1. Introduction
Natural products containing sulfur exhibit significant structural diversities and diverse biological activities [
1,
2]. Among them, thiodiketopiperazine, as an interesting subgroup, is an important class of secondary metabolites of fungi, especially species of
Aspergillus fumigatus,
A. terreus,
A. flavus,
A. niger,
Trichoderma virens,
T. viride, and
Penicillium terlikowskii [
3]. The structures of thiodiketopiperazines are diverse [
4,
5], of which the spirocyclic thiodiketopiperazines are rare in nature [
5,
6]. Many thiodiketopiperazines have been reported to display a wide range of biological properties, including antiproliferative, cytotoxic, antibacterial, antifungal, antiviral, and anti-angiogenic activities [
7,
8]. For examples, brocazine G displayed potent antibacterial activity against
Staphylococcus aureus with a MIC value of 0.25 μg/mL and strong cytotoxicity against human ovarian cancer cells (IC
50 = 0.66 μM) [
8]. Penicisulfuranols A–C showed cytotoxicity towards human cervical carcinoma cell lines and human promyelocytic leukemia cells with IC
50 values of 0.1–3.9 μM [
9]. Penicibrocazine C exhibited antibacterial activity against
Micrococcus luteus with MIC value of 0.25 μg/mL, which was stronger than the positive control, chloromycetin (MIC = 2.0 μg/mL) [
10]. Numerous structurally unique and potential variety activities of the thiodiketopiperazines have drawn much attention from synthetic chemists and pharmacologists.
The fungal genus
Lecanicillium (formerly
Verticillium), including
L. fusisporum,
L. psalliotae, and
L. lecanii, are pathogenic fungi that are widely used as biological pesticides [
11,
12,
13]. The secondary metabolisms of these fungi are rarely reported [
11]. Only a few compounds belonging to indolosesquiterpenoids, phenopicolinic acid derivatives, tetracyclic diterpenoids, pregnanes, and cyclic lipodepsipeptides have been isolated from the genus of
Lecanicillium up to now [
14,
15,
16]. The fungus
L. kalimantanense SCSIO41702 was isolated from a mangrove sediment sample collected from Hainan province. There was no report about the secondary metabolisms of
L. kalimantanense. In order to explore novel, natural compounds from fungi, we investigated the secondary metabolisms of
L. kalimantanense SCSIO41702, which led to the isolation of six new thiodiketopiperazine-class alkaloids, lecanicilliums A-F (
1–
6), together with thirteen known analogues (
Figure 1): emethacin B (
7) [
17], bisdethiobis(methylsulfanyl)acetylaranotin (
8) [
8], bisdethiobis(methylsulfanyl)deacetylaranotin (
9) [
8], bisdethiobis(methylsulfanyl)-aranotin (
10) [
8], 8-acetyl-bisdethiobis(methylsulfanyl)apoaranotin (
11) [
18], bisdethiobis(methylsulfanyl)acetylapoaranotin (
12) [
8], bisdethiobis(methylsulfanyl)apoaranotin (
13) [
19], bisdethiobis(methylsulfanyl)deacetylapoaranotin (
14) [
8], haematocin (
