
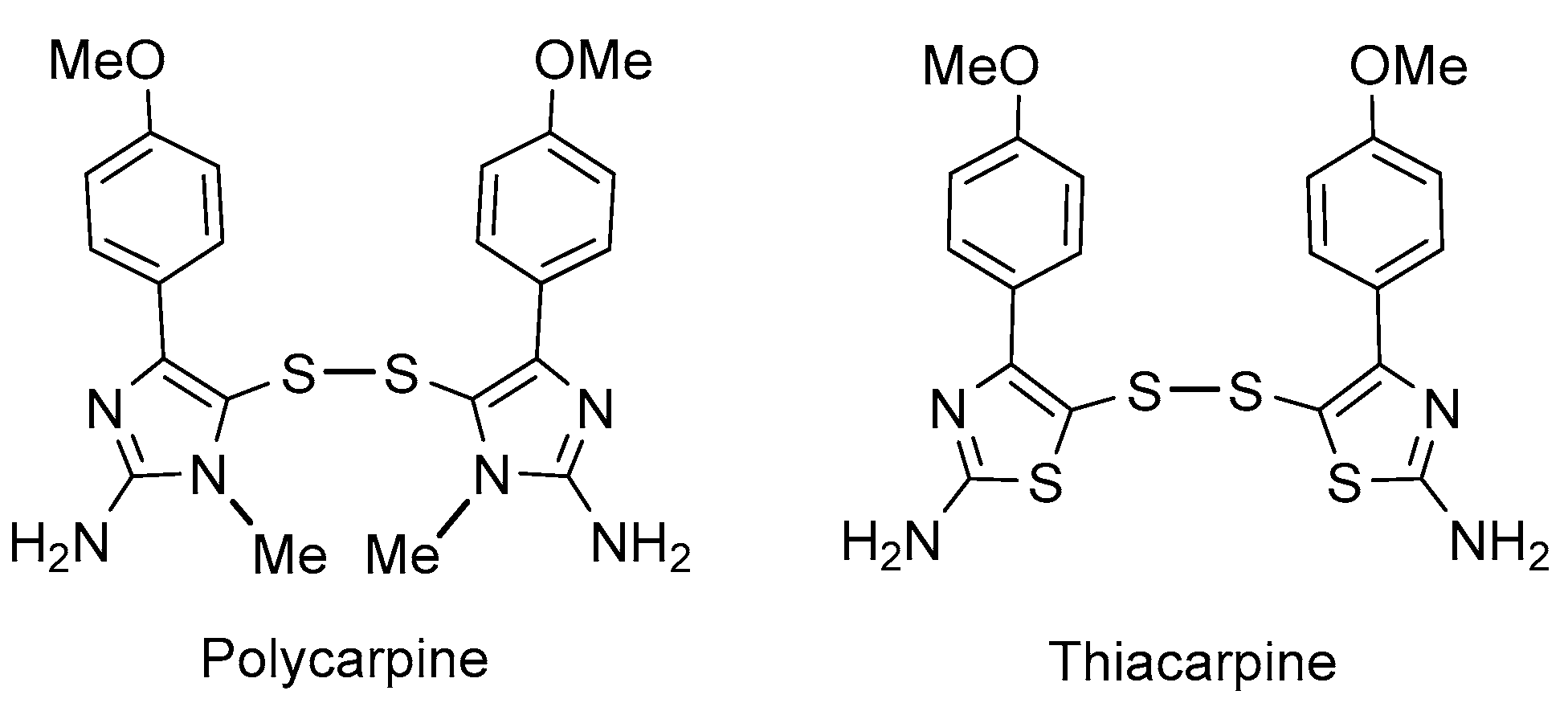
Synthesis and Biological Activity of Glycosyl Thiazolyl Disulfides Based on Thiacarpine, an Analogue of the Cytotoxic Alkaloid Polycarpine from the Ascidian Polycarpa aurata
 , , ,
, , ,
Abstract
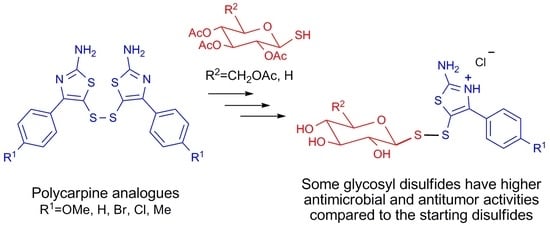
:
1. Introduction
2. Results and Discussion
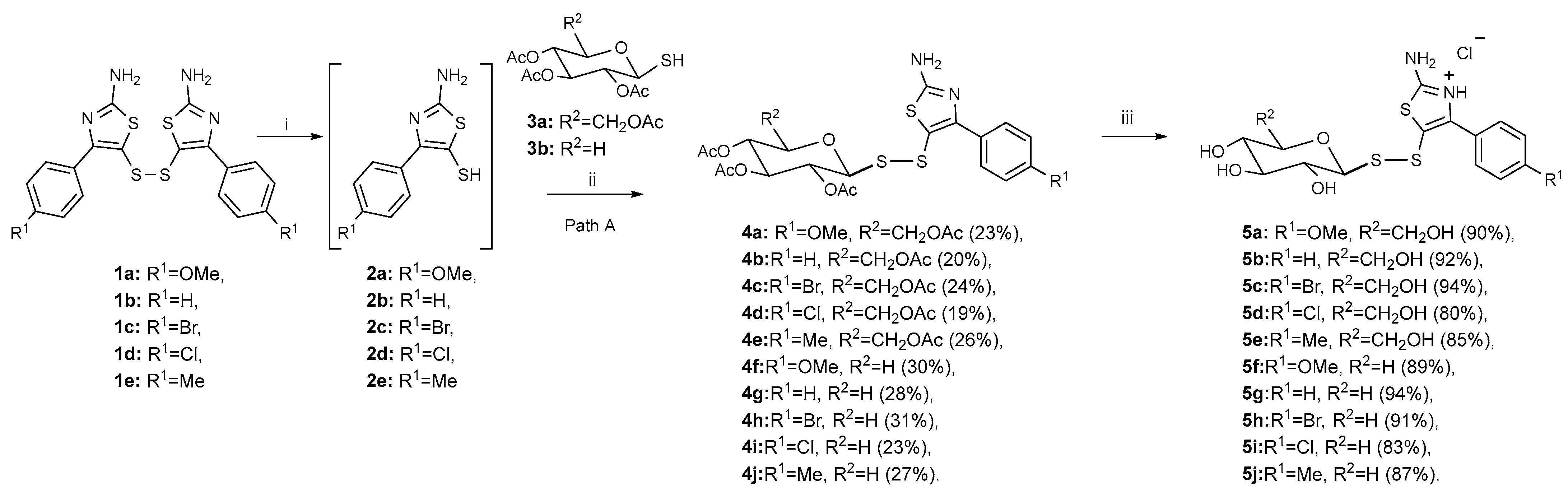
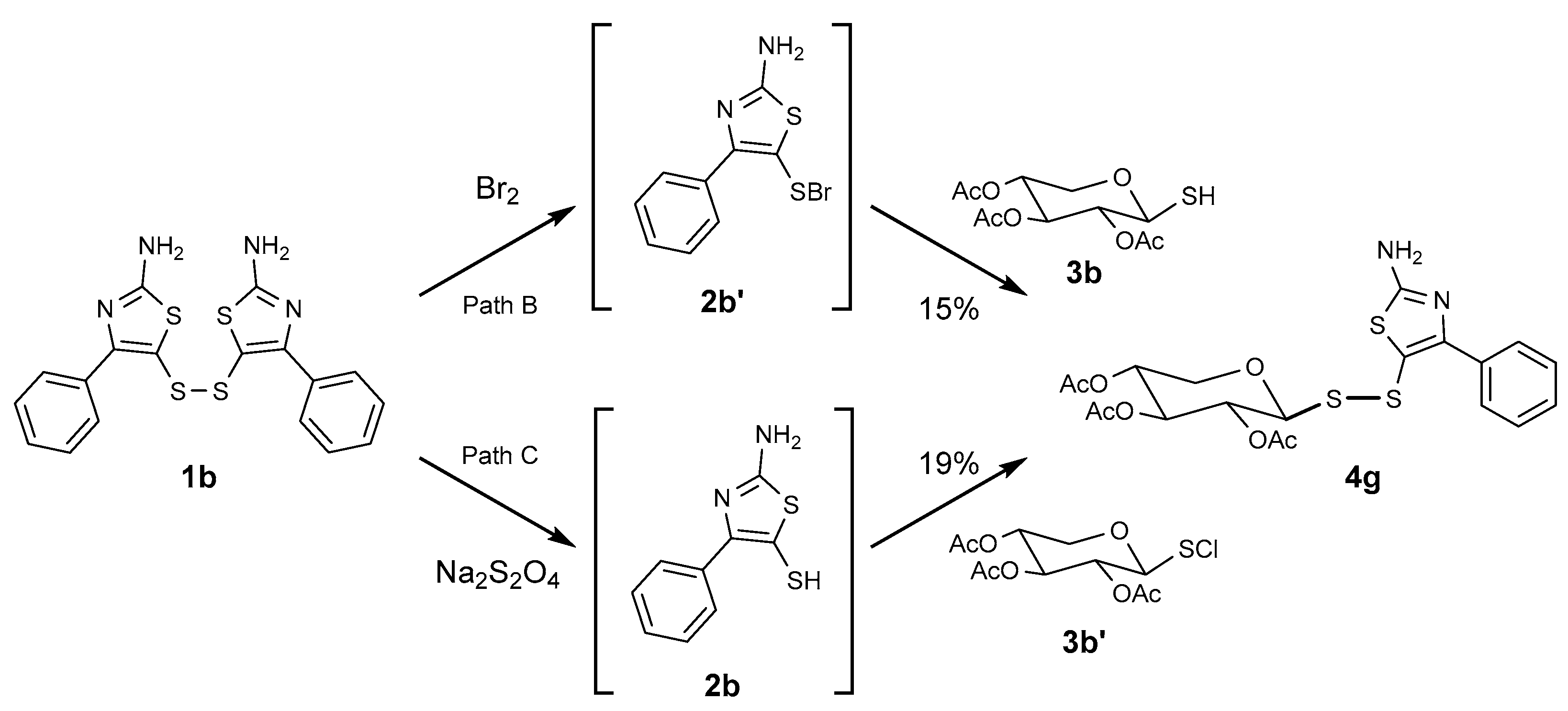
2.1. Synthesis of Glycosyl Disulfides
2.2. Biological Activity of the Compounds
2.2.1. Cytotoxic Activity
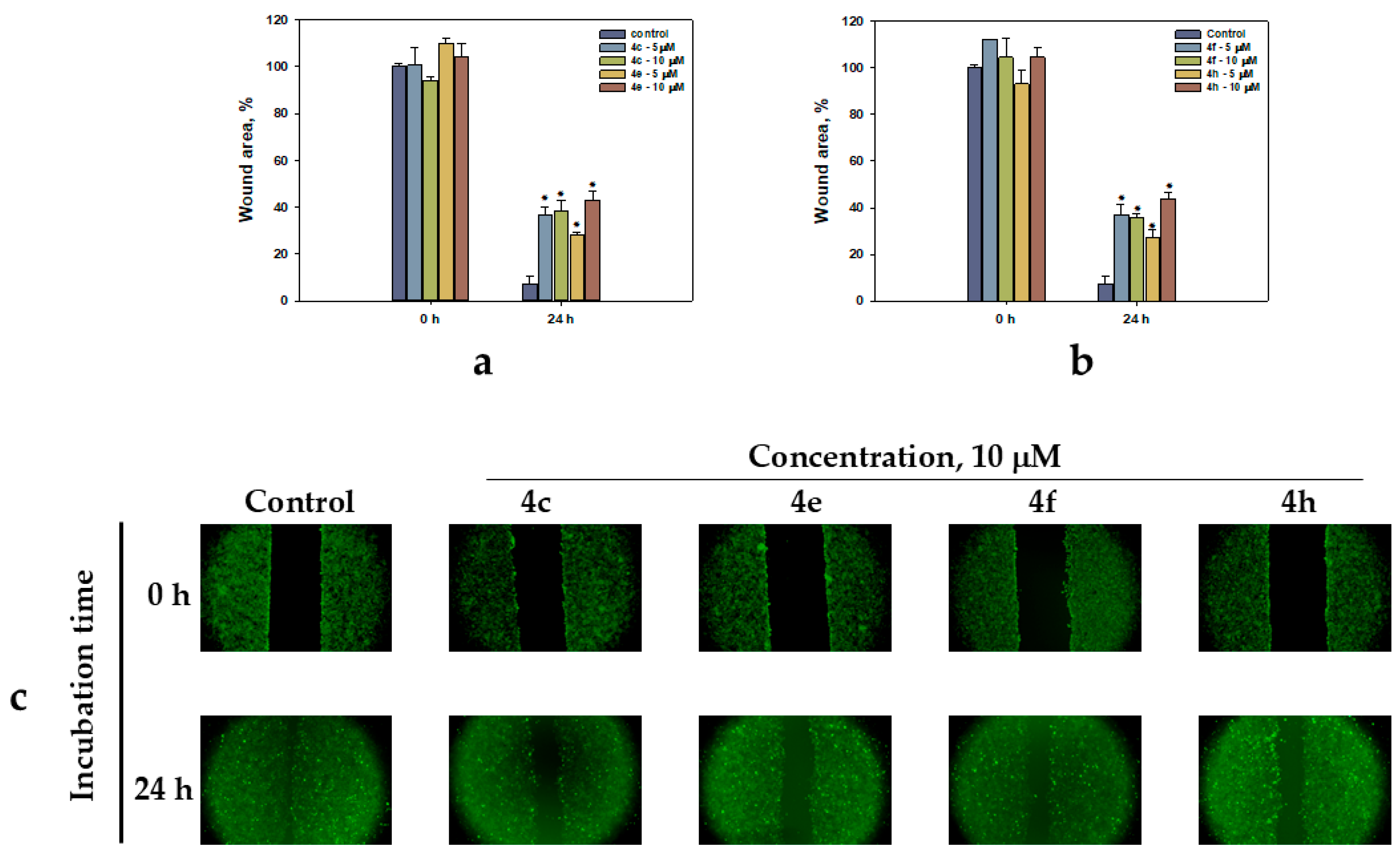
2.2.2. HeLa Cell Migration
2.2.3. Compounds Reduced the Number of Colonies Forming of HeLa Cells
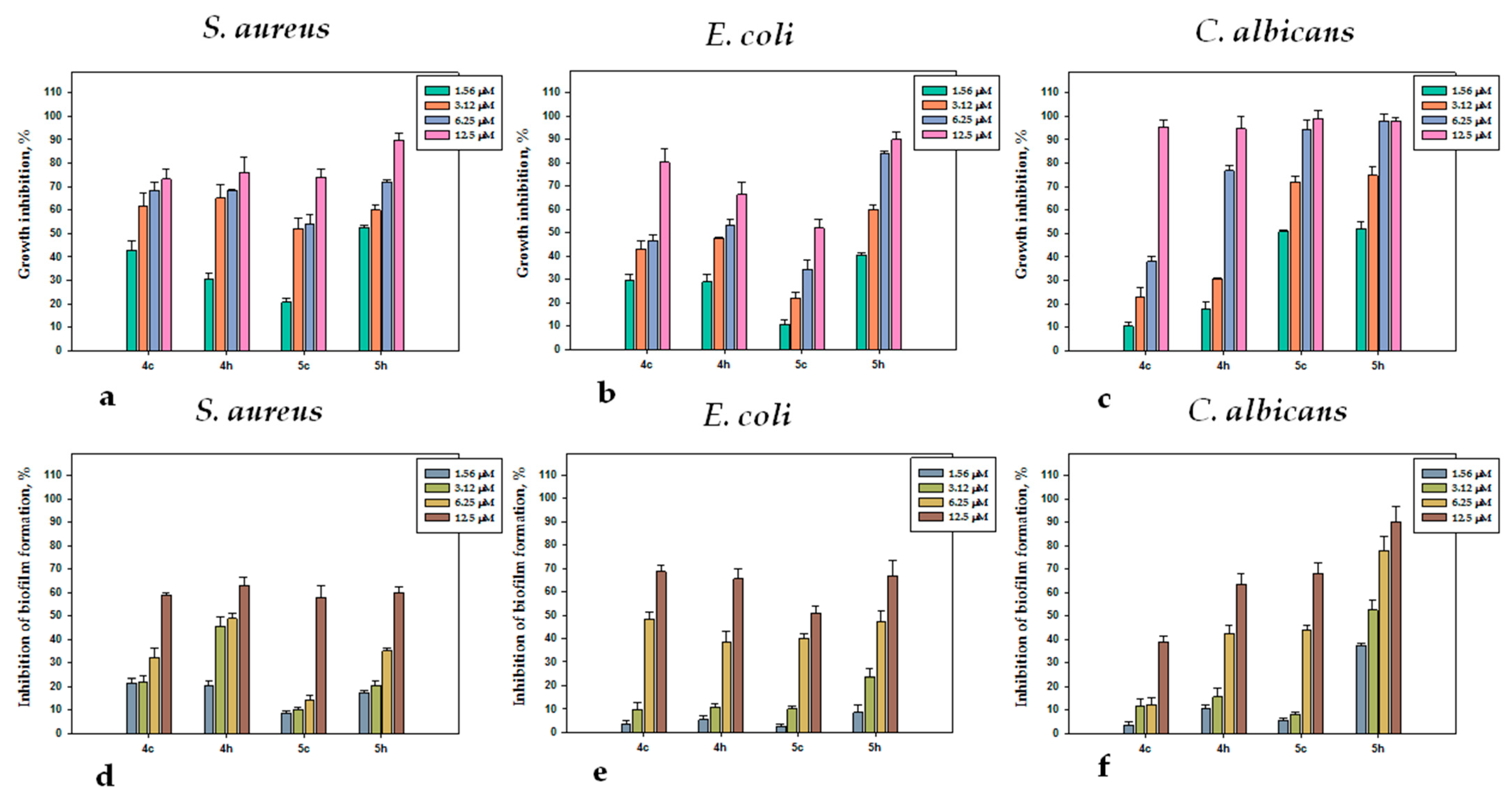
2.2.4. Antimicrobial Activity
3. Materials and Methods
3.1. General Information
3.1.1. General Procedure for the Synthesis of Acetylated Conjugates 4a–j
3.1.2. General Procedure for the Synthesis of Deacetylated Derivatives 5a–j
3.2. Cell Lines
3.3. MTT Assay to Determine the Effect on Cell Viability
3.4. Wound Scratch Migration Assay
3.5. Colony Formation Assay to Determine the Effect on Cell Proliferation
3.6. Antimicrobial Activity
3.7. Inhibition of Biofilm Formation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kang, H.; Fenical, W. Polycarpine dihydrochloride: A cytotoxic dimeric disulfide alkaloid from the Indian Ocean ascidian Polycarpa clavate. Tetrahedron Lett. 1996, 37, 2369–2372. [Google Scholar] [CrossRef]
- Fedoreyev, S.A.; Radchenko, O.S.; Novikov, V.L.; Isakov, V.V.; Ilyin, S.G.; Popov, A.M.; Elyakov, G.B.; Murphy, P.T.; Willis, R.H.; Baker, J.T. Polycarpine, a novel cytotoxic sulfur-containing alkaloid from the marine ascidian Polycarpa aurata and related compounds. In Proceedings of the Eighth International Symposium on Marine Natural Products, Tenerife, Canary Islands, Spain, 10–15 September 1995; pp. 196–197. [Google Scholar]
- Wang, W.; Oda, T.; Fujita, A.; Mangindaan, R.E.P.; Nakazawa, T.; Ukai, K.; Kobayashi, H.; Namikoshi, M. Three new sulfur-containing alkaloids, polycarpaurines A, B, and C, from an Indonesian ascidian Polycarpa aurata. Tetrahedron 2007, 63, 409–412. [Google Scholar] [CrossRef]
