Role of Manganese Oxyhydroxides in the Transport of Rare Earth Elements Along a Groundwater Flow Path



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Regional Hydrogeological Settings
2.2. Groundwater Sample Collection
2.3. Groundwater Sample Analysis
2.4. Speciation Modeling
3. Results
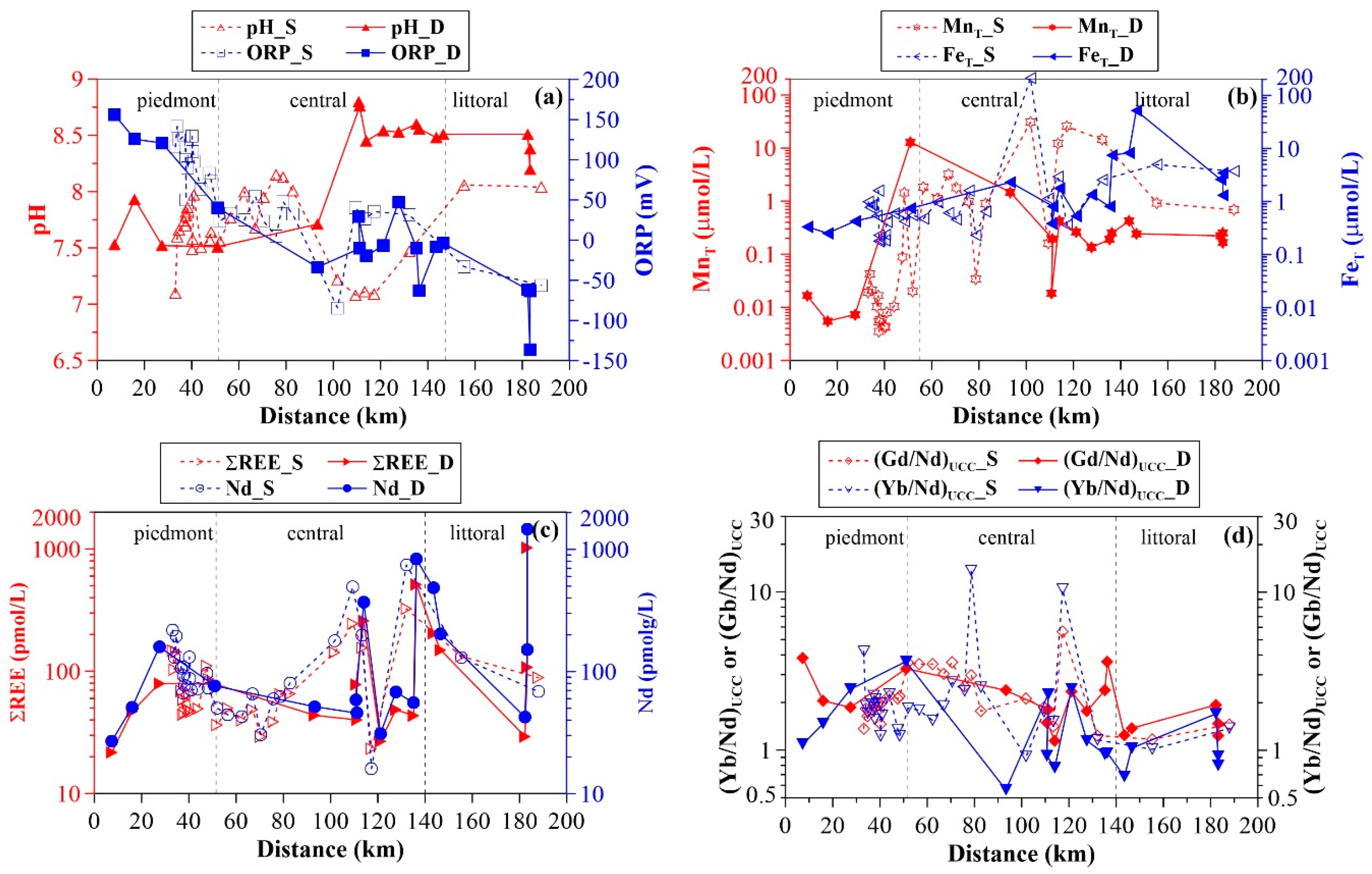
3.1. Groundwater General Chemistry
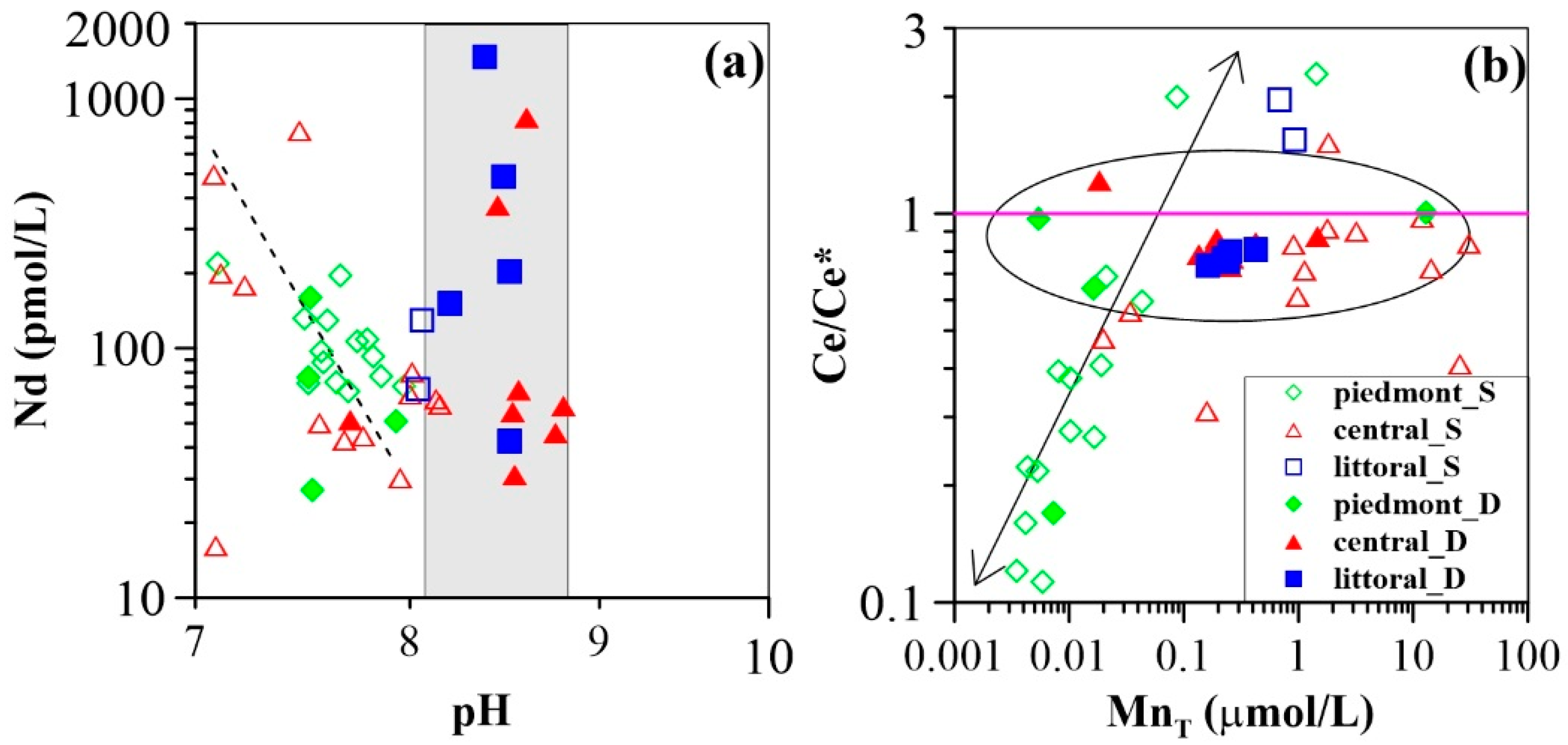
3.2. Groundwater REE Signatures
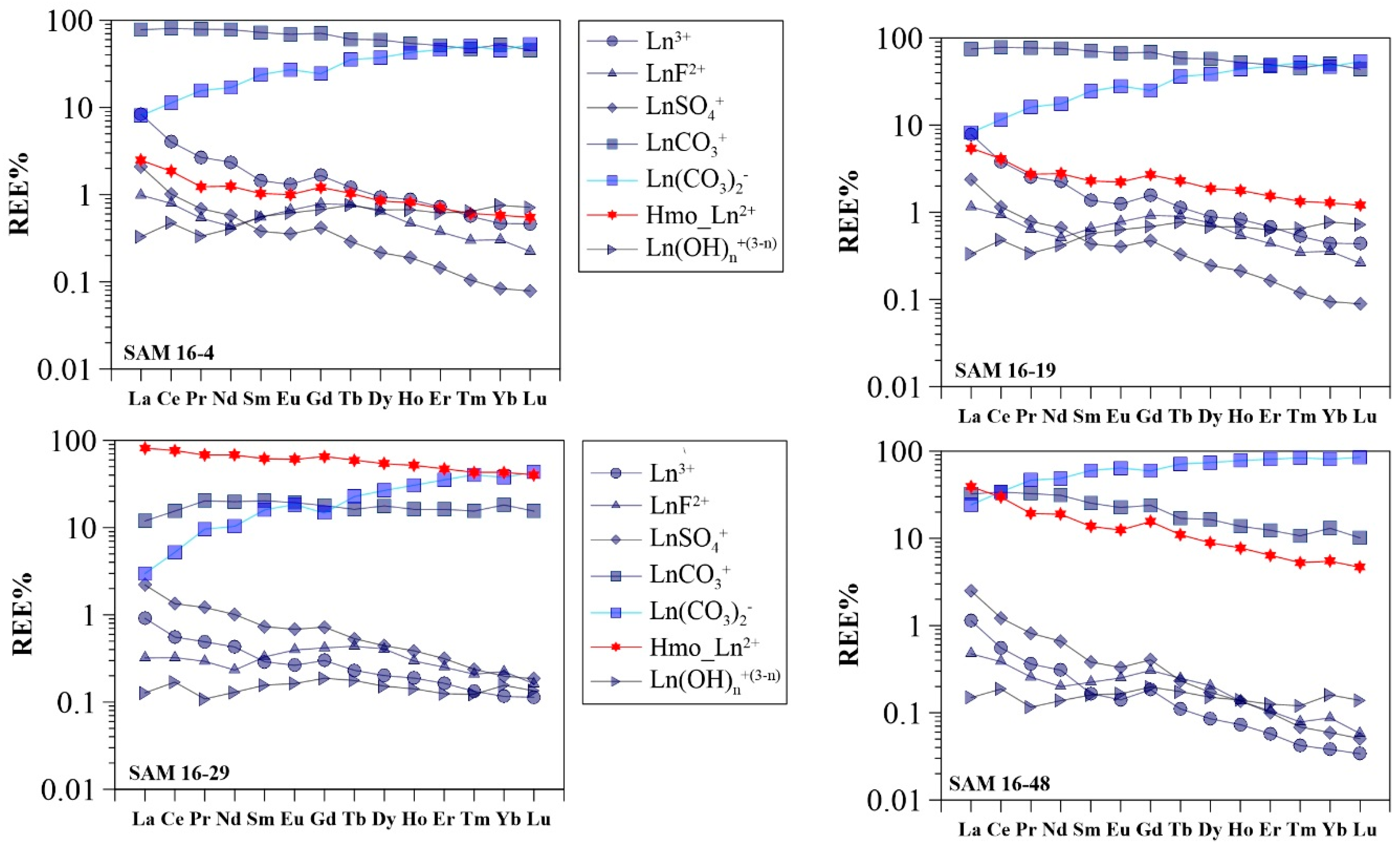
3.3. Modeling Results
4. Discussion
4.1. Groundwater REE Mobility along the Flow Path
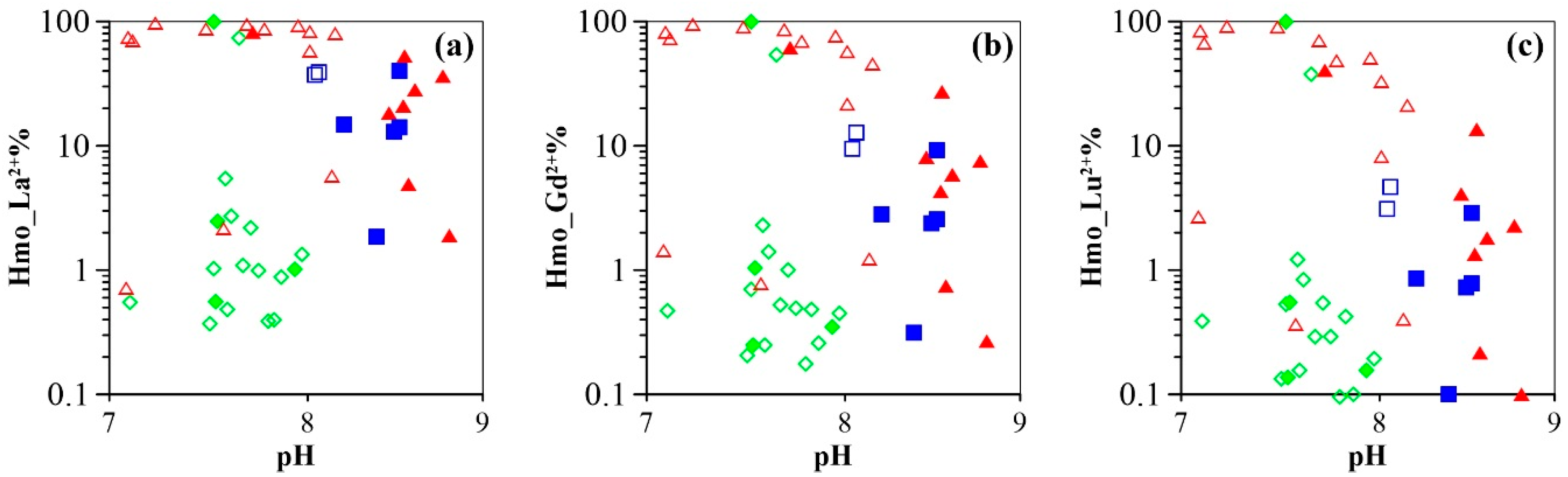
4.2. Controls on Groundwater REE Signatures: Roles of Hydrous Manganese Oxyhydroxides
4.3. Influence of Hydrous Manganese Oxide Content
4.4. Model Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Verplanck, P.P.; Taylor, H.E.; Nordstrom, D.K.; Barber, L.B. Aqueous Stability of Gadolinium in Surface Waters Receiving Sewage Treatment Plant Effluent, Boulder Creek, Colorado. Environ. Sci. Technol. 2005, 39, 6923–6929. [Google Scholar] [CrossRef] [PubMed]
- Hatje, V.; Bruland, K.W.; Flegal, A.R. Increases in anthropogenic gadolinium anomalies and rare earth element concentrations in San Francisco Bay over a twenty-year record. Environ. Sci. Technol. 2016, 50, 4159–4168. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.G.; Ort, C.; Keller, J. Detection of anthropogenic gadolinium in treated wastewater in South East Queensland, Australia. Water Res. 2009, 43, 3534–3540. [Google Scholar] [CrossRef] [PubMed]
- Kulaksız, S.; Bau, M. Anthropogenic gadolinium as a microcontaminant in tap water used as drinking water in urban areas and megacities. Appl. Geochem. 2011, 26, 1877–1885. [Google Scholar] [CrossRef]
- Merschel, G.; Bau, M.; Baldewein, L.; Dantas, E.L.; Walde, D.; Bühn, B. Tracing and tracking wastewater-derived substances in freshwater lakes and reservoirs: Anthropogenic gadolinium and geogenic REEs in Lake Paranoá, Brasilia. C. R. Geosci. 2015, 347, 284–293. [Google Scholar] [CrossRef]
- Kulaksız, S.; Bau, M. Rare earth elements in the Rhine River, Germany: First case of anthropogenic lanthanum as a dissolved microcontaminant in the hydrosphere. Environ. Int. 2011, 37, 973–979. [Google Scholar] [CrossRef]
- Elbaz-Poulichet, F.; Seidel, J.L.; Othoniel, C. Occurrence of an anthropogenic gadolinium anomaly in river and coastal waters of Southern France. Water Res. 2002, 36, 1102–1105. [Google Scholar] [CrossRef]