15) [
8], versicolor A (
16) [
20], 12,12a-anhydro-desacetyl-bis-dethio-7a, 14a-di-(methylmercapto)-apoaranotin (
17) [
21], emestrin H (
18) [
22], and citriperazine B (
19) [
23]. These compounds were evaluated for their cytotoxicity, toxicity against brine shrimps, and antibacterial activity. Herein, we report the isolation and structural elucidation as well as the biological activities of these compounds.
2. Results and Discussion
Lecanicillium A (
1) was obtained as a white powder with the molecular formula C
18H
16N
2O
5S as determined by HRESIMS ([M + H]
+ m/
z 373.0851) and NMR data. The
1H NMR spectrum (
Table 1) displayed the presence of two active hydrogens at
δH 10.06 (
1H, s), 5.31 (
1H, s), four aromatic hydrogens at
δH 7.26 (
1H, d,
J = 7.5 Hz), 7.14 (
1H, t,
J = 8.0 Hz), 6.92 (
1H, t,
J = 7.5 Hz), 6.81 (
1H, d,
J = 8.0 Hz), two olefinic methines at
δH 6.00 (
1H, d,
J = 6.5 Hz), 4.82 (
1H, ddd,
J = 7.8, 6.5, 1.2 Hz), four tertiary methine groups at
δH 6.61 (
1H, d,
J = 6.5 Hz), 4.86 (
1H, ddd,
J = 7.8, 6.5, 1.2 Hz), 3.87 (
1H, d,
J = 6.5 Hz), 3.06 (
1H, t,
J = 6.5 Hz), and two methylenes at
δH 3.94 (
1H, d,
J = 16.4 Hz), 3.26 (
1H, d,
J = 16.4 Hz), 2.71 (
1H, dd,
J = 12.7, 6.5 Hz), 2.21 (
1H, d,
J = 12.7 Hz). The
13C NMR spectrum (
Table 2) displayed 18 carbon signals including two methylenes, ten methines (six aromatic/olefinic and three oxygenated or heteroatomic), and six nonprotonated carbons (two diagnostic amide carbonyl, two olenfinic and two heteroatomic). These NMR data of
1 (
Table 1 and
Table 2) showed similarity to those of spirobrocazines A–C [
5], which suggested that
1 was a thiodiketopiperazine alkaloid. Detailed analysis of HSQC, HMBC and COSY spectra (
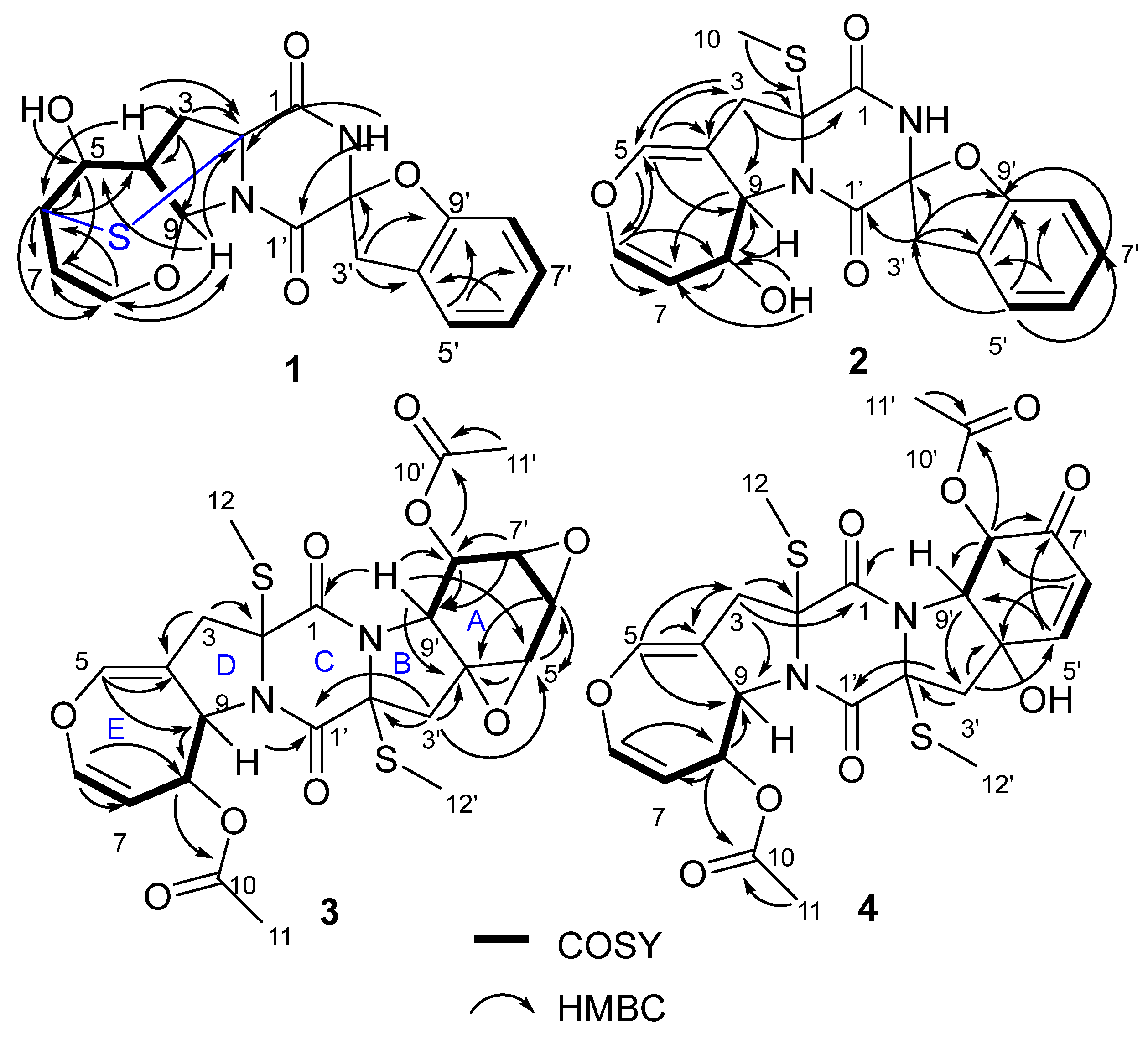
Figure 2) proved that
1 contained the same structural parts of rings A–C as those of spirobrocazines A–C. However, the COSY spectrum (
Figure 2) showing correlations from H-4 (
δH 3.06,
1H, t,
J = 6.5 Hz) to H
b-3 [
δH 2.71 (
1H, dd,
J = 12.7, 6.5 Hz)], H-5 (
δH 3.87,
1H, d,
J = 6.5 Hz) and H-9 (
δH 6.61,
1H, d,
J = 6.5 Hz), from H-5 to H-6 (
δH 4.86,
1H, ddd,
J = 7.8, 6.5, 1.2 Hz), and from H-7 (
δH 4.82,
1H, ddd,
J = 7.8, 6.5, 1.2 Hz) to H-6 and H-8 (
δH 6.00,
1H, d,
J = 6.5 Hz), and the HMBC spectrum (
Figure 2) showing correlations from H-4 to C-2 (
δC 70.8), C-3 (
δC 48.2), C-6 (
δC 52.5), from OH-5 to C-5 (
δC 69.9), from H-6 to C-2, C-4 (
δC 46.3), C-5, C-7, C-8 (
δC 144.9), from H-8 (
δH 6.00,
1H, d,
J = 6.5 Hz) to C-6, C-7 (
δC 100.6), C-9 (
δC 92.9), from H-9 to C-2, C-5, C-8, and from H-3 to C-2, C-4, C-9, suggested the presence and assignment of D/E rings in
1 as shown. Combining the molecular formula and the chemical shifts of C-2 and C-6/H-6, it was reasonable to infer a sulfide bond between C-2 and C-6 to form a sulfide six-membered-ring. Thus, the planar structure of
1 was determined as shown.