- Abas, S.A.; Hossain, M.B.; Helm, D.; Schmitz, F.J.; Laney, M.; Cabuslay, R.; Schatzman, R.C. Alkaloids from the Tunicate Polycarpa aurata from Chuuk Atoll. J. Org. Chem. 1996, 61, 2709–2712. [Google Scholar] [CrossRef]
- Popov, A.M.; Novikov, V.L.; Radchenko, O.S.; Elyakov, G.B. The cytotoxic and antitumor activities of the imidazole alkaloid polycarpin from the ascidian Polycarpa aurata and its synthetic analogues. Dokl. Biochem. Biophys. 2002, 385, 213–218. [Google Scholar] [CrossRef]
- Das, D.; Sikdar, C.; Bairagi, M. Recent developments of 2-aminothiazoles in medicinal chemistry. Eur. J. Med. Chem. 2016, 109, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Elsadek, F.M.; Ahmed, M.B.; Farahat, F.M. An overview on synthetic 2-aminothiazole—Based compounds associated with four biological activities. Molecules 2021, 26, 1449. [Google Scholar] [CrossRef]
- Radchenko, O.S.; Novikov, V.L.; Willis, R.H.; Murphy, P.T.; Elyakov, G.B. Synthesis of polycarpine, a cytotoxic sulfur-containing alkaloid from the ascidian Polycarpa aurata and related compounds. Tetrahedron Lett. 1997, 38, 3581–3584. [Google Scholar] [CrossRef]
- Wan, Y.; Long, J.; Gao, H.; Tang, Z. 2-Aminothiazole: A privileged scaffold for the discovery of anti-cancer agents. Eur. J. Med. Chem. 2021, 210, 112953. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, S.R.; Hashemi, S.M. Development and therapeutic potential of 2-aminothiazole derivatives in anticancer drug discovery. Med. Chem. Res. 2021, 30, 771–806. [Google Scholar] [CrossRef]
- Minickaite, R.; Grybaite, B.; Vaickelioniene, R.; Kavaliauskas, P.; Petraitis, V.; Petraitiene, R.; Tumosiene, I.; Jonuskiene, I.; Mickevicius, V. Synthesis of novel aminothiazole derivatives as promising antiviral, antioxidant and antibacterial candidates. Int. J. Mol. Sci. 2022, 23, 7688. [Google Scholar] [CrossRef]