- Möller, P.; Morteani, G.; Dulski, P. Anomalous gadolinium, cerium, and yttrium contents in the Adige and Isarco river waters and in the water of their tributaries (Provinces Trento and Bolzano/Bozen, NE Italy). Acta Hydrochim. Hydrobiol. 2003, 31, 225–239. [Google Scholar] [CrossRef]
- Möller, P.; Paces, T.; Dulski, P.; Morteani, G. Anthropogenic Gd in Surface Water, Drainage System, and the Water Supply of the City of Prague, Czech Republic. Environ. Sci. Technol. 2002, 36, 2387–2394. [Google Scholar] [CrossRef]
- Nozaki, Y.; Lerche, D.; Alibo, D.S.; Tsutsumi, M. Dissolved indium and rare earth elements in three Japanese rivers and Tokyo Bay: Evidence for anthropogenic Gd and In. Geochim. Cosmochim. Acta 2000, 64, 3975–3982. [Google Scholar] [CrossRef]
- Song, H.; Shin, W.J.; Ryu, J.S.; Shin, H.S.; Chung, H.; Lee, K.S. Anthropogenic rare earth elements and their spatial distributions in the Han River, South Korea. Chemosphere 2017, 172, 155–165. [Google Scholar] [CrossRef] [PubMed]
- National Natural Science Foundation of China. Environmental Chemistry; Science Press: Beijing, China, 1996. [Google Scholar]
- Meryem, B.; Ji, H.B.; Yang, G.; Ding, H.J.; Li, C. Distribution of rare earth elements in agricultural soil and human body (scalp hair and urine) near smelting and mining areas of Hezhang, China. J. Rare Earth 2016, 34, 1156–1167. [Google Scholar] [CrossRef]
- Gwenzi, W.; Mangori, L.; Danha, C.; Chaukura, N.; Dunjana, N.; Sanganyado, E. Sources, behaviour, and environmental and human health risks of high-technology rare earth elements as emerging contaminants. Sci. Total Environ. 2018, 636, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Hatje, V.; Lamborg, C.H.; Boyle, E.A. Trace-Metal Contaminants: Human Footprint on the Ocean. Elements 2018, 14, 403–408. [Google Scholar] [CrossRef]
- Liu, W.S.; Wu, L.L.; Zheng, M.Y.; Chao, Y.Q.; Zhao, C.M.; Zhong, X.; Ding, K.B.; Huot, H.; Zhang, M.Y.; Tang, Y.T.; et al. Controls on rare-earth element transport in a river impacted by ion-adsorption rare-earth mining. Sci. Total Environ. 2019, 660, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Smedley, P.L. The geochemistry of rare earth elements in groundwater from the Carnmenellis area, southwest England. Geochim. Cosmochim. Acta 1991, 55, 2767–2779. [Google Scholar] [CrossRef]
