Abstract
Methyl bromide (CH3Br) is one of the largest natural sources of bromine in the stratosphere, where it leads to ozone depletion. This paper reported the photochemical production of CH3Br from syringic acid (SA) that has been used as an environmentally relevant model compound for terrestrially-derived dissolved organic matter. The formation of CH3Br increased with the increase of bromide ion concentration ranging from 0.8 to 80 mmol L−1. Ferric ions (Fe(III)) enhanced CH3Br production, while chloride inhibited it, with or without Fe(III). Meanwhile, methyl chloride (CH3Cl) was generated in the presence of chloride and was inhibited by Fe(III). The different effects of Fe(III) on the formation of CH3Cl and CH3Br indicate their diverse formation paths. Based on the intermediates identified by liquid chromatography-mass spectrometry and the confirmation of the formation of Fe(III)-SA complexes, it was proposed that there were two formation paths of CH3Br from SA in the bromide-enriched water under simulated sunlight irradiation. One path was via nucleophilic attack of Br− on the excited state protonation of SA; the other was via the combination of methyl radical and bromine radical when Fe(III) was present. This work suggests that the photochemical formation of CH3Br may act as a potential natural source of CH3Br in the bromide-enriched environmental matrix, and helps in better understanding the formation mechanism of CH3Br.
1. Introduction
Methyl bromide (CH3Br) is the most abundant brominated gas in the troposphere, which is transported to the stratosphere, releasing bromine atoms, which then destroy ozone catalytically. The global averaged mixing ratio of CH3Br is 7.9 ppt (parts per trillion), representing >50% of total organic bromine in the troposphere [1]. Although CH3Br has lower atmospheric abundance compared with methyl chloride (CH3Cl) at 545 ppt, release of a Br atom is more destructive to stratospheric ozone than a Cl atom [2]. In the past 20 years, a lot of work has been done to quantify the sources and the sinks of CH3Br. However, large uncertainties in its budgets still remain. Estimates of global sinks of CH3Br (148 Gg yr−1) are significantly unbalanced by ~30% with the estimates of global sources (112 Gg yr−1) [3]. It is estimated that methyl halides originate in large part from natural sources which are believed to account for ~60–80% of the global CH3Br [3]. These natural sources include oceans, biomass burning (which can also be anthropogenic), wetlands, Brassica crops, and fungus [3,4,5,6,7]. The ocean, as one of the major sources and also major sinks for CH3Br, plays an important role in its atmospheric budget [8,9]. While the open ocean is a net sink for CH3Br, the coastal ocean often exhibits significant super saturation in previous studies [10]. For example, investigation of the distributions of halocarbons in the marine boundary of air and surface seawater indicated that the Yangzi River Estuary and adjacent coastal area were net sources of atmospheric CH3Br [11]. Therefore, more attention should be paid to the process of CH3Br production in the estuarial and coastal ocean areas.
In principle, natural processes for CH3Br production proceed through an assortment of biological [12] and/or abiotic pathways. Focusing on the abiotic pathways, one abiotic production pathway of CH3Br was suggested for a methoxy moiety (-OCH3) reacting with halides directly via a nucleophilic displacement reaction [13,14]. In addition, CH3Br production from biomass burning was thought to proceed in a path via methanol production during pyrolysis [15]. In both of these proposed pathways, lignin and pectin components which can provide a large amount of methoxy moieties seem essential for CH3Br production. Keppler et al. found that CH3Br was generated abiotically through alkylation of Br ions during the oxidation of soil organic matter by ferric iron (Fe3+) [16]. Moore reported that some lignin model compounds could act as the carbon precursor to form CH3Cl in saline waters [17]. Considering that humic acid is partially originated from terrestrial plants containing large amounts of lignin [18], it is reasonable to speculate that humic acid could act as the precursor of methyl halides.
Humic acid deriving from terrestrial biota contains a large assemblage of complex chemical structures of polyphenol, carboxyl, methoxyl, and quinone functionalities, and is distributed as a component of dissolved organic matter (DOM) in estuary and coastal ocean areas [19,20]. As we know, DOM can absorb sunlight energy and plays an important role in the photochemical transformation of organic contaminants through producing an excited triplet state, reactive oxygen species, and reactive halogen species, and so on [19,21,22]. In estuarine waters, terrestrial DOM and high levels of halides, e.g., chloride and bromide, occur simultaneously [23], providing a suitable situation for the production of organo-halogens [24]. There are a number of reports on the photochemical production of methyl halides (chiefly methyl iodide (CH3I) and CH3Cl) from DOM in seawater [17,25,26]. The photochemical production of CH3I from DOM which has been demonstrated to be a major source of CH3I in marine environments involved the release of methyl radicals, •CH3 from DOM, and their combination with iodine radicals, I• [25,27,28]. In contrast, the photochemical production of CH3Cl was thought to proceed through a methoxy moiety of DOM directly reacting with chloride ions via a nucleophilic displacement reaction [17,29,30]. Thus it can be seen that the productions of CH3Cl and CH3I are proposed to occur via two different pathways. However, the photochemical generation of CH3Br from DOM is less understood until now.
The present work attempted to investigate the photochemical production of CH3Br from DOM and to gain insight into the path of CH3Br generation. Considering that structural moieties on syringic acid (SA) have been identified in terrestrially derived DOM [17], and Benner and Opsahl have shown that aryl-methoxy groups on syringyl moieties of lignin are photochemically labile and preferentially degraded as terrigenous carbon moves from freshwater to the open ocean [31], SA was used as a model compound for DOM in the experiment. This study first confirmed the formation of CH3Br from SA in the aqueous bromide solutions, then, by comparing the different generation profiles of CH3Br and CH3Cl as a function of ferric ions concentration, proposed two reaction pathways for the photo-initiated formation of CH3Br described.