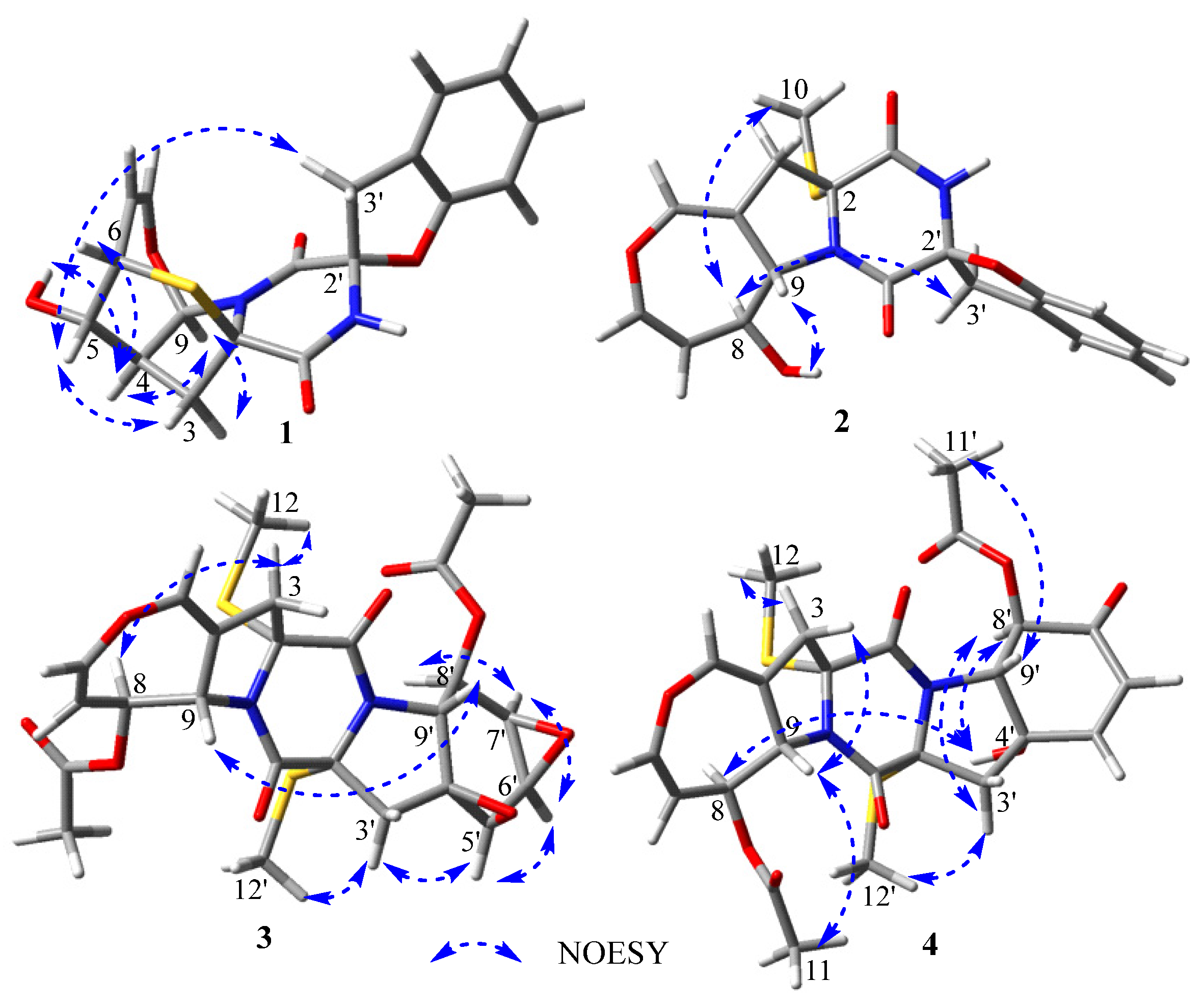
The relative configuration of
1 was elucidated by analysis of the NOESY spectrum (
Figure S6). The NOE correlations of H-4 with OH-5 and H-9, H-9 with H-3 (
δH 2.71) and H-4, and H-6 with H-4 (
Figure 3), indicated their cofacial β-orientations. The NOE correlation of H-5 with H
a-3 (
δH 2.21) indicated their cofacial
α-orientations. The NOE correlations from H-5 to H
2-3′ revealed their cofacial orientations and the CH
2-3′ group at the axial position. The coupling constant of
JH-7-H-8 = 6.5 Hz indicated that the double bond was a
Z-configuration. The absolute configuration of
1 was further determined by electronic circular dichroism (ECD) calculations. The calculated ECD spectrum of (2
R, 4
S, 5
S, 6
S, 9
R, 2′
S)
-1 showed similarity to the experimental ECD spectrum of
1 (
Figure 4), which confirmed that the absolute configuration of
1 was 2
R, 4
S, 5
S, 6
S, 9
R, 2′
S.
A possible biosynthetic pathway of
1 is shown in
Figure S66 [
5,
8]. The biosynthetic pathway of
1 likely starts with the cyclodipeptide (I) composed of two phenylalanines followed by oxidation to afford the key intermediate II, which could be transferred to III by dehydration and oxidation. Then cyclization and oxidation of III via intermediate IV could produce V. Finally, V could be translated into compound
1 by further oxidation, cyclization, and sulfurization.