- Dia, A.; Gruau, G.; Olivié-Lauquet, G.; Riou, C.; Molénat, J.; Curmi, P. The distribution of rare earth elements in groundwaters: Assessing the role of source-rock composition, redox changes and colloidal particles. Geochim. Cosmochim. Acta 2000, 64, 4131–4151. [Google Scholar] [CrossRef]
- Gruau, G.; Dia, A.; Olivie-Lauquet, G.; Davranche, M.; Pinay, G. Controls on the distribution of rare earth elements in shallow groundwaters. Water Res. 2004, 38, 3576–3586. [Google Scholar] [CrossRef]
- Pourret, O.; Gruau, G.; Dia, A.; Davranche, M.; Molenat, J. Colloidal control on the distribution of rare earth elements in shallow groundwaters. Aquat. Geochem. 2010, 16, 31. [Google Scholar] [CrossRef]
- Willis, S. Trace element Geochemistry in Groundwater Flow Systems. Ph.D. Thesis, The University of Texas at Arlington, Arlington, TX, USA, 11 November 2010. [Google Scholar]
- Noack, C.W.; Dzombak, D.A.; Karamalidis, A.K. Rare earth element distributions and trends in natural waters with a focus on groundwater. Environ. Sci. Technol. 2014, 48, 4317–4326. [Google Scholar] [CrossRef]
- Liu, H.Y.; Pourret, O.; Guo, H.M.; Martinez, R.E.; Zouhri, L. Impact of hydrous manganese and ferric oxides on the behavior of aqueous rare earth elements (REE): Evidence from a modeling approach and implication for the sink of REE. Int. J. Environ. Res. Public Health 2018, 15, 2837. [Google Scholar] [CrossRef] [PubMed]
- Patino, L.C.; Velbel, M.A.; Price, J.R.; Wade, J.A. Trace element mobility during spheroidal weathering of basalts and andesites in Hawaii and Guatemala. Chem. Geol. 2003, 202, 343–364. [Google Scholar] [CrossRef]
- Tweed, S.O.; Weaver, T.R.; Cartwright, I.; Schaefer, B. Behavior of rare earth elements in groundwater during flow and mixing in fractured rock aquifers: An example from the Dandenong Ranges, southeast Australia. Chem. Geol. 2006, 234, 291–307. [Google Scholar] [CrossRef]
- Leybourne, M.I.; Johannesson, K.H. Rare earth elements (REE) and yttrium in stream waters, stream sediments, and Fe-Mn oxyhydroxides: Fractionation, speciation, and controls over REE+Y patterns in the surface environment. Geochim. Cosmochim. Acta 2008, 72, 5962–5983. [Google Scholar] [CrossRef]
- Ohta, A.; Kawabe, I. REE (III) adsorption onto Mn dioxide (δ-MnO2) and Fe oxyhydroxide: Ce (III) oxidation by δ-MnO2. Geochim. Cosmochim. Acta 2001, 65, 695–703. [Google Scholar] [CrossRef]
- Bau, M. Scavenging of dissolved yttrium and rare earths by precipitating iron oxyhydroxide: Experimental evidence for Ce oxidation, Y-Ho fractionation, and lanthanide tetrad effect. Geochim. Cosmochim. Acta 1999, 63, 67–77. [Google Scholar] [CrossRef]