- Siddiqui, H.L.; Zia-Ur-Rehman, M.; Ahmad, N.; Weaver, G.W.; Lucas, P.D. Synthesis and antibacterial activity of bis [2-amino-4-phenyl-5-thiazolyl] disulfides. Chem. Pharm. Bull. 2007, 55, 1014–1017. [Google Scholar] [CrossRef]
- Cascioferro, S.; Parrino, B.; Carbone, D.; Schillaci, D.; Giovannetti, E.; Cirrincione, G.; Diana, P. Thiazoles, their benzofused systems, and thiazolidinone derivatives: Versatile and promising tools to combat antibiotic resistance. J. Med. Chem. 2020, 63, 7923. [Google Scholar] [CrossRef] [PubMed]
- Lemilemu, F.; Bitew, M.; Demissie, T.B.; Eswaramoorthy, R.; Endale, M. Synthesis, antibacterial and antioxidant activities of thiazole-based schiff base derivatives: A combined experimental and computational study. BMC Chem. 2021, 15, 67. [Google Scholar] [CrossRef]
- Siddiqui, N.; Ahsan, W. Synthesis, anticonvulsant and toxicity screening of thiazolyl–thiadiazole derivatives. Med. Chem. Res. 2011, 20, 261–268. [Google Scholar] [CrossRef]
- Grewal, A.S.; Kharb, R.; Prasad, D.N.; Dua, J.S.; Lather, V. Design synthesis and evaluation of novel 3,5-disubstituted benzamide derivatives as allosteric glucokinase activators. BMC Chem. 2019, 13, 2. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Godoy, A.; Gever, J.; Fife, K.L.; Silber, B.M.; Prusiner, S.B.; Renslo, A.R. 2-Aminothiazoles as therapeutic leads for prion diseases. J. Med. Chem. 2011, 54, 1010–1021. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.J.; Patel, S.G.; Upadhyay, D.B.; Ravi, L.; Dhanasekaran, A.; Patel, H.M. An efficient, catalyst-free and aqueous ethanol-mediated synthesis of 5-((2-aminothiazol-5-yl)(phenyl)methyl)-6-hydroxypyrimidine-2,4(1H,3H)-dione derivatives and their antioxidant activity. RSC Adv. 2023, 13, 24466. [Google Scholar] [CrossRef]
- Kayagil, I.; Demirayak, S. Synthesis and anticancer activities of some thiazole derivatives. Phosphorus Sulfur Silicon 2009, 184, 2197–2207. [Google Scholar] [CrossRef]
