
Genotypic Diversity among Angolan Children with Sickle Cell Anemia

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Hematological and Biochemical Parameters
2.3. Sequencing Analysis
2.4. Haplotype Analysis
2.5. Statistical Analysis
3. Results
3.1. Clinical Findings
3.2. Genetic Findings
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McGann, P.T. Time to invest in sickle cell anemia as a global health priority. Pediatrics 2016, 137, e20160348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makani, J.; Cox, S.E.; Soka, D.; Komba, A.N.; Oruo, J.; Mwamtemi, H.; Magesa, P.; Rwezaula, S.; Meda, E.; Mgaya, J.; et al. Mortality in sickle cell anemia in africa: A prospective cohort study in Tanzania. PLoS ONE 2011, 6, e14699. [Google Scholar] [CrossRef] [Green Version]
- Mcgann, P.T.; Ferris, M.G.; Ramamurthy, U.; Santos, B.; de Oliveira, V.; Bernardino, L.; Ware, R.E. A prospective newborn screening and treatment program for sickle cell anemia in Luanda, Angola. Am. J. Hematol. 2013, 88, 984–989. [Google Scholar] [CrossRef] [PubMed]
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle Cell Disease. Nat. Rev. Dis. Prim. 2018, 4, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, T.N. Sickle Cell Disease in Sub-Saharan Africa. Hematol. Oncol. Clin. N. Am. 2016, 30, 343–358. [Google Scholar] [CrossRef]
- Chang, A.K.; Ginter Summarell, C.C.; Birdie, P.T.; Sheehan, V.A. Genetic modifiers of severity in sickle cell disease. Clin. Hemorheol. Microcirc. 2018, 68, 147–164. [Google Scholar] [CrossRef]
- Galarneau, G. Genetic Determinants of Clinical Heterogeneity in Sickle Cell Disease. Ph.D. Thesis, Université de Montréal, Montréal, QC, Canada, 2014. [Google Scholar]
- Bernaudin, F.; Arnaud, C.; Kamdem, A.; Hau, I.; Lelong, F.; Epaud, R.; Pondarré, C.; Pissard, S. Biological impact of α genes, β haplotypes, and G6PD activity in sickle cell anemia at baseline and with hydroxyurea. Blood Adv. 2018, 2, 626–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habara, A.; Steinberg, M.H. Minireview: Genetic basis of heterogeneity and severity in sickle cell disease. Exp. Biol. Med. 2016, 241, 689–696. [Google Scholar] [CrossRef] [Green Version]
- Lettre, G.; Sankaran, V.G.; Bezerra, M.A.C.; Araújo, A.S.; Uda, M.; Sanna, S.; Cao, A.; Schlessinger, D.; Costa, F.F.; Hirschhorn, J.N.; et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and β-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc. Natl. Acad. Sci. USA 2008, 105, 11869–11874. [Google Scholar] [CrossRef] [Green Version]