- Kawabe, I.; Ohta, A.; Miura, N. Distribution coefficients of REE between Fe oxyhydroxide precipitates and NaCl solutions affected by REE-carbonate complexation. Geochem. J. 1999, 33, 181–197. [Google Scholar] [CrossRef]
- Ohta, A.; Kawabe, I. Rare earth element partitioning between Fe oxyhydroxide precipitates and aqueous NaCl solutions doped with NaHCO3: Determinations of rare earth element complexation constants with carbonate ions. Geochem. J. 2000, 34, 439–454. [Google Scholar] [CrossRef]
- Quinn, K.A.; Byrne, R.H.; Schijf, J. Sorption of yttrium and rare earth elements by amorphous ferric hydroxide: Influence of solution complexation with carbonate. Geochim. Cosmochim. Acta 2006, 70, 4151–4165. [Google Scholar] [CrossRef]
- Verplanck, P.L.; Nordstrom, D.K.; Taylor, H.E.; Kimball, B.A. Rare earth element partitioning between iron oxyhydroxides and acid mine waters. Appl. Geochem. 2004, 19, 1339–1354. [Google Scholar] [CrossRef]
- Quinn, K.A.; Byrne, R.H.; Schijf, J. Sorption of yttrium and rare earth elements by amorphous ferric hydroxide: Influence of temperature. Environ. Sci. Technol. 2007, 41, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, M.; Stille, P. Controls on transport and fractionation of the rare earth elements in stream water of a mixed basaltic-granitic catchment basin (Massif Central, France). Chem. Geol. 2008, 254, 1–18. [Google Scholar] [CrossRef]
- Schijf, J.; Marshall, K.S. YREE sorption on hydrous ferric oxide in 0.5 M NaCl solutions: A model extension. Mar. Chem. 2011, 123, 32–43. [Google Scholar] [CrossRef]
- Pourret, O.; Davranche, M. Rare earth element sorption onto hydrous manganese oxide: A modeling study. J. Colloid interface Sci. 2013, 395, 18–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- German, C.R.; Elderfield, H. Application of the Ce anomaly as a paleoredox indicator: The ground rules. Paleoceanography 1990, 5, 823–833. [Google Scholar] [CrossRef]
- Koeppenkastrop, D.; De Carlo, E.H. Sorption of rare-earth elements from seawater onto synthetic mineral particles: An experimental approach. Chem. Geol. 1992, 95, 251–263. [Google Scholar] [CrossRef]
- Koeppenkastrop, D.; De Carlo, E.H. Uptake of rare earth elements from solution by metal oxides. Environ. Sci. Technol. 1993, 27, 1796–1802. [Google Scholar] [CrossRef]