- Lee, Y.-S.E.; Chuang, S.-H.; Huang, L.Y.L.; Lai, C.-L.; Lin, Y.-H.; Yang, J.-Y.; Liu, C.-W.; Yang, S.-C.; Lin, H.-S.; Chang, C.-C.; et al. Discovery of 4-aryl-N-arylcarbonyl-2-aminothiazoles as Hec1/Nek2 inhibitors. Part I: Optimization of in vitro potencies and pharmacokinetic properties. J. Med. Chem. 2014, 57, 4098–4110. [Google Scholar] [CrossRef]
- Liao, M.; Huang, D.; Cheng, Y.-X. 2-Amino-4-(aminomethyl)thiazole-based derivatives as potential antitumor agents: Design, synthesis, cytotoxicity and apoptosis inducing activities. Mendeleev Commun. 2023, 33, 73–76. [Google Scholar] [CrossRef]
- Turos, E.; Revell, K.D.; Ramaraju, P.; Gergeres, D.A.; Greenhalgh, K.; Young, A.; Sathyanarayan, N.; Dickey, S.; Lim, D.; Alhamadsheh, M.M.; et al. Unsymmetric aryl-alkyl disulfide growth inhibitors of methicillin-resistant Staphylococcus aureus and Bacillus anthracis. Bioorg. Med. Chem. 2008, 16, 6501–6508. [Google Scholar] [CrossRef]
- Choi, K.J.; Yu, Y.G.; Hahn, H.G.; Choi, J.D.; Yoon, M.Y. Characterization of acetohydroxyacid synthase from Mycobacterium tuberculosis and the identification of its new inhibitor from the screening of a chemical library. FEBS Lett. 2005, 579, 4903–4910. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.J.; Noh, K.M.; Kim, D.E.; Ha, B.H.; Kim, E.E.; Yoon, M.Y. Identification of the catalytic subunit of acetohydroxyacid synthase in Haemophilus influenzae and its potent inhibitors. Arch. Biochem. Biophys. 2007, 466, 24–30. [Google Scholar] [CrossRef]
- Sudarikov, D.V.; Gyrdymova, Y.V.; Borisov, A.V.; Lukiyanova, J.M.; Rumyantcev, R.V.; Shevchenko, O.G.; Baidamshina, D.R.; Zakarova, N.D.; Kayumov, A.R.; Sinegubova, E.O.; et al. Synthesis and Biological Activity of Unsymmetrical Monoterpenylhetaryl Disulfides. Molecules 2022, 27, 5101. [Google Scholar] [CrossRef]
- Sepúlveda, C.S.; García, C.C.; Levingston Macleod, J.M.; López, N.; Damonte, E.B. Targeting of arenavirus RNA synthesis by a carboxamide-derivatized aromatic disulfide with virucidal activity. PLoS ONE 2013, 8, e81251. [Google Scholar] [CrossRef]