2. Materials and Methods
2.1. Reagents
Syringic acid (4-hydroxy-3,5-dimethoxybenzoic acid) was purchased from Molekula Ltd., United Kingdom. Liquid standards of CH3Br and CH3Cl (200 μg mL−1 in methanol) were purchased from Accustandard, USA. Sodium bromide (NaBr), sodium chloride (NaCl), FeCl3·6H2O and other chemicals were reagent grade. Ultrapure water (18 MW cm) was obtained with a Millipore water purification water unit to prepare all aqueous solutions.
2.2. Irradiation Experiments
The irradiation experiments were performed in a solar simulator (Phchem III, Beijing Newbit Technology Co., Ltd, China) equipped with a 500 W xenon arc lamp and filters to cut off light with a wavelength below 290 nm. Considering the attenuation of light in a water body, the average light intensity in the euphotic zone should be much lower than the surface water (around 85 mW cm−2). Thus, the light intensity of the solar simulator was set at 15 mW cm−2. The reactor was a round bottom sealed quartz tube (3.5 cm o.d.; volume ca. 125 mL) with one outlet (4 mm o.d.) in the middle of the bottom. Details are in Text S1, Figure S1, and Figure S2 in the supplementary information. The dark control tubes wrapped in Al foil were also placed in the solar simulator. Two parallel samples were set up in each experiment.
2.3. Analysis Methods
The concentrations of CH3Br were analyzed by gas chromatography-mass spectrometry (GC-MS, Aglient 7890B/5977C, Santa Clara, CA, USA) equipped with a purge-and-trap sample concentrator (Eclipse 4760, College Station, TX, USA). Briefly, aqueous samples were injected into a 25 mL-purge tube, subsequently purged with ultrapure nitrogen at 40 mL min−1 for 11 min. The extracted gases were pre-concentrated in the trap tube containing VOCARB 3000, and then released from the trap column by heating to 240 °C, finally were introduced into GC-MS. A DB-VRX capillary column (60 m × 250 μm × 1.4 μm, Agilent Technologies, Palo Alto, CA, USA) was used. The inlet worked in a split model with a split ratio of 20:1 and the temperature was set at 150 °C. The oven temperature was initially set at 32 °C for 6 min and rose to 180 °C at 20 °C min−1 then held at 180 °C for 4 min. The select ion monitor (SIM) mode was used for quantitative analysis of CH3Br with m/z of 94 and 96, and CH3Cl with m/z of 50 and 52. The detection limits (signal to noise ratio, S/N = 3) of CH3Cl and CH3Br were 62 and 4.1 pmol L−1, respectively, and the relative standard deviations of replicate analyses (n = 6) were within 6% and 8%, respectively.
The photolysis intermediates of SA were characterized by triple quadrupole liquid chromatography-mass spectrometry (LC-MS). After irradiation, in order to maximize the recovery of the acid products, aqueous samples were acidized to pH2.0 using sulfuric acid, then passed through SPE cartridges at a flow rate of 1~2 mL min−1 after the cartridges were activated with 3 mL methanol and 3 mL Milli-Q water in sequence. Then the intermediates were eluted by methanol. The LC-MS system consisted of an Agilent QQQ 6410B MS system equipped with electro spray ionization (ESI) interface and an Agilent 1200SL system (Agilent Technologies, Santa Clara CA, USA). The analytical column was XTERRA® MS C18 (2.1 × 100 mm, 3.5 μm, Waters, Milford, MA, USA). Mobile phases A and B were water with 0.1% HCOOH and acetonitrile, respectively, using a gradient from 10% B at 0.1 min to 60% B at 10 min, which was kept isocratic for 2 min, followed by a gradient back to the initial 10% B at 20 min with a flow rate of 0.25 mL min−1. The injection volume was 10 μL. The source temperature of the heated capillary was set at 350 °C, and the source voltage was 4.0 kV.
The solutions of Fe(III)–SA complex were stirred for 1 h in the dark to reach equilibrium. Their Ultraviolet –visible (UV–Vis) absorption spectra were measured using a spectrophotometer (Hitachi UH5300, Ibaraki, Japan).
3. Results and Discussion
3.1. Formation of CH3Br from SA in Aqueous Bromide Solutions
The formation of CH3Br was first investigated in the presence of 50 μmmol L−1 SA and 8 mmol L−1 bromide ions under simulated sun-light irradiation. As shown in Figure 1, about 220 pmol L−1 CH3Br was generated after irradiation of 30 h, whereas no detectable CH3Br was formed in the dark. This result indicates that CH3Br was produced through a photochemical reaction of SA and bromide, which is in agreement with the previous reports of the formation of CH3Cl by Moore and Dallin et al. [17,29]. In addition, Moore inferred the production of CH3Cl was not from SA directly but an intermediate of SA photolysis, somewhat a quinone derivative [17]. Although the loss of SA was almost negligible during irradiation (Figure S3), CH3Br formation was lowered after long-term irradiation. Here, the slow accumulation of CH3Br is speculated to be related to the slow formation rate of the active intermediate of SA and/or the degradation of CH3Br itself.
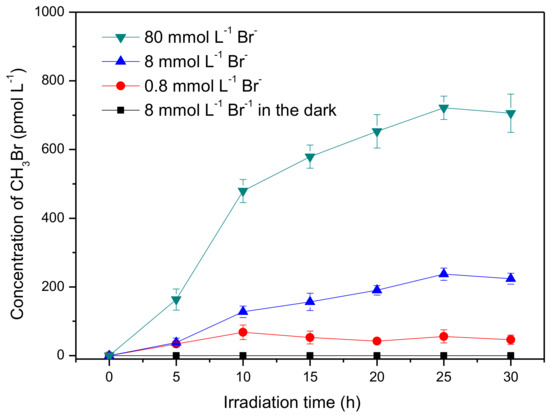
Figure 1.