- De Carlo, E.R.; Wen, X.Y.; Cowen, J.P. Rare earth element fractionation in hydrogenetic Fe-Mn crusts: The influence of carbonate complexation and phosphatization on Sm/Yb ratios. In Marine Authigenesis: From Global to Microbial; Glenn, C.R., Prevot-Lucas, L., Lucas, J., Eds.; Society for Sedimentary Geology Special Publication: Tulsa, OK, USA, 2000; pp. 271–285. [Google Scholar]
- De Carlo, E.H.; Wen, X.Y.; Irving, M. The influence of redox reactions on the uptake of dissolved Ce by suspended Fe and Mn oxide particles. Aquat. Geochem. 1998, 3, 357–389. [Google Scholar] [CrossRef]
- Davranche, M.; Pourret, O.; Gruau, G.; Dia, A.; Le Coz-Bouhnik, M. Adsorption of REE (III)-humate complexes onto MnO2: Experimental evidence for cerium anomaly and lanthanide tetrad effect suppression. Geochim. Cosmochim. Acta 2005, 69, 4825–4835. [Google Scholar] [CrossRef]
- Duncan, T.; Shaw, T.J. The mobility of rare earth elements and redox sensitive elements in the groundwater/seawater mixing zone of a shallow coastal aquifer. Aquat. Geochem. 2003, 9, 233–255. [Google Scholar] [CrossRef]
- Tang, J.; Johannesson, K.H. Controls on the geochemistry of rare earth elements along a groundwater flow path in the Carrizo Sand aquifer, Texas, USA. Chem. Geol. 2006, 225, 156–171. [Google Scholar] [CrossRef]
- Guo, H.M.; Zhang, B.; Wang, G.C.; Shen, Z.L. Geochemical controls on arsenic and rare earth elements approximately along a groundwater flow path in the shallow aquifer of the Hetao Basin, Inner Mongolia. Chem. Geol. 2010, 270, 117–125. [Google Scholar] [CrossRef]
- Chevis, D.A.; Johannesson, K.H.; Burdige, D.J.; Tang, J.; Moran, S.B.; Kelly, R.P. Submarine groundwater discharge of rare earth elements to a tidally-mixed estuary in Southern Rhode Island. Chem. Geol. 2015, 397, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.Y.; Guo, H.M.; Xing, L.N.; Zhan, Y.H.; Li, F.L.; Shao, J.L.; N, H.; L, X.; Li, C.Q. Geochemical behaviors of rare earth elements in groundwater along a flow path in the North China Plain. J. Asian Earth Sci. 2016, 117, 33–51. [Google Scholar] [CrossRef]
- Decrée, S.; Pourret, O.; Baele, J.M. Rare earth element fractionation in heterogenite (CoOOH): Implication for cobalt oxidized ore in the Katanga Copperbelt (Democratic Republic of Congo). J. Geochem. Explor. 2015, 159, 290–301. [Google Scholar] [CrossRef]
- Chen, W.H. Groundwater in Hebei; Seismological Press: Beijing, China, 1999. [Google Scholar]