- Lara, H.H.; Ixtepan-Turrent, L.; Garza-Treviño, E.N.; Flores-Teviño, S.M.; Borkow, G.; Rodriguez-Padilla, C. Antiviral propierties of 5, 5′-dithiobis-2-nitrobenzoic acid and bacitracin against T-tropic human immunodeficiency virus type 1. Virol. J. 2011, 8, 137. [Google Scholar] [CrossRef]
- García, C.C.; Topisirovic, I.; Djavani, M.; Borden, K.L.; Damonte, E.B.; Salvato, M.S. An antiviral disulfide compound blocks interaction between arenavirus Z protein and cellular promyelocytic leukemia protein. Biochem. Biophys. Res. Commun. 2010, 393, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bao, B.; Song, G.; Chen, C.; Zhang, X.; Lu, W.; Wang, Z.; Cai, Y.; Li, S.; Fu, S. Discovery of unsymmetrical aromatic disulfides as novel inhibitors of SARS-CoV main protease: Chemical synthesis, biological evaluation, molecular docking and 3D-QSAR study. Eur. J. Med. Chem. 2017, 137, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Morais, G.R.; Falconer, R.A. Glycosyl disulfides: Importance, synthesis and application to chemical and biological systems. Org. Biomol. Chem. 2021, 19, 82–100. [Google Scholar] [CrossRef]
- Vrudhula, V.M.; MacMaster, J.F.; Li, Z.; Kerr, D.E.; Senter, P.D. Reductively activated disulfide prodrugs of paclitaxel. Bioorg. Med. Chem. Lett. 2002, 12, 3591–3594. [Google Scholar] [CrossRef]
- Calvaresi, E.C.; Hergenrother, P.J. Glucose conjugation for the specific targeting and treatment of cancer. Chem. Sci. 2013, 4, 2319–2333. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, S.; Ahmad, S.; Gao, Q. Targeting key transporters in tumor glycolysis as a novel anticancer strategy. Curr. Top. Med. Chem. 2018, 18, 454–466. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Yang, J.; Seeberger, P.H.; Yin, J. Glycoconjugates for glucose transporter-mediated cancer-specific targeting and treatment. Carbohydr. Res. 2020, 498, 108195. [Google Scholar] [CrossRef] [PubMed]