Lecanicillium B (
2) was obtained as a white powder with the molecular formula C
19H
18N
2O
5S as determined by HRESIMS ([M + H]
+ m/
z 387.1009) and NMR data. The
1H and
13C NMR spectra of
2 (
Table 1 and
Table 2) showed similarity to those of spirobrocazine A [
5]. The obvious difference between them was the chemical shift changes of C-4, C-5, C-6, C-7, C-8, and C-9 (
δC 134.1 (C), 120.3 (CH), 124.0 (CH), 131.6 (CH), 75.1 (CH), and 70.4 (CH) in spirobrocazine A [
5], and correspondingly
δC 109.1 (C), 136.7 (CH), 137.9 (CH), 111.0 (CH), 71.2 (CH) and 64.1 (CH) in
2, respectively). The COSY spectrum (
Figure 2) showing the sequential correlations of H-6/H-7/H-8/H-9, and the HMBC spectrum (
Figure 2) showing correlations from H-5 (
δH 6.69,
1H, t,
J = 2.4 Hz) to C-3 (
δC 40.2), C-4 (
δC 109.1), C-6 (
δC 137.9), C-9 (
δC 64.1), from H-6 (
δH 6.29,
1H, dd,
J = 8.2, 2.4 Hz) to C-5 (
δC 136.7), C-7 (
δC 111.0), C-8 (
δC 71.2), from H-8 (
δH 4.46,
1H, ddt,
J = 7.7, 6.7, 2.1 Hz) to C-7, C-9, from OH-8 (
δH 5.28, d,
J = 6.7 Hz) to C-8, and from H-3 [
δH 2.91 (
1H, dt,
J = 15.3, 1.2 Hz), 3.14 (
1H, dt,
J = 15.3, 2.2 Hz)] to C-2 (
δC 69.2), C-4, C-5, C-9, suggested that the E ring in
2 was a seven-membered 4,5-dihydrooxepine ring, as shown, instead of a six-membered ring. The coupling constant of
JH-6-H-7 = 8.2 Hz indicated that the double bond was a
Z-configuration. In addition, the NOESY sepctrum of
2 (
Figure S15) also showed great similarity to that of spirobrocazine A. The NOE correlation of OH-8 with H-9 indicated their cofacial β-orientations, while the NOE correlations of H-8 with H
3-10 and H
a-3′ (
δH 4.01) indicated their cofacial
α-orientations and the CH
2-3′ group at the axial position (
Figure 3). Furthermore, the calculated ECD spectrum of (2
R, 8
S, 9
S, 2′
S)
-2 showed similarity to the experimental ECD spectrum of
2 (
Figure 4), indicating the absolute configuration of
2, as shown.
Lecanicillium C (
3) was isolated as a white powder with the molecular formula C
24H
26N
2O
9S
2 as determined by HRESIMS ([M + Na]
+ m/
z 573.0969) and NMR. The
1H and
13C NMR spectra of
3 (
Table 1 and
Table 2) showed similarity to those of bisdethiobis(methylsulfanyl)acetylapoaranotin [
8]. The obvious difference between them was the additional presence of three tertiary methine signals (
δH 3.83 (
1H, d,
J = 2.7 Hz), 3.67 (
1H, dd,
J = 4.3, 2.7 Hz), 3.34 (
1H, dd,
J = 4.3, 1.2 Hz);
δC 54.1, 53.6, 47.9) and one oxygenated nonprotonated carbon (
δC 60.9), and the disappearance of the signals for two double bonds in
3. Detailed analysis of HSQC, HMBC and COSY spectra (
Figure 2,
Figures S21–S23) proved that
3 contained the same structural parts of rings B–E as those of bisdethiobis(methylsulfanyl)acetylapoaranotin. Furthermore, the COSY spectrum (
Figure 2) showed correlations from H-5′ (
δH 3.83,
1H, d,
J = 2.7 Hz) to H-6′ (
δH 3.67,
1H, dd,
J = 4.3, 2.7 Hz), from H-6′ to H-7′ (
δH 3.34,
1H, dd,
J = 4.3, 1.2 Hz), and from H-8′ (
δH 5.79,
1H, dd,
J = 9.1, 1.2 Hz) to H-7′ and H-9′ (
δH 4.34,
1H, d,
J = 9.1 Hz), and the HMBC spectrum (
Figure 2) showed correlations from H-5′ to C-4′ (
δC 60.9) and C-6′ (
δC 47.9), from H-6′ to C-4′, C-5′ (
δC 53.6), from H-7′ to C-8′ (
δC 72.6), C-9′ (
δC 57.5), from H-8′ to C-9′, C-10′ (
δC 169.4), from H-9′ to C-1 (
δC 163.5), C-4′, C-5′, C-8′, from H-3′ [
δH 2.85 (
1H, d,
J = 14.4 Hz), 2.43 (
1H, d,
J = 14.4 Hz)] to C-1′ (
δC 163.6), C-2′ (
δC 71.7), C-4′, C-5′, and from H
3-11′ to C-10′. Combining with the molecular formula, these above data suggested that the A ring was a six-membered ring with an acetyl group attached at C-8′ and two oxygenated three-membered rings between C-4′ and C-5′, and between C-6′ and C-7′, respectively. The small coupling constants of
JH-5′-H-6′ (2.7 Hz),
JH-6′-H-7′ (4.3 Hz), and
JH-7′-H-8′ (1.2 Hz) suggested a
cis-diaxial relationship between H-5′ and H-6′, between H-6′ and H-7′, and between H-7′ and H-8′, respectively. NOE correlations of H-5′ with H-6′, H-6′ with H-7′ and H-7′ with H-8′ also indicated H-5′, H-6′, H-7′, and H-8′ on the same face. The large coupling constants of
JH-8-H-9 (8.1 Hz) and
JH-8′-H-9′ (9.1 Hz) suggested a
trans-diaxial relationship between H-8 and H-9, and between H-8′ and H-9′, respectively. Furthermore, the NOE correlations of H-3 (
δH 3.25) with H-8 and H
3-12 indicated the assignment of
α-orientation for H-8 and H
3-12, NOE correlations of H-3′ (
δH 2.43) with H-5′ and H
3-12′ indicated
α-orientation for H
3-12′ and H-5′, and NOE correlations of H-9 with H-9′ indicated β-orientation for H-9 and H-9′ (
Figure 3). Thus, the structure of
3 was inferred as shown. The absolute configuration of
3 was determined as 2
R, 8
S, 9
S, 2′
R, 4′
S, 5′
R, 6′
R, 7′
R, 8′
R, 9′
R by a single crystal X-ray diffraction analysis using Cu K
α radiation (
Figure 5), which was supported by the calculated ECD spectrum of (2
R, 8
S, 9
S, 2′
R, 4′
S, 5′
R, 6′
R, 7′
R, 8′
R, 9′
R)-
3 showing agreement with the experimental ECD spectrum of
3 (
Figure 4).