- Liu, C.F.; Wang, P.Y.; Zhou, L. The environment significance of H, O, C and Cl isotopic composition in groundwater of Hebei Plain. Earth Sci. Front. 1997, 4, 267–274. [Google Scholar]
- Chen, Z.Y.; Nie, Z.L.; Zhang, Z.J.; Qi, J.X.; Nan, Y.J. Isotopes and sustainability of ground water resources, North China Plain. Groundwater 2005, 43, 485–493. [Google Scholar]
- Chen, W.H.; Ni, M.Y. Quaternary Geology in Hebei; Geological Publish House: Beijing, China, 1987. [Google Scholar]
- Zhang, Z.H.; Shen, Z.L.; Xue, Y.Q.; Ren, F.H.; Shi, D.H.; Yin, Z.Z.; Zhong, Z.X.; Sun, X.H. Evolution of Ground Water Environment in the North China Plain; Geological Publish House: Beijing, China, 2000. [Google Scholar]
- Kendy, E.; Zhang, Y.; Liu, C.; Wang, J.; Steenhuis, T. Groundwater recharge from irrigated cropland in the North China Plain: Case study of Luancheng County, Hebei Province, 1949–2000. Hydrol. Process. 2004, 18, 2289–2302. [Google Scholar] [CrossRef]
- Xing, L.N.; Guo, H.M.; Zhan, Y.H. Groundwater hydrochemical characteristics and processes along flow paths in the North China Plain. J. Asian Earth Sci. 2013, 70–71, 250–264. [Google Scholar] [CrossRef]
- Parkhurst, D.L.; Appelo, C.A.J. Description of Input and Examples for PHREEQC Version 3-a Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; US Geological Survey Techniques and Methods; US Geological Survey: Reston, VA, USA, 2013; Volume 6, p. 497.
- Hummel, W.; Berner, U.; Curti, E.; Pearson, F.J.; Thoenen, T. Nagra/PSI Chemical Thermodynamic Data Base 01/01. Radiochim. Acta 2002, 90, 805–813. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Pourret, O.; Guo, H.; Bonhoure, J. Rare earth elements sorption to iron oxyhydroxide: Model development and application to groundwater. Appl. Geochem. 2017, 87, 158–166. [Google Scholar] [CrossRef]
- Bethke, C.M. Geochemical and Biogeochemical Reaction Modeling, 2nd ed.; Cambridge University Press: Cambridge, UK, 2007; 543p, ISBN 9780521875547. [Google Scholar]
- McLennan, S.M. Relationships between the trace element composition of sedimentary rocks and upper continental crust. Geochem. Geophys. Geosyst. 2001, 2, 109. [Google Scholar] [CrossRef]
- Drever, J.I. The Geochemistry of Natural Waters: Surface and Groundwater Environments, 3rd ed.; Prentic Hall: Upper Saddle River, NJ, USA, 1997; 436p. [Google Scholar]
- Nesbitt, H.W. Mobility and fractionation of rare earth elements during weathering of a granodiorite. Nature 1979, 279, 206–210. [Google Scholar] [CrossRef]