- McClintock, M.K.; Zhang, K. Chapter 8: Xylose Metabolism and Its Metabolic Engineering Applications. In Engineering Microbial Metabolism for Chemical Synthesis. Reviews and Perspectives; Yan, Y., Ed.; World Scientific: Singapore, 2018; pp. 209–235. [Google Scholar] [CrossRef]
- Cheng, J.; Miller, C.J. Quantum interference effects in self-assembled asymmetric disulfide monolayers: Comparisons between experiment and ab initio/Monte Carlo theories. J. Phys. Chem. B 1997, 101, 1058–1062. [Google Scholar] [CrossRef]
- Yang, F.; Wang, W.; Li, K.; Zhao, W.; Dong, X. Efficient one-pot construction of unsymmetrical disulfide bonds with TCCA. Tetrhedron Lett. 2017, 58, 218–222. [Google Scholar] [CrossRef]
- Ishigeev, R.S.; Potapov, V.A.; Skurchenko, I.V.; Khabibulina, A.G.; Amosova, S.V. Synthesis of new polycyclic compounds via the reaction of quinoline-8-sulfenyl halides with cyclic alkenes. Chem. Heterocycl. Comp. 2021, 57, 314–319. [Google Scholar] [CrossRef]
- Walencka, E.; Sadowska, B.; Rózalska, S.; Hryniewicz, W.; Rózalska, B. Lysostaphin as a potential therapeutic agent for staphylococcal biofilm eradication. Pol. J. Microbiol. 2005, 54, 191–200. [Google Scholar]
- Fujihira, T.; Chida, M.; Kamijo, H.; Takido, T.; Seno, M. Novel synthesis of 1-thioglycopyranoses via thioiminium salts. J. Carbohyd. Chem. 2002, 21, 287–292. [Google Scholar] [CrossRef]
- Zhuravleva, O.I.; Chingizova, E.A.; Oleinikova, G.K.; Starnovskaya, S.S.; Antonov, A.S.; Kirichuk, N.N.; Menshov, A.S.; Popov, R.S.; Kim, N.Y.; Berdyshev, D.V.; et al. Anthraquinone derivatives and other aromatic compounds from marine fungus Asteromyces cruciatus KMM 4696 and their effects against Staphylococcus aureus. Mar. Drugs 2023, 21, 431. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cell Lines, EC50, µM | |||
|---|---|---|---|---|
| HeLa | Hep G2 | MDA-MB-231 (TNBC) | BEAS 2B | |
| 1a | >25 | >25 | 21.4 | >25 |
| 4a | >25 | >25 | >25 | 19.9 |
| 4b | >25 | >25 | 24.1 | 19.0 |
| 4c | 19.4 | 24.3 | 21.7 | 15.5 |
| 4d | >25 | >25 | 22.9 | 14.1 |
| 4e | 18.5 | >25 | 15.0 | >25 |
| 4f | 20.4 | 22.6 | 22.9 | 23.5 |