Formation of methyl bromide (CH3Br) in aqueous solutions containing syringic acid (50 μmol L−1) and bromide ions (0.8~80 mmol L−1). Error bars represent one standard deviation.
Bromide ions in seawater are at an average concentration of 0.8 mmol L−1, and are further enriched in the nanolayer close to the air–sea interface; they can even reach a level of dozens of mmol L−1 at the marine boundary layer [32,33]. Consequently, the effect of bromide ions on the formation of CH3Br was investigated with concentrations ranging from 0.8 mmol L−1 to 80 mmol L−1. It was found that CH3Br production increased with increasing bromide concentration. The formation rates of CH3Br at the first 10 h in the presence of 0.8, 8.0 and 80 mmol L−1 Br− were estimated to be 6.81, 12.7, and 47.9 pmol L−1h−1, respectively. This result indicates that Br− acted as the limiting factor for the yield of CH3Br, which is consistent with the geochemical behavior of bromide in soil that bromide was the limiting factor for the bromination of soil organic matter [34]. As described above, the bromide concentration is further enhanced in the thin film of the surface seawater; it could be proposed that the emission of CH3Br might be more significant at the air–sea interface [32,33], although, of course, various other anions and cations in seawater may have different impacts on this process as well.
3.2. Effect of Chloride on the Formation of CH3Br
The experiment was then carried out with the addition of 0.5 mol L−1 NaCl, in order to know the effects of chloride on the production of CH3Br. When [Br−] was 8 mmol L−1, the concentration of CH3Br in the presence of Cl− after 30 h of irradiation reduced to 47.3 pmol L−1, meaning about 20% of the sample was without Cl− (Figure 2, blue curves), showing that Cl− inhibited the production of CH3Br. Meanwhile, a significant amount of CH3Cl was generated, which reached 865 pmol L−1 after 30 h of irradiation (Figure 2, red curve). When [Br−] was 0.8 mmol L−1, (the ratio of Cl: Br was 625 close to the natural seawater), the circumstance were similar, where CH3Br decreased distinctly (Figure 2, black curves). These results indicate that chloride was a forceful competitor for bromide to react with SA forming CH3X (X = Cl, Br), and also suggest that CH3Br might be generated through a similar pathway to CH3Cl under the experimental conditions.
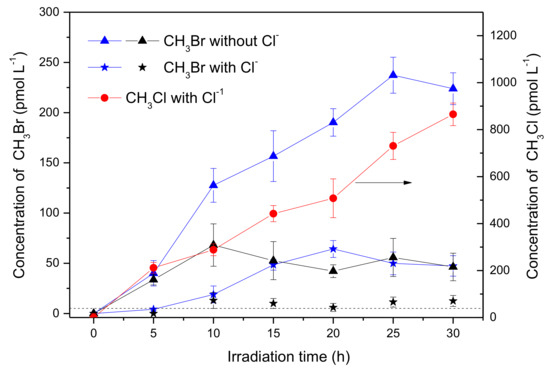
Figure 2.
Formation of CH3Br (triangle and star) and methyl chloride (CH3Cl; circle) in the solutions containing 50 μmol L−1 SA, 8 mmol L −1 Br− (blue) and 0.8 mmol L−1 Br− (black) in the presence or in the absence of 0.5 mol L−1 Cl−. There was no obvious difference for CH3Cl in the presence of 0.8 or 8 mmol L−1 Br−. Dashed line indicates the detection limit of CH3Br. Error bars represent one standard deviation.
To understand the photochemical transformation pathway of SA and the formation mechanism of CH3Br, LC-MS was employed to identify the intermediates of this reaction (Figure S4). The intermediate with molecule weight (MW) 184 confirmed using ESI(−) MS was attributed to 3-methoxy-4,5-dihydroxybenzoic acid, a demethylation product of SA. Hence, it could be assumed that the methyl group of CH3Br originated from the methoxy group of SA. This proposal was consistent with the formation pathway of CH3Cl from SA which has been well demonstrated by Dallin and Moore et al. [17,29]. The reaction proceeds via aromatic ring protonation followed by demethoxylation, as shown in Figure 3. The first step of CH3Br formation is protonation, i.e., SA is initiated by the protonation of excited state benzene ring (ipso positions with OCH3). The arenium ion intermediate has a minor resonance contributor with the positive charge distributed onto the methoxy oxygen. The second step is demethylation, involving nucleophilic attack of Br− onto the methoxy carbon resulting in Cmethyl–O cleavage [29]. When chloride and bromide ions coexist, attacking the methoxy carbon yields both CH3Cl and CH3Br. Since 0.5 mol L−1 chloride ions was more competitive for this process than 0.8 and 8 mmol L−1 bromide, the production of CH3Br decreased.
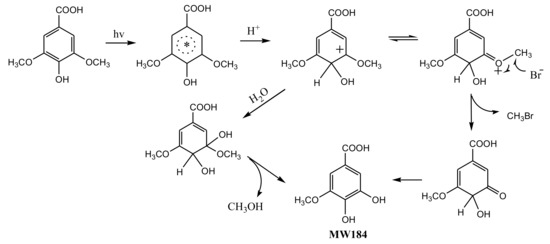
Figure 3.
A possible formation pathway of CH3Br from syringic acid (SA) under irradiation.