Lecanicillium D (
4) was isolated as a white solid with the molecular formula C
24H
26N
2O
9S
2 as determined by HRESIMS ([M + NH
4]
+ m/
z 568.1408). The
1H and
13C NMR spectra of
4 (
Table 1 and
Table 2) showed similarity to those of
3. The obvious difference between them was the presence of an
α, β-unsaturated ketone group (
δH 6.98 (
1H, d,
J = 10.3 Hz), 6.09 (
1H, d,
J = 10.3 Hz);
δC 191.5, 150.2, 125.8) and the disappearance of three tertiary methine signals in
4. The HMBC spectrum (
Figure 2) showing correlations from H-5′ (
δH 6.98,
1H, d,
J = 10.3 Hz) to C-7′ (
δC 191.5) and C-9′ (
δC 69.1), from H-6′ (
δH 6.09,
1H, d,
J = 10.3 Hz) to C-8′ (
δC 74.8) and C-4′ (
δC 75.5), from H-8′ (
δH 5.73,
1H, d,
J = 11.2 Hz) to C-7′ and C-10′, from H-3′ [
δH 2.94 (
1H, d,
J = 15.4 Hz), 3.16 (
1H, dd,
J = 15.4, 2.1 Hz)] to C-4′ and C-5′, and from H
3-11′ to C-10′, and the COSY spectrum (
Figure 2) showing correlations from H-5′ to H-6′, and from H-8′ to H-9′, revealed the existence of an
α, β-unsaturated ketone instead of two oxygenated three-membered rings in
4. The NOE correlation of H-9 with H
3-11 suggested the assignment of
α-oriention for H-8 and β-oriention for H-9, and the NOE correlations of H-3 (
δH 2.92) with H-9, and H-3 (
δH 3.20) with H
3-12 indicated H
3-12 was
α-oriented. The 11.2 Hz coupling constant between H-8′ and H-9′ and the NOE correlation of H-9′ with H
3-11′ suggested the assignment of
α-oriention for H-8′ and β-oriention for H-9′. The NOE correlation of OH-4′ with H-8′ indicated their cofacial
α-orientations. The NOE correlations of H-3′ (
δH 2.94) with H-8′ and H
3-12′ indicated H
3-12′ was also
α-oriented. The NOE correlations of H-8 with OH-4′ indicated that H-8 and OH-4′were
α-orientated. The absolute configuration of
4 was further determined by ECD calculation. The great similarity between the experimental ECD spectrum of
4 and the calculated ECD spectrum of (2
R, 8
S, 9
S, 2′
R, 4′
S, 8′
R, 9′
R)-
4 (
Figure 4) indicated the absolute configuration of
4 as shown.
Lecanicillium E (
5) was obtained as a white powder with the molecular formula C
24H
26N
2O
8S
3 as determined by HRESIMS ([M + NH
4]
+ m/
z 584.1189). The
1H and
13C NMR spectra of
5 (
Table 1 and
Table 2) showed great similarity to those of bisdethiobis(methylthio)acetylaranotin [
8]. The only obvious difference between them was the down-field shifts of H
3-12′ (
δH 2.47,
1H, s), C-12′ (
δC 24.1) and C-2′ (
δC 74.9) in
5. Combined with the molecular formula, these data indicated a disulfide methyl attached at C-2′ instead of a sulfide methyl in
5. Combined with the coupling constants of
JH-8′-H-9′ and
JH-8-H-9 (both 8.2 Hz), the NOE correlations of H-8 with H
3-12, H-9 with H
3-11, H-8′ with H
3-12′, and H-9′ with H
3-11′ indicated that H-8, H-8′, H
3-12′, H
3-12 were
α-oriented, and H-9, H-9′, H
3-11′, H
3-11 were β-oriented (
Figure S65). The absolute configuration of
5 was determined as shown by comparing the experimental ECD spectrum of
5 and the calculated ECD spectrum of (2
R, 8
S, 9
S, 2′
R, 8′
S, 9′
S)-
5 (
Figure 4), which was further supported by the great similarity of the ECD spectra of
5 with bisdethiobis(methylthio)acetylaranotin [
24].
Lecanicillium F (
6) was isolated as a white solid with the molecular formula C
19H
20N
2O
2S as determined by HRESIMS ([M + H]
+ m/
z 341.1316). The
1H and
13C NMR spectra of
6 (
Table 1 and
Table 2) exhibited great similarity to those of emethacin B [
17]. The only obvious difference between them was the disappearance of a sulfide methyl and the additional presence of a methine (
δC 54.8,
δH 3.26) instead of a nonprotonated carbon in
6. The COSY correlations of
δH 3.26 with H-4 and NH-1′ (
δH 8.23), and the HMBC correlations from
δH 3.26 to C-2 (
δC 166.2), C-4 (
δC 37.2), C-5 (
δC 135.9), from NH-1 (
δH 8.66) to C-2′ (
δC 164.8), C-3′ (
δC 68.2), C-4′ (
δC 44.4), and from NH-1′ to C-2, C-3′ (
Figure S63), suggested the methine was C-3 (
δC 54.8,
δH 3.26) in
6. The NOE correlations of H-3 with H
3-11′, and H-4 (
δH 2.70) with H-4′ (
δH 3.28) (
Figure S65) indicated the cofacial orientations for H-3 and H
3-11′, and H-4 (
δH 2.70) and H-4′ (
δH 3.28), respectively. The similarity between the experimental ECD spectrum of
6 and the calculated ECD spectrum of (3
S,3′
R)-
6 (
Figure 4) indicated the absolute configuration of
6 as shown.