| 4h | 21.3 | 24.6 | 21.1 | 19.9 |
| 4j | >25 | >25 | 23.7 | >25 |
| 5a | >25 | >25 | 24.9 | >25 |
| 5b | >25 | >25 | >25 | 20.8 |
| 5e | >25 | >25 | 23.5 | 22.6 |
| 5g | >25 | >25 | >25 | 22.0 |
| 5h | >25 | >25 | >25 | 23.5 |
| cisplatin | 10.2 | 14.8 | 81.5 | 63.3 |
| Compound | Inhibition of Microbial Growth MIC50, µM | Inhibition of Biofilm Formation MIC50, µM | ||||
|---|---|---|---|---|---|---|
| S. aureus | E. coli | C. albicans | S. aureus | E. coli | C. albicans | |
| 1a | 10.3 ± 1.1 | >50 | 38.2 ± 3.2 | 34.5 ± 3.7 | >50 | 35.3 ± 3.4 |
| 1b | 5.2 ± 0.5 | 13.9 ± 1.3 | 6.2 ± 0.6 | 17.3 ± 1.1 | 5.9 ± 0.5 | 11.5 ± 1.1 |
| 1c | 3.4 ± 0.4 | 15.7 ± 2.3 | 3.1 ± 0.4 | >50 | >50 | 12.4 ± 2.5 |
| 1d | 2.8 ± 0.2 | 30.9 ± 3.0 | 5.0 ± 0.4 | >50 | >50 | 25.1 ± 3.1 |
| 1e | 30.7 ± 2.1 | 33.9 ± 3.9 | 19.5 ± 2.0 | 34.0 ± 3.1 | 31.6 ± 3.6 | 23.4 ± 2.6 |
| 4a | 32.5 ± 3.2 | >50 | 31.5 ± 3.1 | 36.2 ± 3.1 | >50 | 22.1 ± 2.4 |
| 4b | 28.4 ± 3.1 | 31.2 ± 2.0 | 20.5 ± 2.7 | 27.4 ± 1.5 | 34.3 ± 3.4 | 15.9 ± 1.0 |
| 4c | 2.3 ± 0.3 | 6.9 ± 0.5 | 7.1 ± 0.7 | 7.9 ± 0.5 | 8.0 ± 0.8 | 14.1 ± 1.3 |
| 4d | 19.2 ± 2.0 | 16.1 ± 1.7 | 6.9 ± 0.9 | 16.7 ± 1.1 | 17.0 ± 1.2 | 7.0 ± 0.3 |
| 4e | >50 | >50 | 17.7 ± 0.3 | 25.1 ± 2.3 | 27.1 ± 2.1 | 18.3 ± 1.5 |
| 4f | 32.1 ± 3.7 | 39.5 ± 4.0 | 34.1 ± 2.1 | 35.4 ± 2.1 | 23.2 ± 2.0 | 22.1 ± 2.1 |
| 4g | 34.0 ± 1.6 | 42.1 ± 4.1 | 35.7 ± 3.0 | 36.0 ± 3.1 | 38.1 ± 3.0 | 37.5 ± 4.0 |
| 4h | 2.4 ± 0.2 | 5.8 ± 0.4 | 4.3 ± 0.5 | 6.3 ± 0.5 | 10.0 ± 0.9 | 7.1 ± 0.4 |
| 4i | 3.1 ± 0.3 | 8.7 ± 0.9 | 2.8 ± 0.3 | 6.3 ± 0.4 | 13.5 ± 1.2 | 4.1 ± 0.1 |
| 4j | 25.0 ± 1.4 | 25.5 ± 2.0 | 18.1 ± 1.0 | 19.4 ± 1.3 | 14.2 ± 1.0 | 13.5 ± 1.6 |
| 5c | 3.0 ± 0.2 | 5.0 ± 0.5 | 1.6 ± 0.2 | 10.7 ± 1.1 | 12.4 ± 1.1 | 8.7 ± 0.7 |
| 5d | 2.5 ± 0.2 | 6.6 ± 0.5 | 3.1 ± 2.3 | 18.6 ± 1.3 | 6.0 ± 0.6 | 10.9 ± 1.0 |
| 5e | >50 | >50 | 38.5 ± 3.4 | >50 | >50 | 17.3 ± 0.4 |
| 5f | >50 | >50 | 36.2 ± 2.1 | >50 | >50 | 16.1 ± 0.4 |
| 5g | >50 | 24.9 ± 2.2 | 25.3 ± 1.0 | >50 | 42.1 ± 3.5 | 38.1 ± 3.0 |