- Thein, S.L. Genetic association studies in β-hemoglobinopathies. Hematology 2013, 2013, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Akinsheye, I.; Alsultan, A.; Solovieff, N.; Ngo, D.; Baldwin, C.T.; Sebastiani, P.; Chui, D.H.K.; Steinberg, M.H. Fetal hemoglobin in sickle cell anemia. Blood 2011, 118, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Gueye Tall, F.; Martin, C.; Ndour, E.H.M.; Renoux, C.; Ly, I.D.; Connes, P.; Gueye, P.M.; Diallo, R.N.; Diagne, I.; Diop, P.A.; et al. Combined and differential effects of alpha-thalassemia and HbF-quantitative trait loci in Senegalese hydroxyurea-free children with sickle cell anemia. Pediatr. Blood Cancer 2019, 66, e27934. [Google Scholar] [CrossRef]
- Thein, S.L. Genetic Basis and Genetic Modifiers of β-Thalassemia and Sickle Cell Disease. In Gene and Cell Therapies for Beta-Globinopathies; Malik, P., Tisdale, J., Eds.; Springer: New York, NY, USA, 2017; Volume 1013, ISBN 978-1-4939-7297-5. [Google Scholar]
- Manco, L.; Bento, C.; Relvas, L.; Cunha, E.; Pereira, J.; Moreira, V.; Alvarez, M.; Maia, T.; Ribeiro, M.L. Multi-Locus Models to Address Hb F Variability in Portuguese β-Thalassemia Carriers. Hemoglobin 2020, 44, 113–117. [Google Scholar] [CrossRef]
- Thein, S.L.; Menzel, S.; Lathrop, M.; Garner, C. Control of fetal hemoglobin: New insights emerging from genomics and clinical implications. Hum. Mol. Genet. 2009, 18, 216–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menzel, S.; Thein, S.L. Genetic Modifiers of Fetal Haemoglobin in Sickle Cell Disease. Mol. Diagnosis Ther. 2018, 23, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Bitoungui, V.J.N.; Pule, G.D.; Hanchard, N.; Ngogang, J.; Wonkam, A. Beta-globin gene haplotypes among cameroonians and review of the global distribution: Is there a case for a single sickle mutation origin in Africa? OMICS 2015, 19, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Cruz, P.R.S.; Ananina, G.; Gil-da-Silva-Lopes, V.L.; Simioni, M.; Menaa, F.; Bezerra, M.A.C.; Domingos, I.F.; Araújo, A.S.; Pellegrino, R.; Hakonarson, H.; et al. Genetic comparison of sickle cell anaemia cohorts from Brazil and the United States reveals high levels of divergence. Sci. Rep. 2019, 9, 10896. [Google Scholar] [CrossRef]
- Abou-Elew, H.H.; Youssry, I.; Hefny, S.; Hashem, R.H.; Fouad, N.; Zayed, R.A. βS globin gene haplotype and the stroke risk among Egyptian children with sickle cell disease. Hematology 2018, 23, 362–367. [Google Scholar] [CrossRef] [Green Version]