Compounds
2–
19 were tested for their cytotoxicity towards human lung adenocarcinoma cell line H1975 and human hepatocellular carcinoma cell line HepG-2, and toxicity towards brine shrimps. The cytotoxicity results (
Table 3) showed that
5,
7, and
16 displayed significant cytotoxicity against H1975, with IC
50 values of 7.2~16.9 μM,
4,
11,
17, and
18 displayed mild cytotoxicity against H1975, with IC
50 values of 35.2~71.5 μM; only
18 had mild cytotoxicity against HepG-2, with an IC
50 value of 41.2 μM. The results indicated that the disulfide bond unit in
5 was an active group for its cytotoxicity, which is consistent with the conclusions reported in the literatures [
25,
26]. A comparison of the structures and cytotoxicities of
6,
7, and
19 suggested that lacking thiomethyl and benzene could significantly decrease the cytotoxicity of this type of alkaloids. In addition, a comparison of the structures and cytotoxicity of
2,
3,
4,
11, and
16 suggests the skeleton of A/B/C ring fragment also could significantly affect the cytotoxicity of this type of alkaloids. Furthermore, brine shrimp lethality assays (
Table 3) showed that only
4 and
15 exhibited medium toxicity, with TC
50 values of 40–50 μM, towards brine shrimps.
Compounds
1–
19 were also evaluated for their antibacterial activity towards eight pathogens:
Bacillus subtilis,
Micrococcus luteus,
Escherichia coli,
Staphylococcus aureus,
S. aureus MRSA,
Streptococcus agalactiae,
S. iniae, and
Pseudomonas aeruginosa. Antibacterial assays (
Table 3) exhibited that only
5 had significant antibacterial activity against
B. subtilis,
M. luteus,
S. agalactiae, and
S. iniae, with MIC values of 10~40 μg/mL. Other compounds showed mild or no obvious antibacterial activity. The results indicated that a disulfide bond unit at C-2′ was crucial for the antibacterial activity of this type of alkaloids.
3. Experimental Section
3.1. General Experimental Procedure
UV spectra were measured using a UV-2600 spectrophotometer (Shimadzu). IR spectra were obtained on an IR Affinity-1 Fourier transform infrared spectrophotometer (Shimadzu, Kyoto, Japan). ECD spectra were acquired on a Chirascan circular dichroism spectrometer (Applied Photophysics Ltd., Graz, Austria). Optical rotations were recorded using a MCP 500 polarimeter (Anton Paar). Melting points were recorded with a digital display microscopic melting point instrument (SGW X-5). NMR data were acquired with a Bruker AVANCE III HD 700 MHz NMR spectrometer (Bruker) with TMS as reference. HRESIMS spectroscopic data were obtained on a MaXis quadrupole-time-of-flight mass spectrometer (Bruker, Karlsruhe, Germany). Preparative reversed-phase HPLC was performed on a Shimadzu LC-20A preparative liquid chromatography system with a YMC-Pack ODS column (250 × 20 mm, S-5 μm, 12 nm). Sephadex LH-20 (GE Healthcare) was used for the chromatographic column (CC). RP-MPLC (reversed-phase-medium pressure preparative liquid chromatography) was carried out using the CHEETAH MP200 system (Agela Technologies, Tianjin, China) and Claricep Flash columns filled with ODS (40-63 μm, YMC). Silica gel (200–300 mesh) for CC and GF254 for TLC were purchased from Yantai Jiangyou Silica Gel Development Co., Ltd. Sea salts were commercially obtained from Guangzhou Hai Li Aquarium Technology Co., Ltd., Guangzhou, China.
3.2. Fungal Material
The fungus Lecanicillium kalimantanense was isolated from a mangrove sediment sample collected in the Bailu park, Sanya city, Hainan province. The strain was identified as Lecanicillium kalimantanense by internally transcribed spacer (ITS) region sequence data of the rDNA and given the Genbank accession number KM264285. The fungus L. kalimantanense was deposited in the RNAM Center, South China Sea Institute of Oceanology, Chinese Academy of Science.
3.3. Fermentation and Extraction
The spores of the fungus L. kalimantanense were added to 5 × 500 mL Erlenmeyer flasks, each containing 200 mL potato dextrose (PD) medium, and fermented for 3 days at 28 °C. Then 3 mL of spore suspension was transferred into 267 × 1 L Erlenmeyer flasks, each containing 300 mL culture media (glucose 1%, D-mannitol 2%, maltose 2%, corn meal 0.05%, monosodium glutamate 1%, KH2PO4 0.05%, MgSO4·7H2O 0.03%, yeast extract 0.3%, sea salt 3%). Static fermentation was performed for 26 days at 28 °C. After fermentation, the broth and mycelia were separated with gauze. The broth was extracted with EtOAc to obtain crude extract (28.9 g). The mycelia were extracted three times with acetone, and further extracted three times with EtOAc to yield a crude extract (58.3 g). Then the two crude extracts (28.9 g and 58.3 g) were combined for further isolation.