3.3. Formation of CH3Br and CH3Cl in the Presence of Ferric Ions
Iron is a universal element in the natural aqueous environment and plays an important role in the photochemical transformation of pollutants [35]. Here, the experiments were carried out by adding ferric ions, Fe(III), with concentrations ranging from 0 to 600 μmol L−1, into the solution containing 50 μmol L−1 SA and 8 mmol L−1 Br−. The pH of SA solutions with 0, 100, 200, 400, and 600 μmol L−1 Fe(III) was 5.3, 3.9, 3.5, 3.2, and 3.0, respectively. Figure 4 shows that Fe(III) had a significant promotion effect on the formation of CH3Br. The concentration of CH3Br in the presence of 600 μmol L−1 Fe(III) reached around 930 pmol L−1 after 20 h of irradiation, which was about four times higher than that without Fe(III).
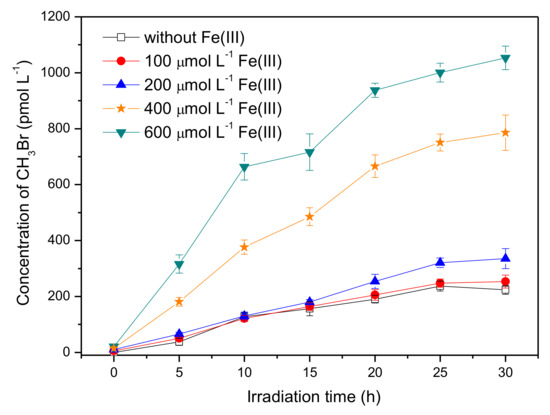
Figure 4.
Effect of Fe(III) on the formation of CH3Br from 50 μmol L−1 SA with 8 mmol L−1 NaBr. Error bars represent one standard deviation.
In general, Fe(III) in the solution exists as iron-hydroxyl complexes including Fe(OH)2+, Fe(OH)2+, Fe2(OH)24+, etc. Among them, Fe(OH)2+ is the most photochemical active species that has been identified to produce •OH when irradiated (Equation (1)) [35]. As we know, •OH is a strong oxidant and can oxidize X− to produce reactive radical species, X•/ X2•−, where X=Br, Cl (Equations (2)–(4)) [36,37].
Fe(OH)2+ + hν→ Fe2+ + •OH
•OH + X− ⇄ HXO•−
HXO•·− + H+ ⇄ X• + H2O
X•+ X− →X2•−
Considering the presence of reactive radical species in the systems containing Fe(III), bromine radical species were proposed to be the active intermediate for the production of CH3Br, which was a different formation pathway from CH3Cl. Previous studies on the formation of CH3Cl have demonstrated that CH3Cl generation occurred via a nucleophilic substitution by chloride ions, and the reactive radical species had no effect on CH3Cl formation [17,30]. In order to figure out the distinction between the generation of CH3Br and CH3Cl, the concentration of CH3Cl in the presence of Fe(III) was monitored as well. The result is displayed in Figure 5. It is notable that the formation profile of CH3Cl was quite different from CH3Br; that Fe(III) reduced the formation of CH3Cl sharply. The concentration of CH3Cl upon 20 h of irradiation decreased from 507 pmol L−1 to undetectable with Fe(III) increasing from 0 to 600 μmol L−1. The disparate effects of Fe(III) on the formation of CH3Cl and CH3Br suggests their different formation pathways in the presence of Fe(III).
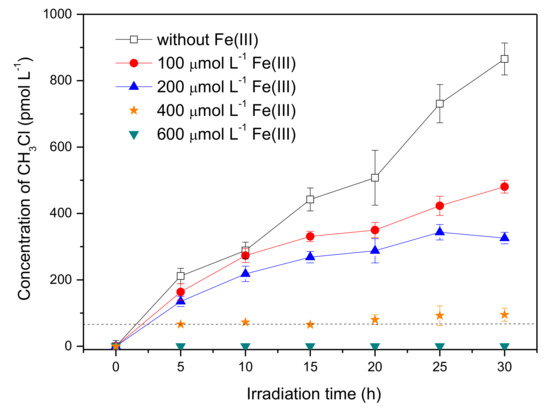
Figure 5.
Effect of Fe(III) on the formation of CH3Cl from 50 μmol L−1 SA with 0.5 mol L−1 NaCl. Dashed line indicates the detection limit of CH3Cl. Error bars represent one standard deviation.
Fe(III) is liable to be chelated by high affinity carboxylates, such as citrate and oxalate, to form Fe(III)-ligand complexes that exhibit appreciable reactivity to give out reactive species, such as •OH, and ferrous ions, Fe(II), upon irradiation [35,38]. SA contains -COOH and -OH groups, and can act as a ligand to complex with Fe(III), and hence it promotes the formation of reactive species. The UV–Vis absorption spectra of SA with different concentration of Fe(III) are shown in Figure 6. SA exhibited a peak at 260 nm whereas the spectrum of Fe(III) showed a peak around 300 nm. When 100 μmol L−1 Fe(III) interacted with SA, the absorption profile exhibited a strong peak at 275 nm. This red shifted spectrum (large differences greater than 15 nm among peaks) was clearly distinguishable. According to Singh and Kumar’s study on the complex formation of Fe(III) and SA, the red shift of the spectrum provided the evidence that chelation of Fe(III) took place where SA acted as a ligand, forming Fe(III)-SA complex [39]. Fe(III)-ligand complexes can undergo ligand-to-metal charge transfer (LMCT) upon irradiation and consequently result in the enhancement of •OH generation [35,38]. Considering the promotion effect of Fe(III) on the production of CH3Br, it is speculated that Fe(III)-SA and the corresponding •OH are essential for the generation of CH3Br.
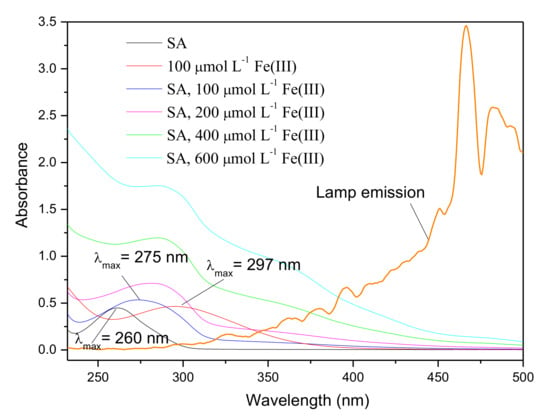
Figure 6.
