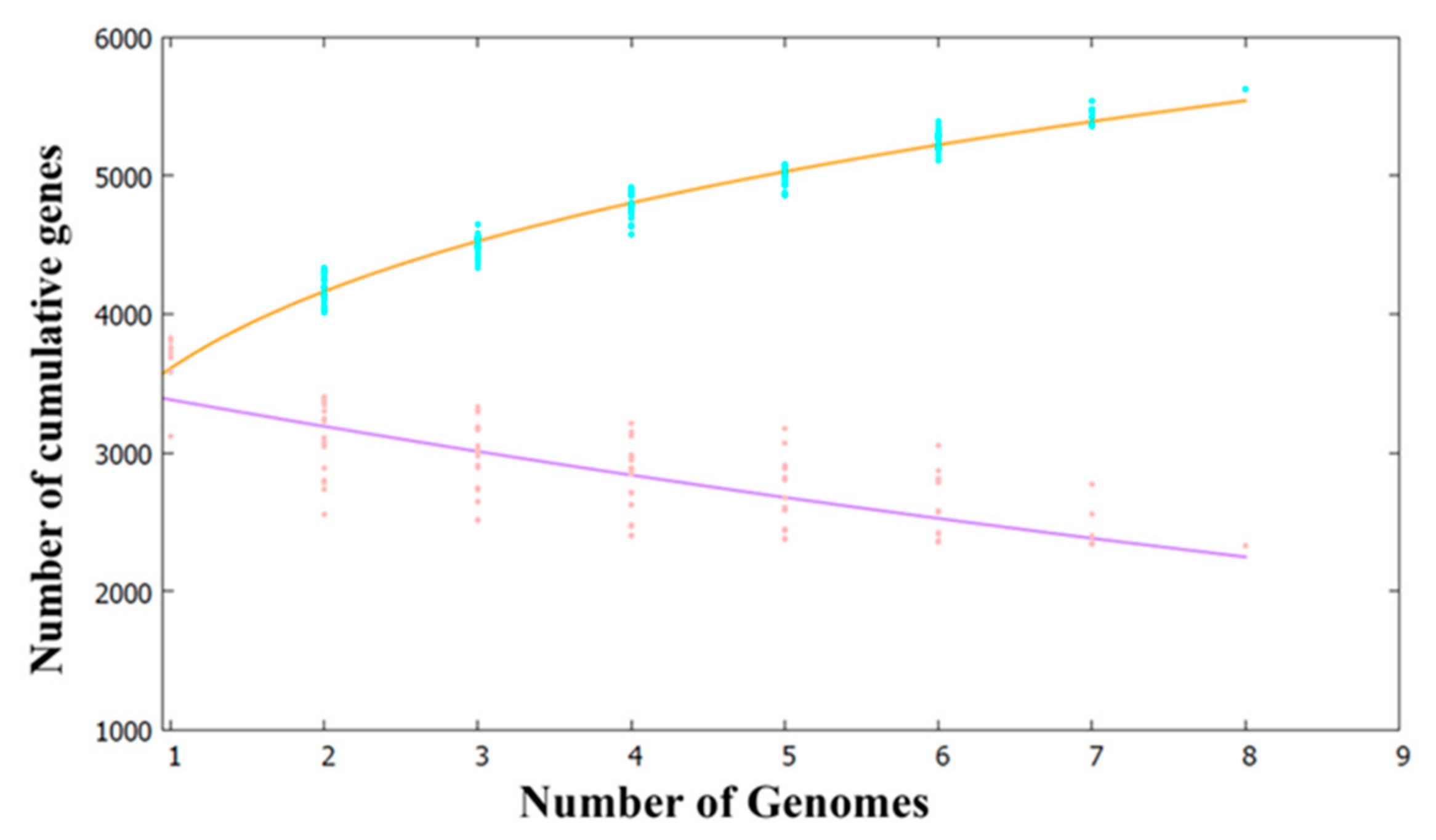
2.2. Pan- and Core-Genome Analyses of Pseudoalteromonas Strains
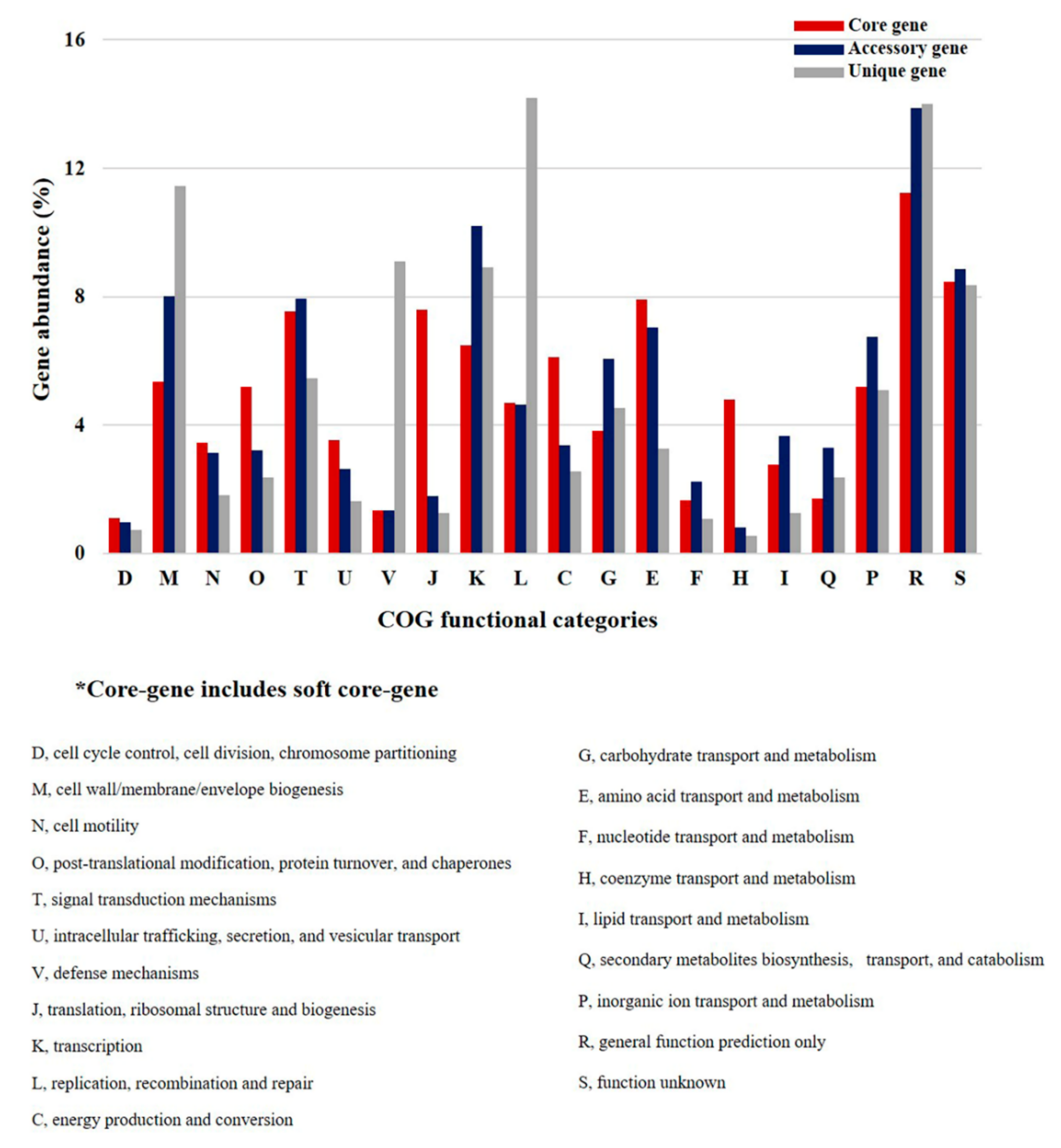
2.3. Clusters of Orthologous Groups (COG) Analyses of P. agarivorans
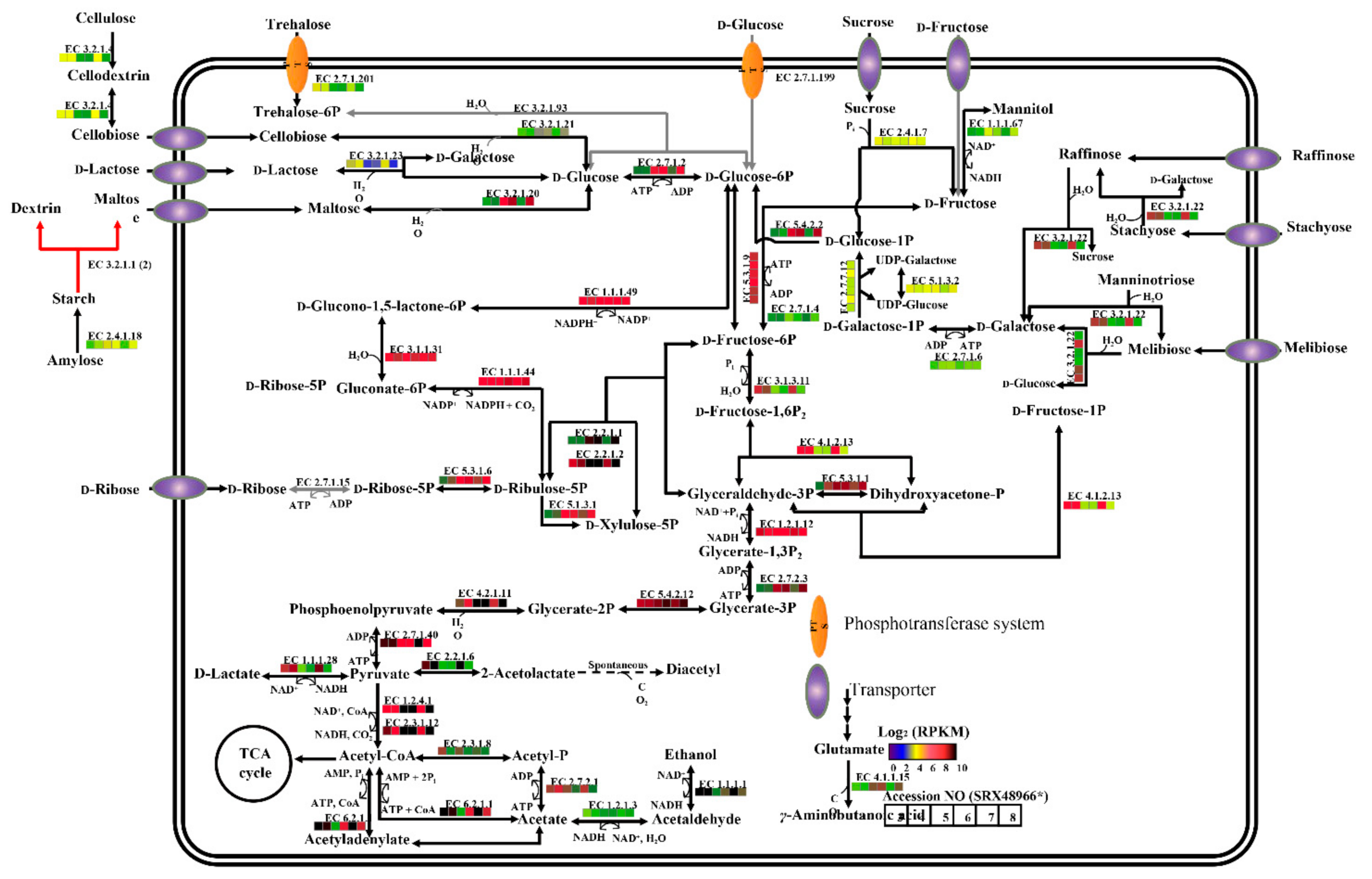
2.4. KEGG and Carbohydrate Metabolism Regulation Analyses of P. agarivorans Hao 2018
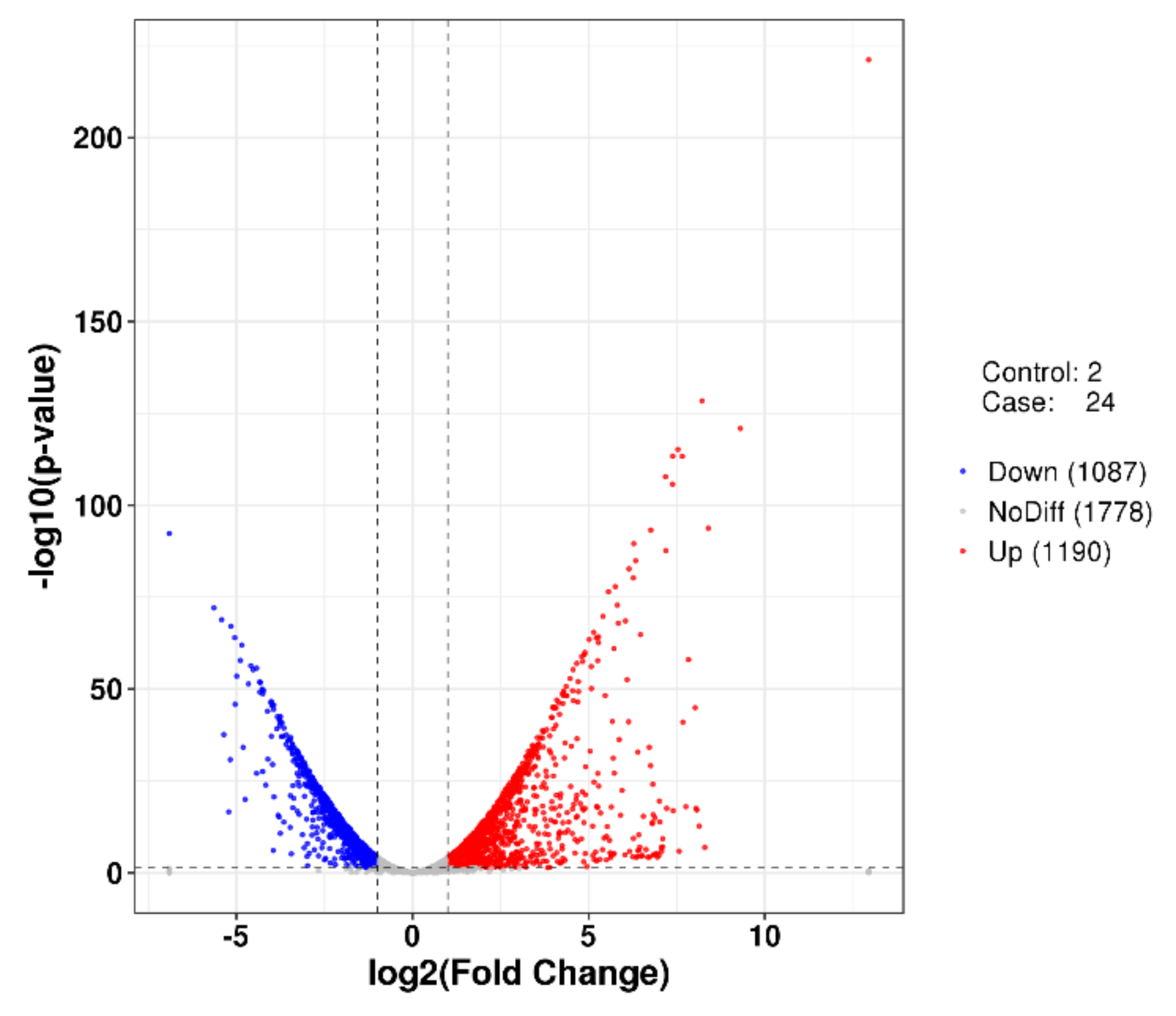
2.5. Differentially Expressed Genes (DEGs) Analyses of P. agarivorans Hao 2018
3. Discussion
4. Materials and Methods
4.1. Strain Collection and Phylogenetic Analysis
4.2. Sample Collection and Processing
4.3. Average Nucleotide Identity (ANI), In Silico DDH, and Pan- and Core-Genome Analyses
4.4. Clusters of Orthologous Groups (COG) Analysis
4.5. Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analyses
4.6. Transcriptome Sequencing and Data Analyses
4.7. Transcriptome Data Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gauthier, G.; Gauthier, M.R. Phylogenetic analysis of the genera Alteromonas, Shewanella, and Moritella using