3.4. Isolation and Purification
The combined crude extract (87.2 g) was fractionated on a normal-phase column using a stepped gradient elution with CH2Cl2/MeOH (v/v, 100:0, 98:2, 95:5, 90:10, 85:15, 80:20, 70:30, 50:50, 0:100) to obtain eight fractions (Fr.1–Fr.8). Fr.3 (10.9 g) was separated with Sephadex LH-20 eluting with CH2Cl2/MeOH (1:1) to obtain eleven subfractions (Fr.3.1–Fr.3.11). Fr.3.3 was further purified by HPLC eluting with MeOH/H2O (v/v 7:3, 3 mL/min) to give 12 (9.0 mg, tR = 13.0 min). Fr.3.4 was further purified by HPLC eluting with MeOH/H2O (v/v 6:4, 3 mL/min) to give 8 (3.5 mg, tR = 29.0 min), 10 (11.0 mg, tR = 22.0 min), and 13 (9.0 mg, tR = 19.0 min). Fr.3.5 was further purified by HPLC eluting with MeOH/H2O (v/v 55:45, 3 mL/min) to give 5 (1.7 mg, tR = 43.0 min), 7 (1.1 mg, tR = 35.5 min), and 15 (2.6 mg, tR = 44.0 min). Fr.3.9 was further purified by HPLC eluting with MeOH/H2O (v/v 7:3, 3 mL/min) to give 9 (20.0 mg, tR = 10.0 min). Then Fr.3.6–Fr.3.8 were combined and further separated by ODS column eluting with MeOH/H2O (v/v 10:90–100:0) to obtain subfractions (Fr.3.6.1–Fr.3.6.22). Fr.3.6.6 was purified by HPLC eluting with MeOH/H2O (v/v 42.5:57.5, 2.5 mL/min) to give 6 (2.3 mg, tR = 32.0 min). Fr.3.6.8 was purified by HPLC eluting with MeOH/H2O (v/v 55:45, 2.5 mL/min) to give 3 (1.6 mg, tR = 26.0 min), 2 (1.5 mg, tR = 30.0 min), and 4 (2.6 mg, tR = 24.0 min). Fr.3.2.10 was purified by HPLC eluting with MeOH/H2O (v/v 58.5:41.5, 3 mL/min) to give 14 (3.0 mg, tR = 33.0 min). Fr.3.6.12 was purified by HPLC eluting with MeOH/H2O (v/v 59:41, 3 mL/min) to give 11 (6.5 mg, tR = 40.0 min) and 18 (3.4 mg, tR = 45.0 min). Fr.3.6.15 was further purified by HPLC eluting with MeOH/H2O (v/v 6:4, 2.5 mL/min) to give 16 (0.8 mg, tR = 75.0 min) and 17 (1.4 mg, tR = 68.0 min). Fr.4 (8.6 g) was separated with Sephadex LH-20 eluting with CH2Cl2/MeOH (1:1) to obtain eleven subfractions (Fr.4.1–Fr.4.3). Fr.4.2 was further separated by ODS column eluting with MeOH/H2O (v/v 10:90–100:0) to obtain subfractions (Fr.4.2.1–Fr.4.2.33). Fr.4.2.8 was purified by HPLC eluting with MeOH/H2O (v/v 9:11, 3 mL/min) to give 19 (2.0 mg, tR = 20.0 min). Fr.4.2.10 was purified by HPLC eluting with MeOH/H2O (v/v 9:11, 3 mL/min) to give 1 (1.5 mg, tR = 26.0 min).
Lecanicillium A (
1). White powder; [
α −86 (
c 0.10, MeOH); UV (MeOH)
λmax (log
ε) 212 (4.30), 277 (3.70), 283 (3.60) nm; ECD (0.33 mM, MeOH)
λmax (∆
ε) 200 (−9.00), 223 (16.86), 258 (−6.02) nm; IR (film)
νmax 3746, 3433, 3267, 2922, 2855, 2365, 2322, 2257, 1676, 1458, 1389, 1236, 1190, 1109, 1084, 1022, 995, 862, 760, 644, 596 cm
−1;
1H and
13C NMR data,
Table 1 and
Table 2; HR-ESIMS
m/
z 373.0851 [M + H]
+ (calcd for C
18H
17N
2O
5S, 373.0853).
Lecanicillium B (
2). White powder; [
α −168 (
c 0.10, MeOH); UV (MeOH)
λmax (log
ε) 210 (4.30), 276 (3.53), 283 (3.47) nm; ECD (0.32 mM, MeOH)
λmax (∆
ε) 200 (21.64), 201 (34.30), 229 (−34.46) nm; IR (film)
νmax 3341, 2922, 2841, 1676, 1649, 1545, 1514, 1460, 1410, 1113, 1018, 671, 598 cm
−1;
1H and
13C NMR data,
Table 1 and
Table 2; HR-ESIMS
m/
z 387.1009 [M + H]
+ (calcd for C
19H
19N
2O
5S, 387.1009).
Lecanicillium C (
3). colorless crystals; mp 172–174 °C; [
α −213 (
c 0.10, CH
3CN); UV (CH
3CN)
λmax (log
ε) 206 (4.50) nm; ECD (0.45 mM, CH
3CN)
λmax (∆
ε) 200 (−24.56), 201 (−17.74), 203 (−25.16), 214 (−11.98), 227 (−31.84), 247 (−3.68), 257 (−6.35) nm; IR (film)
νmax 3861, 3744, 3618, 2922, 2853, 1738, 1674, 1514, 1377, 1236, 1194, 1138, 1034, 972, 731, 648, 602 cm
−1;
1H and
13C NMR data,
Table 1 and
Table 2; HR-ESIMS
m/
z 573.0969 [M + Na]
+ (calcd for C
24H
26N
2NaO
9S
2, 573.0972).
Lecanicillium D (
4). White powder; [
α −206 (
c 0.10, MeOH); UV (MeOH)
λmax (log
ε) 207 (4.35) nm; ECD (0.45 mM, MeOH)
λmax (∆
ε) 200 (3.58), 226 (−33.79), 247 (−8.64), 256 (−10.55) nm; IR (film)
νmax 3331, 2945, 2920, 2841, 1740, 1659, 1535, 1514, 1460, 1398, 1113, 1018, 671, 598 cm
−1;
1H and
13C NMR data,
Table 1 and
Table 2; HR-ESIMS
m/
z 568.1408 [M + NH
4]
+ (calcd for C
24H
30N
3O
9S
2, 568.1418).
Lecanicillium E (
5). White powder; [
α −282 (
c 0.10, MeOH); UV (MeOH)
λmax (log
ε) 207 (4.36) nm; ECD (0.44 mM, MeOH)
λmax (∆
ε) 200 (9.37), 202 (−2.30), 225 (−61.07), 247 (−17.03), 256 (−19.43) nm; IR (film)
νmax 3347, 2951, 2924, 2843, 1734, 1670, 1375, 1302, 1236, 1192, 1130, 1018, 669, 598 cm
−1;
1H and
13C NMR data,
Table 1 and
Table 2; HR-ESIMS
m/
z 584.1189 [M + NH
4]
+ (calcd for C
24H
30N
3O
8S
3, 584.1190).