UV–Vis spectra of 50 μmol L−1 SA, Fe(III) and Fe(III)-SA complexes with different concentration of Fe(III), and the emission spectrum of the Xenon lamp.
The reaction products in Fe(III)-SA system were analyzed using LC-MS. Besides 3-methoxy-4,5-dihydroxybenzoic acid, two new intermediates were detected. One product with MW 168 identified by ESI(+) MS was attributed to be 3,5-dimethoxy-1,4-benzoquinone, and the other one with MW 308 was attributed to be the dimer product (Figure S5 and S6). The proposed reaction pathway is displayed in Figure 7. Route I, an electron transferring from SA to Fe(III) in Fe(III)-SA complex resulted in Fe(II) and SA radical, SA•, with unpaired electron distributed on the carboxyl oxygen. Then SA• released CO2 through decarboxylation reaction forming 2,6-dimethoxyl-phenol radical with the unpaired electron distributed on the aromatic carbon (1), or its resonance contributor with the unpaired electron distributed onto the hydroxyl oxygen, i.e., 2,6-dimethoxyl-phenoxy radical (2). The cleavage of Cmethoxyl–O resulted in methoxy-quinone (3) and methyl radical, •CH3, which then recombined with Br• to produce CH3Br. As the formation of CH3 radical was a minor reaction, the methoxy-quinone was not detectable by LC-MS. Route II, another reaction path, was dominated by •OH where SA reacted with •OH, forming 2,6-dimethoxyl-1,4-hydroquinone (4), which was then oxidized by oxygen forming 3,5-dimethoxy-1,4-benzoquinone (MW168). The product with MW308 might be generated by the combination of 4 with 1 or 2.
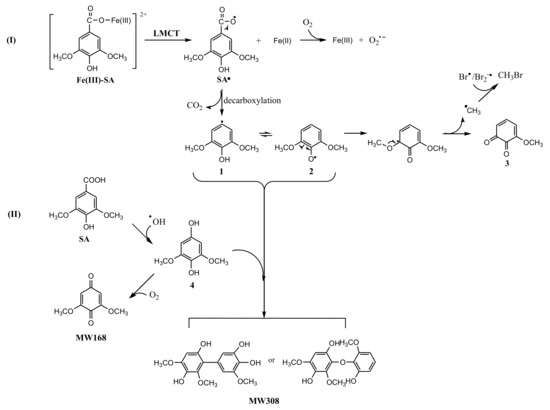
Figure 7.
A possible formation pathway of CH3Br in the Fe(III)-SA system.
From the results of the above analysis, it could be hypothesized that CH3Br was primarily formed through a radical combination pathway in the presence of Fe(III). However, it was not able to be ruled out that the nucleophilic substitution path as shown in Figure 3 still existed, but played a minor role. Based on this proposal, it was not difficult to understand why Fe(III) enhanced the production of CH3Br. On the one hand, Fe(III) led to the generation of •OH that can oxidize Br− to produce reactive bromine radical species (Equations (2)–(4)); on the other hand, the LMCT of Fe(III)-SA resulted in the formation of methyl radical. That is, Fe(III) enhanced the formation of CH3Br by providing both •Br and •CH3 moieties. However, CH3Cl formation decreased sharply after adding Fe(III). The reason for this is that CH3Cl is generated through the nucleophilic substitution of Cl− where protonation of excited-state benzene ring plays an important role [29,30]. The addition of Fe(III) lowering the solution pH could weaken the protonation of the excited state SA, and consequently reduce the formation of CH3Cl [29,40].
One more thing that should be examined is why CH3Cl was not produced through the radical combination pathway? This can be explained from the perspective of the formation rate of chlorine radicals from •OH. Actually, Cl− is an ineffective •OH scavenger because the intermediate HClO•− primarily reverts to •OH and Cl− (Equation (2)). For example, the rate constant for Equation (2) where X = Cl, k+ (forward) is 4.0 × 109 M−1s−1, and k− (backward) is 6.0 × 109 M−1s−1; while X = Br, the k+ is 1.1 × 1010 M−1s−1, and k− is 3.3 × 107 M−1s−1 [37]. Consequently, about >99.98% HClO•− will reform •OH and Cl−, and will not form chlorine radical; in contrast, only 24% of HBrO•− will reform •OH and Br−. Therefore, Br• has an order of magnitude dominance over Cl•. In addition, according to the calculation results of Parker et al., Cl• is only present at a higher concentration relative to •OH in more acidic solutions with pH lower than 3.0 [37]. Furthermore, Cl• is more reactive than Br•, and can react with H2O more easily (backward reaction of Equation (3)). For example, k for Cl• and H2O is 1.3 × 103 M−1s−1, while the k for Br• and H2O is 1.36 M−1s−1 [37]. Therefore, chlorine radical was hardly generated under the experimental conditions.
The effect of chloride on the formation of CH3Br and CH3Cl in the presence of Fe(III) is demonstrated in Figure 8. For CH3Br, chloride decreased its formation nearly from 1000 to 400 pmol L−1, which was similar as that in the absence of Fe(III) (Figure 2). Meanwhile, there was formation of CH3Cl in the presence of chloride. It is noteworthy that no detectable CH3Cl was generated in the coexistence of 600 μmol L−1 Fe(III) and Cl− (Figure 5); however, CH3Cl was generated with a concentration around 300 pmol L−1 in the coexistence of Fe(III), Br− and Cl− (Figure 8, black square). This is probably due to the existence of the different halogen radicals in the reaction systems containing Fe(III), Br− and Cl−. In fact, the mixed-halogen radical (BrCl•−) should be the major halogen radical that could be formed through the following Equations. [37]. The unit of the rate constant is M−1s−1.