genes coding for small-subunit rRNA sequences and division of the genus Alteromonas into two genera, Alteromonas (emended) and Pseudoalteromonas gen. nov., and proposal of twelve new species combinations. Int. J. Syst. Bacteriol. 1995, 45, 755–761. [Google Scholar]
- Yoon, J.-H.; Kang, S.-J.; Lee, M.-H.; Oh, T.-K. Tamlana agarivorans sp. nov., isolated from seawater off Jeju Island in Korea. Int. J. Syst. Evol. Microbiol. 2008, 58, 1892–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romanenko, L.A.; Zhukova, N.V.; Rohde, M.; Lysenko, A.M.; Mikhailov, V.V.; Stackebrandt, E. Pseudoalteromonas agarivorans sp. nov., a novel marine agarolytic bacterium. Int. J. Syst. Evol. Microbiol. 2003, 53, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, J.P. Bioactive compound synthetic capacity and ecological significance of marine bacterial genus Pseudoalteromonas. Mar. Drugs 2007, 5, 220–241. [Google Scholar] [CrossRef]
- Nichols, C.A.; Guezennec, J.; Bowman, J.P. Bacterial exopolysaccharides from extreme marine environments with special consideration of the southern ocean, sea ice, and deep-sea hydrothermal vents: A review. Mar. Biotechnol. 2005, 7, 253–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitova, M.; Tutino, M.L.; Infusini, G.; Marino, G.; Rosa, S.D. Exocellular peptides from Antarctic psychrophile Pseudoalteromonas haloplanktis. Mar. Biotechnol. 2005, 7, 523–531. [Google Scholar] [CrossRef]
- Carrión, O.; Delgado, L.; Mercade, E. New emulsifying and cryoprotective exopolysaccharide from Antarctic Pseudomonas sp. ID1. Carbohydr. Polym. 2015, 117, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Zhou, W.Z.; Zhang, Y.Z.; Wang, J.; Zhu, X.B. Flocculation behavior and mechanism of an exopolysaccharide from the deep-sea psychrophilic bacterium Pseudoalteromonas sp. SM9913. Bioresour. Technol. 2008, 99, 6893–6899. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Liu, W.; Liu, K.; Shan, K.; Wang, C.L.; Xi, C.X.; Liu, J.; Fan, Q.; Zhang, X.; Lu, X.; et al. Isolation, optimization of fermentation conditions, and