- Loggetto, S.R. Sickle cell anemia: Clinical diversity and beta S-globin haplotypes. Rev. Bras. Hematol. Hemoter. 2013, 35, 155–157. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, M.H.; Sebastiani, P. Genetic modifiers of sickle cell disease. Am. J. Hematol. 2012, 87, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Shaikho, E.M.; Farrell, J.J.; Alsultan, A.; Qutub, H.; Al-Ali, A.K.; Figueiredo, M.S.; Chui, D.H.K.; Farrer, L.A.; Murphy, G.J.; Mostoslavsky, G.; et al. A phased SNP-based classification of sickle cell anemia HBB haplotypes. BMC Genom. 2017, 18, 608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Ghamrawy, M.; Yassa, M.E.; Tousson, A.M.S.; El-hady, M.A.; Mikhaeil, E.; Mohamed, N.B.; Khorshied, M.M. Association between BCL11A, HSB1L-MYB, and XmnI γG-158 (C/T) gene polymorphism and hemoglobin F level in Egyptian sickle cell disease patients. Ann. Hematol. 2020, 99, 2279–2288. [Google Scholar] [CrossRef]
- McGann, P.T.; Williams, T.N.; Olupot-Olupot, P.; Tomlinson, G.A.; Lane, A.; Luís Reis da Fonseca, J.; Kitenge, R.; Mochamah, G.; Wabwire, H.; Stuber, S.; et al. Realizing effectiveness across continents with hydroxyurea: Enrollment and baseline characteristics of the multicenter REACH study in Sub-Saharan Africa. Am. J. Hematol. 2018, 93, 537–545. [Google Scholar] [CrossRef] [Green Version]
- Borges, E.; Tchonhi, C.; Couto, C.S.B.; Gomes, V.; Amorim, A.; Prata, M.J.; Brito, M. Unusual β-Globin Haplotype Distribution in Newborns from Bengo, Angola. Hemoglobin 2019, 43, 149–154. [Google Scholar] [CrossRef]
- Chamouine, A.; Saandi, T.; Muszlak, M.; Larmaraud, J.; Lambrecht, L.; Poisson, J.; Balicchi, J.; Pissard, S.; Elenga, N. High fetal hemoglobin level is associated with increased risk of cerebral vasculopathy in children with sickle cell disease in Mayotte. BMC Pediatr. 2020, 20, 302. [Google Scholar] [CrossRef]
- Piel, F.B.; Steinberg, M.H.; Rees, D.C. Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 1561–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, B.; Delgadinho, M.; Ferreira, J.; Germano, I.; Miranda, A.; Arez, A.P.; Faustino, P.; Brito, M. Co-Inheritance of alpha-thalassemia and sickle cell disease in a cohort of Angolan pediatric patients. Mol. Biol. Rep. 2020, 47, 5397–5402. [Google Scholar] [CrossRef]