Cl• + Br− ⇄ BrCl•− k+ = 1.2 × 1010, k− = 1.9 × 103
Br• + Cl− ⇄ BrCl•− k+ = 2.3 × 108, k− = 6.1 × 104
HClO•− + Br− ⇄ BrCl•− + OH− k+ = 1.0 × 109, k− = 3.0 × 106
HBrO•− + Cl− ⇄ BrCl•− + OH− k+ = 1.9 × 108, k− = 2.0 × 107
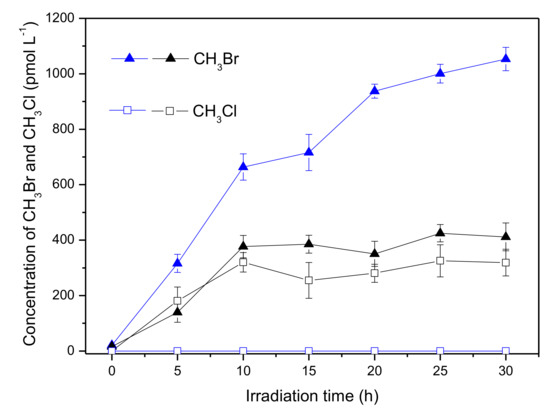
Figure 8.
Formation of CH3Br (triangle) and CH3Cl (square) in the solutions containing 50 μmol L−1 SA, 600 μmol L−1 Fe(III) and 8 mmol L−1 Br− in the presence (black) and in the absence (blue) of 0.5 mol L−1 Cl−. Error bars represent one standard deviation.
As discussed above, Cl• hardly existed in the reaction system and most of the HClO•− was prone to reform •OH and Cl−, so Equation (5) and Equation (7) could be neglected. Because of the close k+ and k− of Equation (8), formation of BrCl•− through Equation (8) was not the major path. Focusing on Equation (6), it could be seen that Br• was consumed to generate BrCl•−, hence the formation of CH3Br decreased. Although no data is available for reactions involving BrCl•−, it is typically assumed to react with rate constants intermediate between or similar to single-halogen radicals [37,41]. Consequently, BrCl•− would react with •CH3 forming CH3Br or CH3Cl (Equation (9)). Different with chlorine radical that was hardly generated, BrCl•− could be formed in the coexistence of Fe(III), Br− and Cl−; therefore, CH3Cl was produced.
BrCl•− + •CH3 → CH3Br + Cl− or CH3Cl + Br−
Halide oxidation by •OH has long been recognized as a source of halogen radicals in seawater [42,43]. The concentration of Br2•− and BrCl•− may exceed •OH concentrations by ~3–4 orders of magnitude. Although Cl− occurs at 670-fold higher concentrations than Br−, Br− oxidation by •OH drives halogen radical production [43]. Even, Br2•− concentration may exceed those of BrCl•− by ~2.5-fold, since Br2•− arises from further reactions of BrCl•− with Br− [37,41]. Consequently, bromide oxidation by •OH and the further production of bromine radicals play an important role for the generation of CH3Br. In addition, based on results in this paper, rates of Br converting to CH3Br ranging from 10−9 (in the coexistence of Br− and Cl−) to 10−7 (in the presence of Fe(III)) are equivalent to the level of biological conversion rates (10−7 for Schizochytrium sp., 10−9 for Ulkenia amoeboidea, and 10−8 for Aurantiochytrium sp. and Phaeocystis globose) [44,45]. Therefore, photochemical formation of CH3Br may partly account for the generation of CH3Br in the marine environmental matrix.
4. Conclusions
The photochemical formation of CH3Br from SA in aqueous bromide solutions indicates a potential natural source of CH3Br in the bromide-enriched environmental matrix. The inhibiting effect of chloride on the formation of CH3Br in the absence of Fe(III) and the simultaneous generation of CH3Cl from SA demonstrates the competition of Cl− and Br− with SA to form CH3X (X = Cl, Br), which also suggests that CH3Br was generated via the nucleophilic substitution reaction. The different effects of Fe(III) on the formation of CH3Br and CH3Cl illustrate an alternative path for CH3Br formation, i.e., combination of •CH3 and •Br. These results suggest that there are two formation pathways for CH3Br from SA, i.e., nucleophilic substitution and radical recombination, which may be in concurrence in the natural environment. This study provides an insight into the pathways of CH3Br formation in an aquatic environment.
Supplementary Materials
The following are available online at https://www.mdpi.com/1660-4601/17/6/2081/s1, Figure S1: Schematic of the device used for irradiation, Figure S2: Temperature change of the reaction solution during irradiation, Figure S3: Photodegradation of SA in aqueous bromide solutions in the presence or absence of Fe(III), Figure S4: Photolysis intermediates of SA analyzed by LC-ESI(−)-MS, Figure S5: Photolysis intermediates of SA in the presence of Fe(III) analyzed by LC-ESI(−)-MS, Figure S6: Photolysis intermediates of SA in the presence of Fe(III) analyzed by LC-ESI(+)-MS.
Author Contributions
Conceptualization, H.L.; methodology, H.L. and X.Z.; formal analysis, H.L. and X.Z.; investigation, T.T. and Y.P.; resources, B.S. and Z.Y.; writing—original draft preparation, T.T. and Y.P.; writing—review and editing, H.L.; funding acquisition, H.L. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (Nos. 41576111, 11975063, 41206095), and Fundamental Research Funds for the Central Universities (No. 3132019329).
Conflicts of Interest
There are no conflicts to declare.