Lecanicillium G (
6). White powder; [
α −5 (
c 0.10, MeOH); UV (MeOH)
λmax (log
ε) 206 (4.08) nm; ECD (0.44 mM, MeOH)
λmax (∆
ε) 200 (−19.33), 201 (16.40), 204 (21.49), 228 (−10.86) nm; IR (film)
νmax 3354, 2943, 2833, 1670, 1653, 1558, 1541, 1506, 1472, 1456, 1418, 1115, 1020, 667, 599 cm
−1;
1H and
13C NMR data,
Table 1 and
Table 2; HR-ESIMS
m/
z 341.1316 [M + H]
+ (calcd for C
19H
21N
2O
2S, 341.1318).
3.5. X-ray Crystallographic Analysis of 3
Crystallographic data were collected on a Rigaku MicroMax 007 diffractometer (Rigaku Corporation, Tokyo, Japan) equipped with Cu Kα radiation and a graphite monochromator. The structure was solved by direct methods with the SHELXTL and refined by full-matrix, least-squares techniques. Crystallographic data for 3 were deposited with the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 2298244.
Crystal data for 3: C24H26N2O9S2·CH3OH, FW = 582.63; colorless crystal from MeOH; crystal size = 0.15 × 0.12 × 0.1 mm3; T = 100.00 (10) K; monoclinic, space group P21 (no. 4); unit cell parameters: a = 7.14328(5) Å, b = 16.25526(10) Å, c = 11.41675(7) Å, α = 90°, β = 93.4877(6)°, γ = 90°, V = 1323.211(14) Å3, Z = 2, Dcalc = 1.462 g/cm3, F (000) = 612.0, μ (CuKα)= 2.357 mm−1; 25678 reflections measured (7.758° ≤ 2θ ≤ 148.752°), 5292 unique (Rint = 0.0323, Rsigma = 0.0216), which were used in all calculations. The final R1 was 0.0242 (I > 2σ(I)) and wR2 was 0.0641 (all data). Flack parameter = −0.001(4).
3.6. ECD Calculations
The ECD calculations for
1–
6 were performed using the Gaussian 16 program package. The procedures were the same as described in our previous studies [
27]. The Spartan 14 program (Wavefunction Inc., Tokyo, Japan) was used for calculating the molecular Merck force field (MMFF). Density functional theory (DFT) and time-dependent density functional theory (TDDFT) calculations were performed using the Gaussian 16 program package. An MMFF model was used for the conformational search, then the conformers with lower relative energies (<10 kcal/mol) were subjected to geometry optimization with the DFT method at the B3LYP/6-31g (d) level in MeOH or CH
3CN. Vibrational frequency calculations were carried out at the same level to evaluate their relative thermal (ΔE) and free energies (ΔG) at 298.15 K. The geometry optimized conformers were further calculated at the M062X/def2TZVP level and the solvent (MeOH or CH
3CN) effects were taken into consideration by using SMD. The optimized conformers with a Boltzmann distribution of more than 1% population were further subjected to ECD calculation, which were performed by TDDFT methodology at the PBE1PBE/6-311g(d) level. The number of excited states was 60 for
1,
2, and
6, 85 for
3 and
4, and 102 for
5. The ECD spectra were generated by the software SpecDis using a Gaussian band shape with 0.20–0.30 eV exponential half-width from dipole-length dipolar and rotational strengths. The equilibrium population of each conformer at 298.15 K was calculated from its relative free energies using Boltzmann statistics. The calculated spectra of compounds
1–
6 were generated from the low-energy conformers according to the Boltzmann weighting of each conformer in MeOH solution for
1,
2,
4,
5, and
6 and CH
3CN for
3, respectively.
3.7. Cytotoxicity
Cytotoxic activity was evaluated using the human lung adenocarcinoma cell line H1975 and human hepatocellular carcinoma cell line HepG-2, using the CCK-8 method as described in our previous study [
28]. Briefly, each of the test compounds was dissolved in DMSO and further diluted to give final concentrations of 80, 40, 20, 10, 5, 2.5, and 1.25 μM, respectively. H1975 cells or HepG-2 cells (5
× 10
3 cells/plate) were seeded in 96-well plates and treated with compounds at the indicated concentration for 24 hours and then 10 μL CCK-8 reagent was added to each well. The plates were incubated at 37 °C for another 4 hours. Finally, the optical density was measured at a wavelength of 450 nm with a Bio-Rad microplate reader. Dose–response curves were generated, and the IC
50 values were calculated from the linear portion of log dose–response curves.
3.8. Brine Shrimp Lethality Bioassays
The methods were the same as those used in our previous studies [
27].
3.9. Antibacterial Assays
The micro broth dilution method [
29] was used to evaluate the antibacterial activities of compounds
1–
19 against the growth of eight common pathogens:
Bacillus subtilis BS01,
Micrococcus luteus, Escherichia coli ATCC 25922,
Staphylococcus aureus ATCC 6538,
S. aureus MRSA,
Streptococcus agalactiae ATCC 13813,
S. iniae, and
Pseudomonas aeruginosa ATCC 9027 in 96-well polystyrene plates. Vancomycin and ciprofloxacin were used as positive controls. Briefly, wells containing 100 μL bacteria dilutions (OD
600 = 0.01) in Luria–Bertani (LB) medium were supplemented with different concentrations of
1–
19 (80, 40, 20, 10, 5, 2.5, 1.25, and 0.625 μg/mL), vancomycin, and ciprofloxacin (40, 20, 10, 5, 2.5, 1.25, 0.625, and 0.3125 μg/mL), respectively. The MICs were determined after 24 hours’ incubation at 37 °C with the tested compounds.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}