| 5h | 1.2 ± 0.1 | 3.1 ± 0.3 | 1.6 ± 0.1 | 11.4 ± 1.0 | 8.9 ± 0.7 | 3.2 ± 0.3 |
| 5i | 13.7 ± 1.1 | 21.0 ± 3.0 | 13.8 ± 1.2 | 20.1 ± 2.0 | 12.1 ± 1.1 | 25.0 ± 2.0 |
| 5j | 34.5 ± 3.0 | 38.0 ± 3.0 | 15.9 ± 1.1 | 38.0 ± 3.6 | 5.6 ± 0.6 | 24.1 ± 2.1 |
| Gentamicin | 4.3 ± 0.8 | 5.1 ± 0.7 | – | 4.6 ± 0.5 | 5.0 ± 0.5 | – |
| Nitrofungin | – | – | 4.1 ± 0.5 | – | – | 4.3 ± 0.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pelageev, D.N.; Sabutski, Y.E.; Kovach, S.M.; Balaneva, N.N.; Menchinskaya, E.S.; Chingizova, E.A.; Burylova, A.L.; Anufriev, V.P. Synthesis and Biological Activity of Glycosyl Thiazolyl Disulfides Based on Thiacarpine, an Analogue of the Cytotoxic Alkaloid Polycarpine from the Ascidian Polycarpa aurata. Mar. Drugs 2025, 23, 117. https://doi.org/10.3390/md23030117
Pelageev DN, Sabutski YE, Kovach SM, Balaneva NN, Menchinskaya ES, Chingizova EA, Burylova AL, Anufriev VP. Synthesis and Biological Activity of Glycosyl Thiazolyl Disulfides Based on Thiacarpine, an Analogue of the Cytotoxic Alkaloid Polycarpine from the Ascidian Polycarpa aurata. Marine Drugs. 2025; 23(3):117. https://doi.org/10.3390/md23030117
Chicago/Turabian StylePelageev, Dmitry N., Yuri E. Sabutski, Svetlana M. Kovach, Nadezhda N. Balaneva, Ekaterina S. Menchinskaya, Ekaterina A. Chingizova, Anna L. Burylova, and Victor Ph. Anufriev. 2025. "Synthesis and Biological Activity of Glycosyl Thiazolyl Disulfides Based on Thiacarpine, an Analogue of the Cytotoxic Alkaloid Polycarpine from the Ascidian Polycarpa aurata" Marine Drugs 23, no. 3: 117. https://doi.org/10.3390/md23030117
APA StylePelageev, D. N., Sabutski, Y. E., Kovach, S. M., Balaneva, N. N., Menchinskaya, E. S., Chingizova, E. A., Burylova, A. L., & Anufriev, V. P. (2025). Synthesis and Biological Activity of Glycosyl Thiazolyl Disulfides Based on Thiacarpine, an Analogue of the Cytotoxic Alkaloid Polycarpine from the Ascidian Polycarpa aurata. Marine Drugs, 23(3), 117. https://doi.org/10.3390/md23030117