characterization of an exopolysaccharide from Pseudoalteromonas agarivorans Hao 2018. Mar. Drugs 2019, 17, 703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmstrom, C.; Kjelleberg, S. Marine Pseudoalteromonas species are associated with higher organisms and produce biologically active extracellular agents. FEMS Microbiol. Ecol. 1999, 30, 285–293. [Google Scholar] [CrossRef]
- Komandrova, N.A.; Isakov, V.V.; Tomshich, S.V.; Romanenko, L.A.; Perepelov, A.V.; Shashkov, A.S. Structure of an acidic O-specific polysaccharide of the marine bacterium Pseudoalteromonas agarivorans KMM 232 (R-form). Biochemistry 2010, 75, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Komandrova, N.A.; Kokoulin, M.S.; Kalinovskiy, A.I.; Tomshich, S.V.; Romanenko, L.A.; Vaskovsky, V.E. The O-specific polysaccharide from the marine bacterium Pseudoalteromonas agarivorans KMM 255T. Carbohydr. Res. 2015, 414, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q. The Research of Exopolysaccharide Produced by Pesudoalteromonas issachenkonii HZ. Master’s Thesis, Qilu University of Technology, Jinan, China, 2016. Available online: https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CMFD&dbname=CMFD201701&filename=1016321192.nh&uniplatform=NZKPT&v=NcDgVoUuD7KmgPVJkfyDDCTeJb2ZH4UHnDme8GcL4KGa7Pj9_u60AQWN2Ui9oAJy (accessed on 5 May 2018).
- Wang, C.; Fan, Q.; Zhang, X.; Lu, X.; Xu, Y.; Zhu, W.; Zhang, J.; Hao, W.; Hao, L. Isolation, characterization, and pharmaceutical applications of an exopolysaccharide from Aerococcus uriaeequi. Mar. Drugs 2018, 16, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X. Effects of Environmental Factors on Exopolysaccharide Synthesis and Related Gene Expression Regulation in Pseudoalteromonas issachenkonii HZ. Master’s Thesis, Qilu University of Technology, Jinan, China, 2017. Available online: https://kns.cnki.net/kcms/detail/detail.aspx?dbcode=CMFD&dbname=CMFD201801&filename=1018703649.nh&uniplatform=NZKPT&v=uotrxgIozy2cpACdUesnWcgqjXK8NiVBX7c-UTOigtP_HpiVQzR3Xrq96g2bqwwo (accessed on 5 May 2018).