- Adeyemo, T.A.; Ojewunmi, O.O.; Oyetunji, I.A.; Rooks, H.; Rees, D.C.; Akinsulie, A.O.; Akanmu, A.S.; Thein, S.L.; Menzel, S. A survey of genetic fetal-haemoglobin modifiers in Nigerian patients with sickle cell anaemia. PLoS ONE 2018, 13, e0197927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sales, R.R.; Belisário, A.R.; Faria, G.; Mendes, F.; Luizon, M.R.; Viana, M.B. Functional polymorphisms of BCL11A and HBS1L-MYB genes affect both fetal hemoglobin level and clinical outcomes in a cohort of children with sickle cell anemia. Ann. Hematol. 2020, 99, 1453–1463. [Google Scholar] [CrossRef]
- Huang, P.; Keller, C.A.; Giardine, B.; Grevet, J.D.; Davies, J.O.J.; Hughes, J.R.; Kurita, R.; Nakamura, Y.; Hardison, R.C.; Blobel, G.A. Comparative analysis of three-dimensional chromosomal architecture identifies a novel fetal hemoglobin regulatory element. Genes Dev. 2017, 31, 1704–1713. [Google Scholar] [CrossRef] [Green Version]
- Ivaldi, M.S.; Diaz, L.F.; Chakalova, L.; Lee, J.; Krivega, I.; Dean, A. Fetal g-globin genes are regulated by the BGLT3 long noncoding RNA locus. Blood 2018, 132, 1963–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manca, L.; Masala, B. Disorders of the synthesis of human fetal hemoglobin. IUBMB Life 2008, 60, 94–111. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, Z.; Karimi, M.; Haghshenass, M.; Merat, A. β-Globin Gene Cluster Haplotypes in Sickle Cell Patients From Southwest Iran. Am. J. Hematol. 2003, 74, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Al-Ali, A.K.; Alsulaiman, A.; Alzahrani, A.J.; Obeid, O.T.; Vatte, C.B.; Cyrus, C.; Alnafie, A.N.; Alali, R.A.; Alfarhan, M.; Mozeleski, B.; et al. Prevalence and Diversity of Haplotypes of Sickle Cell Disease in the Eastern Province of Saudi Arabia. Hemoglobin 2020, 44, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Nemati, H.; Rahimi, Z.; Bahrami, G. The Xmn1 polymorphic site 5′ to the Gγ gene and its correlation to the Gγ:Aγ ratio, age at first blood transfusion and clinical features in β-Thalassemia patients from Western Iran. Mol. Biol. Rep. 2010, 37, 159–164. [Google Scholar] [CrossRef] [PubMed]
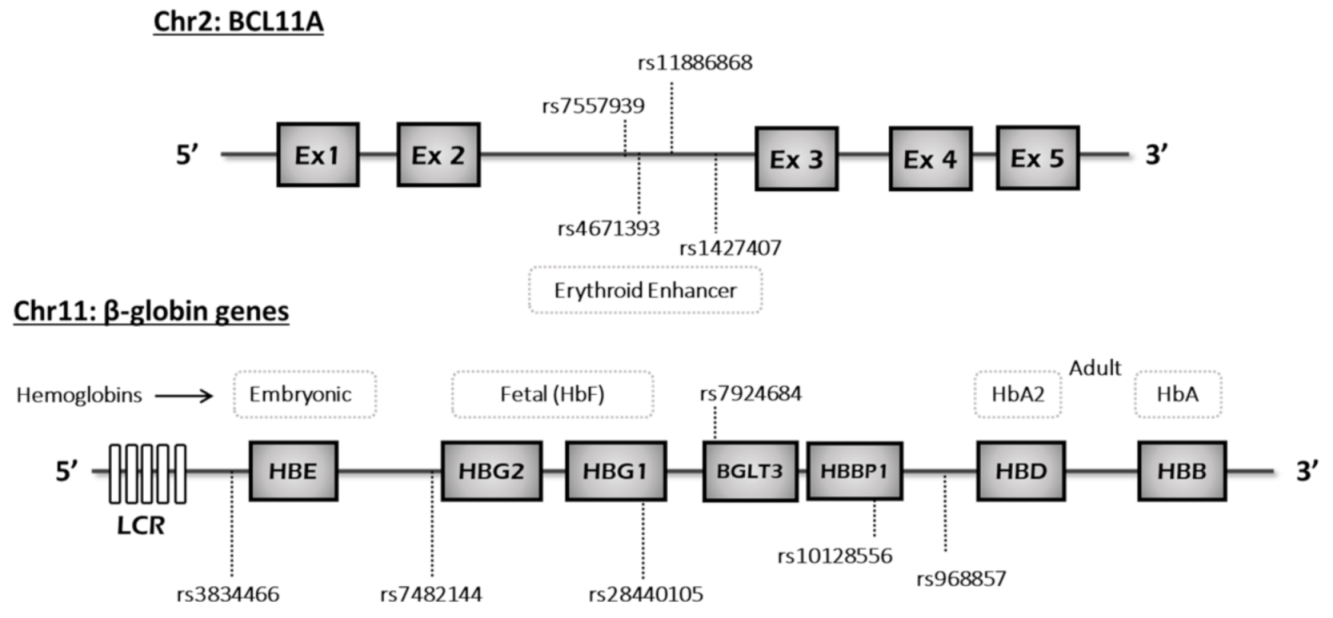

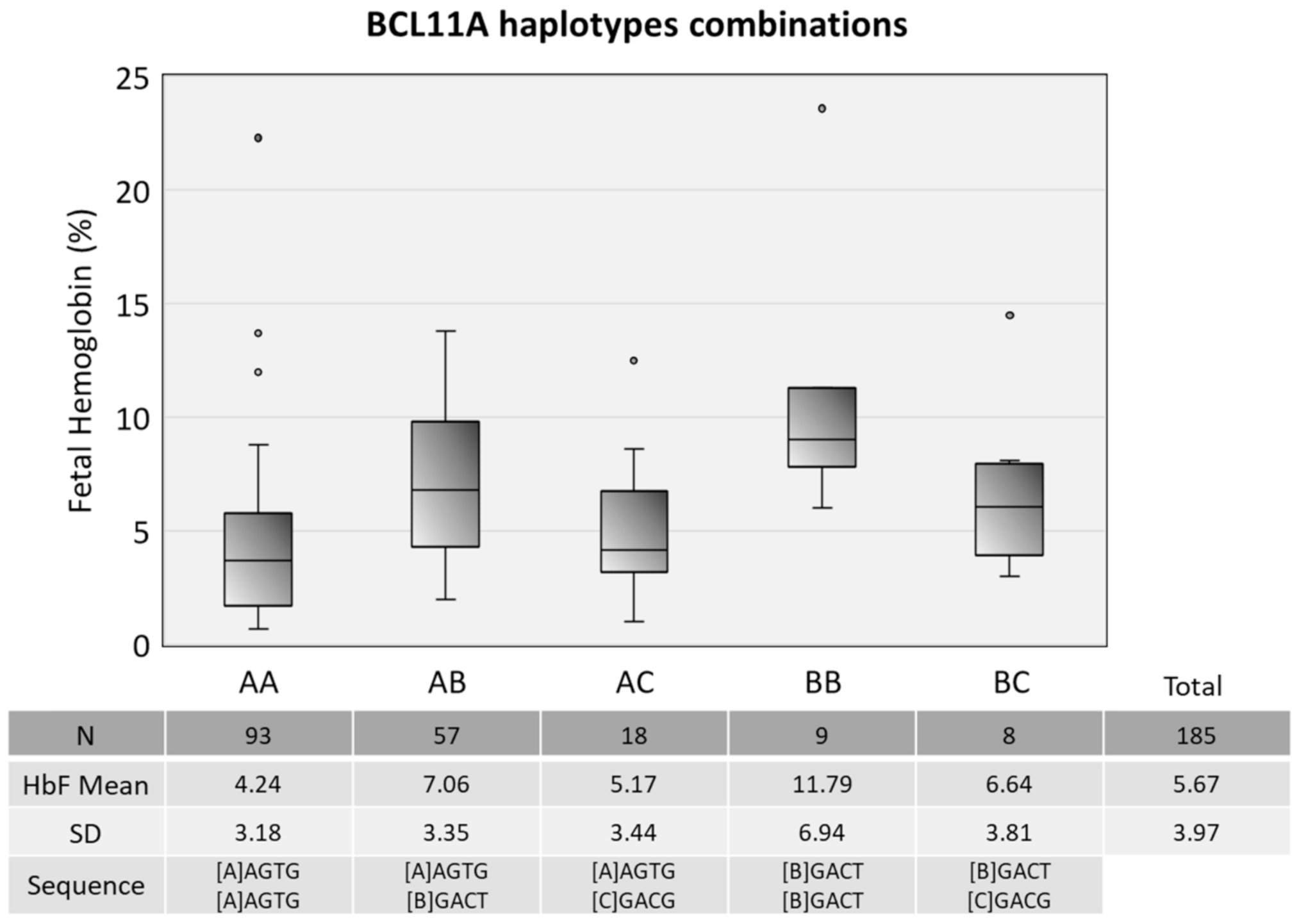

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | CAR/CAR | CAR/SEN | CAR/CAM | CAR/BEN | p-Value ** | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n = 176 | n = 2 | n = 2 | n = 11 | ||||||||||
| Mean | SD | p-Value * | Mean | SD | p-Value * | Mean | SD | p-Value * | Mean | SD | p-Value * | ||