References
- Schauffler, S.M.; Atlas, E.L.; Blake, D.R.; Flocke, F.; Lueb, R.A.; Lee-Taylor, J.M.; Stroud, V.; Travnicek, W. Distributions of brominated organic compounds in the troposphere and lower stratosphere. J. Geophys. Res. 1999, 104, 513–535. [Google Scholar] [CrossRef]
- Butler, J.H. Better budgets for methyl halides. Nature 2000, 403, 260–261. [Google Scholar] [CrossRef] [PubMed]
- WMO. Scientific Assessment of Ozone Depletion: 2010. In Global Ozone Research and Monitoring Project-Report No. 52; WMO: Geneva, Switzerland, 2011. [Google Scholar]
- Yvon-Lewis, S.A.; Saltzman, E.S.; Montzka, S.A. Recent trends in atmospheric methyl bromide: Analysis of post-Montreal Protocol variability. Atmos. Chem. Phys. 2009, 9, 5963–5974. [Google Scholar] [CrossRef]
- Rhew, R.C.; Miller, B.R.; Weiss, R.F. Natural methyl bromide and methyl chloride emissions from coastal salt marshes. Nature 2000, 403, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Redeker, K.R.; Wang, N.Y.; Low, J.C.; McMillan, A.; Tyler, S.C.; Cicerone, R.J. Emissions of methyl halides and methane from rice paddies. Science 2000, 290, 966–969. [Google Scholar] [CrossRef] [PubMed]
- Andreae, M.O.; Merlet, P. Emission of trace gases and aerosols from biomass burning. Glob. Biogeochem. Cycles 2001, 15, 955–966. [Google Scholar] [CrossRef]
- King, D.B.; Butler, J.H.; Yvon-Lewis, S.A.; Cotton, S.A. Predicting oceanic methyl bromide saturation from SST. Geophys. Res. Lett. 2002, 29, 2199. [Google Scholar] [CrossRef]
- Butler, J.H.; King, D.B.; Lobert, J.M.; Montzka, S.A.; Yvon-Lewis, S.A.; Hall, B.D.; Warwick, N.J.; Mondeel, D.J.; Aydin, M.; Elkins, J.W. Oceanic distributions and emissions of short-lived halocarbons. Glob. Biogeochem. Cycles 2007, 21, GB1023. [Google Scholar] [CrossRef]
- Hu, L.; Yvon-Lewis, S.A.; Liu, Y.; Salisbury, J.E.; Julia, E.O. Coastal emissions of methyl bromide and methyl chloride along the eastern Gulf of Mexico and the east coast of the United States. Glob. Biogeochem. Cycles 2010, 24, GB1007. [Google Scholar] [CrossRef]
- Yuan, D.; Zhen, H.; Yang, G.P. Spatiotemporal distributions of halocarbons in the marine boundary air and surface seawater of the Changjiang estuary and its adjacent East China Sea. Mar. Pollut. Bull. 2019, 140, 227–240. [Google Scholar] [CrossRef]
- Butler, A.; Sandy, M. Mechanistic considerations of halogenating enzymes. Nature 2009, 460, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.T.G.; McRoberts, W.C.; Keppler, F.; Kalin, R.M.; Harper, D.B. Chloride methylation by plant pectin: An efficient environmentally significant process. Science 2003, 301, 206–209. [Google Scholar] [CrossRef]
- Horst, A.; Holmstrand, H.; Andersson, P.; Thornton, B.F.; Wishkerman, A.; Keppler, F.; Gustafsson, O. Stable bromine isotopic composition of methyl bromide released from plant matter. Geochim. Cosmochim. Acta 2014, 125, 186–195. [Google Scholar] [CrossRef]
- Wishkerman, A.; Gebhardt, S.; McRoberts, C.W.; Hamilton, J.T.G.; Williams, J.; Keppler, F. Abiotic methyl bromide formation from vegetation, and its strong dependence on temperature. Environ. Sci. Technol. 2008, 42, 6837–6842. [Google Scholar] [CrossRef] [PubMed]
- Keppler, F.; Eiden, R.; Niedan, V.; Pracht, J.; Schöler, H.F. Halocarbons produced by natural oxidation processes during degradation of organic matter. Nature 2000, 403, 298–301. [Google Scholar] [CrossRef]
- Moore, R.M. A photochemical source of methyl chloride in saline waters. Environ. Sci. Technol. 2008, 42, 1933–1937. [Google Scholar] [CrossRef]
- Wang, W.; He, C.; Gao, Y.; Zhang, Y.; Shi, Q. Isolation and characterization of hydrophilic dissolved organic matter in waters by ion exchange solid phase extraction followed by high resolution mass spectrometry. Environ. Chem. Lett. 2019, 17, 1857–1866. [Google Scholar] [CrossRef]
- McNeill, K.; Canonica, S. Triplet state dissolved organic matter in aquatic photochemistry: Reaction mechanisms, substrate scope, and photophysical properties. Environ. Sci. Process. Impacts 2016, 18, 1381–1399. [Google Scholar] [CrossRef]
- Sandron, S.; Rojas, A.; Wilson, R.; Davies, N.W.; Haddad, P.R.; Shellie, R.A.; Nesterenko, P.N.; Kelleher, B.P.; Paull, B. Chromatographic methods for the isolation, separation and characterisation of dissolved organic matter. Environ. Sci. Process. Impacts 2015, 17, 1531–1567. [Google Scholar] [CrossRef]
- Glover, C.M.; Rosario-Ortiz, F.L. Impact of halides on the photoproduction of reactive intermediates from organic matter. Environ. Sci. Technol. 2013, 47, 13949–13956. [Google Scholar] [CrossRef]