- Lu, X.X.; Fan, Q.; Hao, L. Effects of different environmental factors on the expression of glycosyltransferase gene in marine bacteria Pseudoalteromonas issachenkonii HZ. Sci. Technol. Food Ind. 2016, 37, 212–216. [Google Scholar]
- Chun, B.H.; Kim, K.H.; Jeong, S.E.; Jeon, C.O. Genomic and metabolic features of the Bacillus amyloliquefaciens group-B. amyloliquefaciens, B. velezensis, and B. siamensis- revealed by pan-genome analysis. Food Microbiol. 2019, 77, 146–157. [Google Scholar] [CrossRef]
- Chun, B.H.; Kim, K.H.; Jeon, H.H.; Lee, S.H.; Jeon, C.O. Pan-genomic and transcriptomic analyses of Leuconostoc mesenteroides provide insights into its genomic and metabolic features and roles in kimchi fermentation. Sci. Rep. 2017, 7, 11504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S.E.; Chun, B.H.; Kim, K.H.; Park, D.; Roh, S.W.; Lee, S.H.; Jeon, C.O. Genomic and metatranscriptomic analyses of Weissella koreensis reveal its metabolic and fermentative features during kimchi fermentation. Food Microbiol. 2018, 76, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Massonnet, M.; Morales-Cruz, A.; Minio, A.; Figueroa-Balderas, R.; Lawrence, D.P.; Travadon, R.; Rolshausen, P.E.; Baumgartner, K.; Cantu, D. Whole-genome resequencing and pan-transcriptome reconstruction highlight the impact of genomic structural variation on secondary metabolite gene clusters in the grapevine Esca pathogen Phaeoacremonium minimum. Front. Microbiol. 2018, 9, 1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medigue, C.; Krin, E.; Pascal, G.; Barbe, V.; Bernsel, A.; Bertin, P.N.; Cheung, F.; Cruveiller, S.; D’Amico, S.; Duilio, A.; et al. Coping with cold: The genome of the versatile marine Antarctica bacterium Pseudoalteromonas haloplanktis TAC125. Genome Res. 2005, 15, 1325–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhury, J.D.; Pramanik, A.; Webster, N.S.; Llewellyn, L.E.; Gachhui, R.; Mukherjee, J. The pathogen of the great barrier reef sponge Rhopaloeides odorabile is a new strain of Pseudoalteromonas agarivorans containing abundant and diverse virulence-related genes. Mar. Biotechnol. 2015, 17, 463–478. [Google Scholar] [CrossRef] [PubMed]
- John, M.S.; Nagoth, J.A.; Ramasamy, K.P.; Ballarini, P.; Mozzicafreddo, M.; Mancini, A.; Telatin, A.; Liò, P.; Giuli, G.; Natalello, A.; et al. Horizontal gene transfer and silver nanoparticles production in a new Marinomonas strain isolated from the Antarctic psychrophilic ciliate Euplotes focardii. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Shan, K.; Wang, C.; Liu, W.; Liu, K.; Jia, B.; Hao, L. Genome sequence and transcriptomic profiles of a marine bacterium, Pseudoalteromonas agarivorans Hao 2018. Sci. Data 2019, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for theprokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [Green Version]
- Rossello-Mora, R.; Amann, R. Past and future species definitions for Bacteria and Archaea. Syst. Appl. Microbiol. 2015, 38, 209–216. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Voulhoux, R.; Ball, G.; Ize, B.; Vasil, M.L.; Lazdunski, A.; Wu, L.F.; Filloux, A. Involvement of the twin-arginine translocation system in protein secretion via the type II pathway. EMBO J. 2001, 20, 6735–6741. [Google Scholar] [CrossRef] [PubMed]
- Kolter, R.; Siegele, D.A.; Tormo, A. The stationary phase of the bacterial life cycle. Annu. Rev. Microbiol. 1993, 47, 855–874. [Google Scholar] [CrossRef]
- Pirog, T.P.; Kovalenko, M.A.; Kuz’minskaya, Y.V. Exopolysaccharide production and peculiarities of C6-metabolism in Acinetobacter sp. grown on carbohydrate substrates. Microbiology 2002, 71, 182–188. [Google Scholar] [CrossRef]
- Torino, M.I.; Taranto, M.P.; Sesma, F.; De Valdez, G.F. Heterofermentative pattern and exopolysaccharide production by Lactobacillus helveticus ATCC 15807 in response to environmental pH. J. Appl. Microbiol. 2001, 91, 846–852. [Google Scholar] [CrossRef] [Green Version]