- Sholkovitz, E.R.; Landing, W.M.; Lewis, B.L. Ocean particle chemistry: The fractionation of rare earth elements between suspended particles and seawater. Geochim. Cosmochim. Acta 1994, 58, 1567–1579. [Google Scholar] [CrossRef]
- Stumm, W.; Morgan, J.J. Aquatic Chemistry, 3rd ed.; Wiley Intersciences: New York, NY, USA, 1996. [Google Scholar]
- Appelo, C.A.J.; Postma, D. Geochemistry, Groundwater and Pollution; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Davranche, M.; Pourret, O.; Gruau, G.; Dia, A.; Jin, D.; Gaertner, D. Competitive binding of REE to humic acid and manganese oxide: Impact of reaction kinetics on development of cerium anomaly and REE adsorption. Chem. Geol. 2008, 247, 154–170. [Google Scholar] [CrossRef]
- Pourret, O.; Tuduri, J. Continental shelves as potential resource of rare earth elements. Sci. Rep. 2017, 7, 5857. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom, D.K.; Plummer, L.N.; Langmuir, D.; Busenberg, E.; May, H.M.; Jones, B.F.; Parkhurst, D.L. Revised Chemical Equilibrium Data for Major Water—Mineral Reactions and their Limitations. In Chemical Modeling of Aqueous Systems II; American Chemical Society: Washington, DC, USA, 1990; Volume 416, pp. 398–413. [Google Scholar]
- Cancès, B.; Ponthieu, M.; Castrec-Rouelle, M.; Aubry, E.; Benedetti, M.F. Metal ions speciation in a soil and its solution: Experimental data and model results. Geoderma 2003, 113, 341–355. [Google Scholar] [CrossRef]
- Schneider, A.R.; Ponthieu, M.; Cancès, B.; Conreux, A.; Morvan, X.; Gommeaux, M.; Marin, B.; Benedetti, M.F. Influence of dissolved organic matter and manganese oxides on metal speciation in soil solution: A modelling approach. Environ. Pollut. 2016, 213, 618–627. [Google Scholar] [CrossRef]
- Kim, I.; Kim, G. Role of colloids in the discharge of trace elements and rare earth elements from coastal groundwater to the ocean. Mar. Chem. 2015, 176, 126–132. [Google Scholar] [CrossRef]
- Tonkin, J.W.; Balistrieri, L.S.; Murray, J.W. Modeling sorption of divalent metal cations on hydrous manganese oxide using the diffuse double layer model. Appl. Geochem. 2004, 19, 29–53. [Google Scholar] [CrossRef]
- Pourret, O.; Davranche, M.; Gruau, G.; Dia, A. Rare earth elements complexation with humic acid. Chem. Geol. 2007, 243, 128–141. [Google Scholar] [CrossRef] [Green Version]