- Brigante, M.; Minella, M.; Mailhot, G.; Maurino, V.; Minero, C.; Vione, D. Formation and reactivity of the dichloride radical (Cl2−•) in surface waters: A modelling approach. Chemosphere 2014, 95, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Mylon, S.E.; Chen, K.L.; Elimelech, M. Influence of natural organic matter and ionic composition on the kinetics and structure of hematite colloid aggregation: Implications to iron depletion in estuaries. Langmuir 2004, 20, 9000–9006. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Díaz, J.D.; Shimabuku, K.K.; Ma, J.; Enumah, Z.O.; Pignatello, J.J.; Mitch, W.A.; Dodd, M.C. Sunlight-driven photochemical halogenation of dissolved organic matter in seawater: A natural abiotic source of organobromine and organoiodine. Environ. Sci. Technol. 2014, 48, 7418–7427. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.M.; Zafiriou, O.C. Photochemical production of methyl iodide in seawater. J. Geophys. Res. 1994, 99, 16415–16420. [Google Scholar] [CrossRef]
- Moore, R.M. Methyl halide production and loss rates from field incubation experiments. Mar. Chem. 2006, 101, 213–219. [Google Scholar] [CrossRef]
- Richter, U.; Wallace, D.W.R. Production of methyl iodide in the tropical Atlantic Ocean. Geophys. Res. Lett. 2004, 31, L23S03. [Google Scholar] [CrossRef]
- Stemmler, I.; Hense, I.; Quack, B.; Maier-Reimer, E. Methyl iodide production in the open ocean. Biogeosciences 2014, 11, 4459–4476. [Google Scholar] [CrossRef][Green Version]
- Dallin, E.; Wan, P.; Krogh, E.; Gill, C.; Moore, R.M. New pH-dependent photosubstitution pathways of syringic acid in aqueous solution: Relevance in environmental photochemistry. J. Photochem. Photobiol. A Chem. 2009, 207, 297–305. [Google Scholar] [CrossRef]
- Yang, Q.; Guo, Y.; Yue, E.; Zhang, S.; Blatchley, E.R., III; Li, J. Methyl chloride produced during UV254 irradiation of saline water. J. Hazard. Mater. 2020, 384, 121263. [Google Scholar] [CrossRef]
- Benner, R.; Opsahl, S. Molecular indicators of the sources and transformations of dissolved organic matter in the Mississippi river plume. Org. Geochem. 2001, 32, 597–611. [Google Scholar] [CrossRef]
- Mekic, M.; Loisel, G.; Zhou, W.; Jiang, B.; Vione, D.; Gligorovski, S. Ionic-strength effects on the reactive uptake of ozone on aqueous pyruvic acid: Implications for air-sea ozone deposition. Environ. Sci. Technol. 2018, 52, 12306–12315. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, L.J.; Nightingale, P.D. Chemistry and release of gases from the surface ocean. Chem. Rev. 2015, 115, 4015–4034. [Google Scholar] [CrossRef] [PubMed]
- Flury, M.; Papritz, A. Bromide in the natural environment: Occurrence and toxicity. J. Environ. Qual. 1993, 22, 747–758. [Google Scholar] [CrossRef]
- Voelker, B.M.; Morel, F.M.M.; Sulzberger, B. Iron redox cycling in surface waters: Effects of humic substances and light. Environ. Sci. Technol. 1997, 31, 1004–1011. [Google Scholar] [CrossRef]
- Yang, Y.; Pignatello, J.J. Participation of the halogens in photochemical reactions in natural and treated waters. Molecules 2017, 22, 1684. [Google Scholar] [CrossRef]
- Zhang, K.; Parker, K.M. Halogen radical oxidants in natural and engineered aquatic systems. Environ. Sci. Technol. 2018, 52, 9579–9594. [Google Scholar] [CrossRef]
- Debbache, I.G.N.; Dekkiche, B.A.; Seraghni, N.; Sehili, T.; Marín, Z.; Santaballa, J.A.; Canle, M. Fe (III)-citrate enhanced sunlight-driven photocatalysis of aqueous Carbamazepine. J. Photochem. Photobiol. A Chem. 2019, 378, 147–155. [Google Scholar]
- Singh, K.; Kumar, A. Kinetics of complex formation of Fe (III) with syringic acid: Experimental and theoretical study. Food Chem. 2018, 265, 96–100. [Google Scholar] [CrossRef]
- Liu, H.; Pu, Y.; Tong, T.; Zhu, X.; Sun, B.; Zhang, X. Photochemical generation of methyl chloride from humic aicd: Impacts of precursor concentration, solution pH, solution salinity and ferric ion. Int. J. Environ. Res. Public Health 2020, 17, 503. [Google Scholar] [CrossRef]
- Parker, K.M.; Mitch, W.A. Halogen radicals contribute to photooxidation in coastal and estuarine waters. Proc. Natl. Acad. Sci. USA 2016, 113, 5868–5873. [Google Scholar] [CrossRef]
- Zafiriou, O.C. Sources and reactions of OH and daughter radicals in seawater. J. Geophys. Res. 1974, 79, 4491–4497. [Google Scholar] [CrossRef]
- Zhou, X.; Mopper, K. Determination of photochemically produced hydroxyl radicals in seawater and freshwater. Mar. Chem. 1990, 30, 71–88. [Google Scholar] [CrossRef]
- Sato, N.; Hamamoto, K.; Kurihara, M.; Abe, M.; Hashimoto, S. Methyl halide production by cultures of marine thraustochytrids, Aurantiochytrium sp., Botryochytrium radiatum, and Schizochytrium sp. Mar. Chem. 2019, 208, 95–102. [Google Scholar] [CrossRef]
- Yan, G.; Ding, Q.; Gao, X. The effect of temperature, salinity and light Intensity on growth and methyl halides production of Phaeocystis globose. Period. Ocean. Univ. China 2019, 49, 67–73. [Google Scholar]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).