- Endo, A.; Tanizawa, Y.; Tanaka, N.; Maeno, S.; Kumar, H.; Shiwa, Y.; Okada, S.; Yoshikawa, H.; Dicks, L.; Nakagawa, J.; et al. Comparative genomics of Fructobacillus spp. and Leuconostoc spp. reveals niche-specific evolution of Fructobacillus spp. BMC Genom. 2015, 16, 1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernikos, G.; Medini, D.; Riley, D.R.; Tettelin, H. Ten years of pan-genome analyses. Curr. Opin. Microbiol. 2015, 23, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Fondi, M.; Fani, R. The horizontal flow of the plasmid resistome: Clues from inter-generic similarity networks. Environ. Microbiol. 2010, 12, 3228–3242. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Dewar, K.; Katsanis, N.; Reiter, L.T.; Lander, E.S.; Devon, K.L.; Wyman, D.W.; Lupski, J.R.; Birren, B. The 1.4-Mb CMT1A duplication/HNPP deletion genomic region reveals unique genome architectural features and provides insights into the recent evolution of new genes. Genome Res. 2001, 11, 1018–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pramateftaki, P.V.; Kouvelis, V.N.; Lanaridis, P.; Typas, M.A. The mitochondrial genome of the wine yeast Hanseniaspora uvarum: A unique genome organization among yeast/fungal counterparts. FEMS Yeast Res. 2006, 6, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Laing, C.R.; Whiteside, M.D.; Gannon, V.P.J. Pan-genome analyses of the species Salmonella enterica, and identification of genomic markers predictive for species, subspecies, and serovar. Front. Microbiol. 2017, 8, 1345. [Google Scholar] [CrossRef] [Green Version]
- Ju, Y.; Shan, K.; Liu, W.; Xi, C.; Zhang, Y.; Wang, W.; Wang, C.; Cao, R.; Zhu, W.; Hao, L. Effect of Different Initial Fermentation pH on Exopolysaccharides Produced by Pseudoalteromonas agarivorans Hao 2018 and Identification of Key Genes Involved in Exopolysaccharide Synthesis via Transcriptome Analysis. Mar. Drugs 2022, 20, 89. [Google Scholar] [CrossRef]
- Hou, S.; Saw, J.H.; Lee, K.S.; Freitas, T.A.; Belisle, C.; Kawarabayasi, Y.; Donachie, S.P.; Pikina, A.; Galperin, M.Y.; Koonin, E.V. Genome sequence of the deep-sea γ-proteobacterium Idiomarina loihiensis reveals amino acid fermentation as a source of carbon and energy. Proc. Natl. Acad. Sci. USA 2004, 101, 18036–18041. [Google Scholar] [CrossRef] [Green Version]
- Roca, C.; Lehmann, M.; Torres, C.A.V.; Baptista, S.; Gaudencio, S.P.; Freitas, F.; Reis, M.A.M. Exopolysaccharide production by a marine Pseudoalteromonas sp. strain isolated from Madeira Archipelago ocean sediments. New Biotechnol. 2016, 33, 460–466. [Google Scholar] [CrossRef]
- Ivars-Martinez, E.; Martin-Cuadrado, A.B.; D’Auria, G.; Mira, A.; Ferriera, S.; Johnson, J.; Friedman, R.; Rodriguez-Valera, F. Comparative genomics of two ecotypes of the marine planktonic copiotroph Alteromonas macleodii suggests alternative lifestyles associated with different kinds of particulate organic matter. ISME J. 2008, 2, 1194–1212. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.; Ouk Kim, Y.; Park, S.C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinf. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 12, 472–477. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; Von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by Eggnog-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [Green Version]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Letunic, I.; Okuda, S.; Kanehisa, M.; Bork, P. iPath2.0: Interactive pathway explorer. Nucleic Acids Res. 2011, 39, W412–W415. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, T.P.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Nat. Preced. 2010. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Name in GenBank (Accession No.) | Genome Status a (No. of Contigs) | Total Size (Mb) | G + C Contents (%) | No. of Genes | Completeness (%) b | Contamination Rate (%) b | Isolation Sources and Geographic Location |
|---|---|---|---|---|---|---|---|
| P. agarivorans DSM 14585T (NZ_CP011011–2) | C (2) | 4.54 | 40.87 | 4119 | 99.56 | 0.76 | Seawater and Ascidians; Pacific Ocean |