- Pourret, O.; Davranche, M.; Gruau, G.; Dia, A. New insights into cerium anomalies in organic-rich alkaline waters. Chem. Geol. 2008, 251, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Tani, Y.; Takahashi, Y.; Tanimizu, M.; Suzuki, Y.; Kozai, N.; Ohnuki, T. A specific Ce oxidation process during sorption of rare earth elements on biogenic Mn oxide produced by Acremonium sp. strain KR21-2. Geochim. Cosmochim. Acta 2010, 74, 5463–5477. [Google Scholar] [CrossRef]
- Ohnuki, T.; Jiang, M.; Sakamoto, F.; Kozai, N.; Yamasaki, S.; Yu, Q.; Tanaka, K.; Utsunomiya, S.; Xia, X.; Yang, K.; et al. Sorption of trivalent cerium by a mixture of microbial cells and manganese oxides: Effect of microbial cells on the oxidation of trivalent cerium. Geochim. Cosmochim. Acta 2015, 163, 1–13. [Google Scholar] [CrossRef]
- Yu, C.; Drake, H.; Mathurin, F.A.; Åström, M.E. Cerium sequestration and accumulation in fractured crystalline bedrock: The role of Mn-Fe (hydr-) oxides and clay minerals. Geochim. Cosmochim. Acta 2017, 199, 370–389. [Google Scholar] [CrossRef]
- Kraemer, D.; Tepe, N.; Pourret, O.; Bau, M. Negative cerium anomalies in manganese (hydr) oxide precipitates due to cerium oxidation in the presence of dissolved siderophores. Geochim. Cosmochim. Acta 2017, 196, 197–208. [Google Scholar] [CrossRef]
- Tang, J.; Johannesson, K.H. Speciation of rare earth elements in natural terrestrial waters: Assessing the role of dissolved organic matter from the modeling approach. Geochim. Cosmochim. Acta 2003, 67, 2321–2339. [Google Scholar] [CrossRef]
- Marsac, R.; Réal, F.; lal Banik, N.; Pedrot, M.; Pourret, O.; Vallet, V. Aqueous chemistry of Ce (IV): Estimations using actinide analogues. Dalton Trans. 2017, 46, 13553–13561. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Guo, H.; Pourret, O.; Chen, Y.; Yuan, R. Role of Manganese Oxyhydroxides in the Transport of Rare Earth Elements Along a Groundwater Flow Path. Int. J. Environ. Res. Public Health 2019, 16, 2263. https://doi.org/10.3390/ijerph16132263
Liu H, Guo H, Pourret O, Chen Y, Yuan R. Role of Manganese Oxyhydroxides in the Transport of Rare Earth Elements Along a Groundwater Flow Path. International Journal of Environmental Research and Public Health. 2019; 16(13):2263. https://doi.org/10.3390/ijerph16132263
Chicago/Turabian StyleLiu, Haiyan, Huaming Guo, Olivier Pourret, Yi Chen, and Rongxiao Yuan. 2019. "Role of Manganese Oxyhydroxides in the Transport of Rare Earth Elements Along a Groundwater Flow Path" International Journal of Environmental Research and Public Health 16, no. 13: 2263. https://doi.org/10.3390/ijerph16132263