| P. agarivorans Hao 2018 (NZ_CP033065–7) | C (3) | 4.56 | 40.83 | 4105 | 99.98 | 0.40 | Surface of abalone seedlings; China: Penglai |
| P. agarivorans NW 4327 (NZ_AZIO00000000) | D (129) | 4.48 | 40.90 | 4035 | 99.94 | 3.91 | Sponge; Great Barrier Reef |
| P. agarivorans S816 (NZ_APME00000000) | D (133) | 4.42 | 40.90 | 4062 | 100 | 0.25 | Seawater; Sierra Leone: Waterberg |
| P. atlantica ECSMB14104 (NZ_CP023464) | C (1) | 3.66 | 41.00 | 3331 | 91.92 | 0.25 | Seawater; East China Sea |
| P. sp. P1–11 (NZ_LJSP00000000) | D (31) | 4.38 | 41.00 | 3923 | 100 | 0.56 | Hydractinia echinate; USA: Woods Hole |
| P. sp. P1–30 (NZ_LKBC00000000) | D (35) | 4.33 | 40.90 | 3869 | 100 | 0.13 | Hydractinia echinate; USA: Woods Hole |
| P. sp. TB51 (NZ_AUTO00000000) | D (369) | 4.63 | 40.90 | 4394 | 99.25 | 1.01 | Marine sponge; Antarctic |
| P. espejiana DSM 9414T (NZ_CP011028–9) | C (2) | 4.50 | 40.30 | 4051 | 99.75 | 0.51 | Coastal sea Water; USA: Northern California |
| P. atlantica T6c (NC_008228) | C (1) | 5.18 | 44.60 | 4319 | 99.92 | 0.13 | Seawater; Atlantic |
| Annotation | Locus Tag | 2 h_RPKM | 24 h_RPKM |
|---|---|---|---|
| glucose/galactose MFS transporter | D9T18_18920 | 210.32 | 90.40 |
| MFS transporter | D9T18_05400 | 150.59 | 42.61 |
| Annotation | Locus Tag | 2 h_RPKM | 24 h_RPKM |
|---|---|---|---|
| UDP-glucuronate-epimerase | D9T18_02060 | 188.19 | 45.88 |
| dTDP-4-dehydrorhamnose 3,5-epimerase | D9T18_02110 | 657.00 | 66.08 |
| dTDP-4-dehydrorhamnose reductase | D9T18_02115 | 582.10 | 45.39 |
| dTDP-glucose 4,6-dehydratase | D9T18_02120 | 800.04 | 66.56 |
| UTP--glucose-1-phosphate uridylyltransferase | D9T18_02300 | 2430.93 | 533.75 |
| UDP-glucose 6-dehydrogenase | D9T18_02305 | 838.21 | 375.42 |
| UTP--glucose-1-phosphateuridylyltransferase | D9T18_06755 | 324.19 | 271.59 |
| glucokinase | D9T18_07205 | 261.87 | 106.27 |
| phosphoglucomutase | D9T18_08400 | 476.66 | 74.74 |
| UTP--glucose-1-phosphate uridylyltransferase | D9T18_11960 | 678.51 | 358.89 |
| Annotation | Locus Tag | 2 h_RPKM | 24 h_RPKM |
|---|---|---|---|
| glycosyl transferase | D9T18_02030 | 156.40 | 15.57 |
| glycosyl transferase family 2 | D9T18_02205 | 145.28 | 48.82 |
| rhamnosyltransferase | D9T18_02235 | 435.05 | 33.75 |
| glycosyl transferase | D9T18_03335 | 8.95 | 37.95 |
| glucosyl transferase family 2 | D9T18_06790 | 166.71 | 43.81 |
| glucosyl transferase family 2 | D9T18_06795 | 162.09 | 27.37 |
| glycosyl hydrolase family 16 | D9T18_17140 | 9.01 | 15.14 |
| glycosyl transferase | D9T18_17235 | 23.92 | 15.30 |
| glycosyl transferase family 2 | D9T18_19775 | 150.85 | 44.41 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jv, Y.; Xi, C.; Zhao, Y.; Wang, W.; Zhang, Y.; Liu, K.; Liu, W.; Shan, K.; Wang, C.; Cao, R.; et al. Pan-Genomic and Transcriptomic Analyses of Marine Pseudoalteromonas agarivorans Hao 2018 Revealed Its Genomic and Metabolic Features. Mar. Drugs 2022, 20, 248. https://doi.org/10.3390/md20040248
Jv Y, Xi C, Zhao Y, Wang W, Zhang Y, Liu K, Liu W, Shan K, Wang C, Cao R, et al. Pan-Genomic and Transcriptomic Analyses of Marine Pseudoalteromonas agarivorans Hao 2018 Revealed Its Genomic and Metabolic Features. Marine Drugs. 2022; 20(4):248. https://doi.org/10.3390/md20040248
Chicago/Turabian StyleJv, Yuhao, Chenxiang Xi, Yanqiu Zhao, Wei Wang, Yiling Zhang, Kai Liu, Wenlin Liu, Kai Shan, Chunlei Wang, Ruiwen Cao, and et al. 2022. "Pan-Genomic and Transcriptomic Analyses of Marine Pseudoalteromonas agarivorans Hao 2018 Revealed Its Genomic and Metabolic Features" Marine Drugs 20, no. 4: 248. https://doi.org/10.3390/md20040248
APA StyleJv, Y., Xi, C., Zhao, Y., Wang, W., Zhang, Y., Liu, K., Liu, W., Shan, K., Wang, C., Cao, R., Dai, C., Jv, Y., Zhu, W., Wang, H., He, Q., & Hao, L. (2022). Pan-Genomic and Transcriptomic Analyses of Marine Pseudoalteromonas agarivorans Hao 2018 Revealed Its Genomic and Metabolic Features. Marine Drugs, 20(4), 248. https://doi.org/10.3390/md20040248