| Hemoglobin (g/dL) | 7.29 | ±0.95 | 0.057 | 7.52 | ±1.2 | 0.792 | 9.19 | ±1.15 | 0.031 | 7.64 | ±0.93 | 0.256 | 0.103 |
| Fetal Hemoglobin (%) | 5.43 | ±3.70 | 0.013 | 13.45 | ±0.35 | 0.022 | 15.10 | ±12.30 | 0.126 | 6.60 | ±3.49 | 0.235 | 0.025 |
| Erythrocytes (1012 L) | 2.92 | ±0.6 | 0.092 | 3.42 | ±1.11 | 0.546 | 4.24 | ±0.16 | 0.030 | 3.11 | ±0.74 | 0.463 | 0.124 |
| MCV (fL) | 77.17 | ±8.70 | 0.432 | 69.43 | ±14.88 | 0.403 | 65.26 | ±4.44 | 0.062 | 78.00 | ±8.03 | 0.785 | 0.236 |
| MCH (pg) | 25.47 | ±2.97 | 0.130 | 22.66 | ±3.91 | 0.212 | 21.80 | ±3.54 | 0.100 | 25.15 | ±2.77 | 0.629 | 0.202 |
| White blood cells count (109 L) | 14.06 | ±4.75 | 0.827 | 12.83 | ±0.56 | 0.847 | 8.76 | ±1.41 | 0.048 | 14.49 | ±4.01 | 0.486 | 0.223 |
| Neutrophil count (109 L) | 5.90 | ±2.34 | 0.764 | 4.11 | ±2.31 | 0.265 | 4.95 | ±1.57 | 0.610 | 6.67 | ±2.70 | 0.291 | 0.468 |
| Platelet count (109 L) | 438.0 | ±174.78 | 0.474 | 433.6 | ±99.11 | 0.949 | 342.0 | ±125.87 | 0.396 | 415.3 | ±139.40 | 0.629 | 0.807 |
| Reticulocyte count (%) | 10.36 | ±4.69 | 0.884 | 8.51 | ±5.81 | 0.690 | 5.00 | ±2.81 | 0.059 | 10.68 | ±4.30 | 0.406 | 0.226 |
| N° of transfusions/year | 0.41 | ±0.55 | 0.169 | 0.00 | ±0.00 | 0.075 | 0.25 | ±0.35 | 0.740 | 0.27 | ±0.32 | 0.508 | 0.282 |
| N° of hospitalizations/year | 0.49 | ±0.51 | 0.524 | 0.25 | ±0.35 | 0.486 | 0.25 | ±0.35 | 0.486 | 0.49 | ±0.54 | 0.899 | 0.799 |
| G gamma:A gamma ratio | 0.53 | ±0.22 | 0.181 | 1.38 | ±0.08 | 0.016 | 0.69 | ±0.50 | 0.786 | 0.55 | ±0.19 | 0.707 | 0.108 |
| LDH (U/L) | 436.72 | ±154.19 | 0.105 | 742.85 | 0.109 | 300.00 | 0.264 | 344.14 | ±128.26 | 0.057 | 0.061 | ||
| Polymorphisms | Hemoglobin (g/dL) | Fetal Hemoglobin (%) | Erythrocytes (1012 L) | MCV (fL) | MCH (pg) | White Blood Cells Count (109 L) | Neutrophil Count (109 L) | Platelet Count (109 L) | Reticulocyte Count (%) | Ratio G Gamma: A Gamma | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr2 BCL11A | rs4671393 | GG (n = 95) | 7.14 ± 0.94 | 4.17 ± 3.18 | 2.90 ± 0.62 | 76.46 ± 9.08 | 25.14 ± 3.10 | 13.65 ± 3.96 | 5.43 ± 1.92 | 416.48 ± 156.66 | 10.36 ± 4.74 | 0.49 ± 0.25 |
| GA (n = 79) | 7.43 ± 0.90 | 6.71 ± 3.42 | 2.95 ± 0.59 | 77.83 ± 8.29 | 25.64 ± 2.83 | 14.44 ± 5.45 | 6.43 ± 2.52 | 455.98 ± 192.38 | 10.52 ± 4.52 | 0.58 ± 0.23 | ||
| AA (n= 18) | 7.88 ± 1.20 | 8.93 ± 6.22 | 3.14 ± 0.76 | 76.73 ± 9.07 | 25.65 ± 3.01 | 14.07 ± 4.54 | 6.13 ± 3.07 | 450.05 ± 138.66 | 9.16 ± 4.90 | 0.64 ± 0.20 | ||
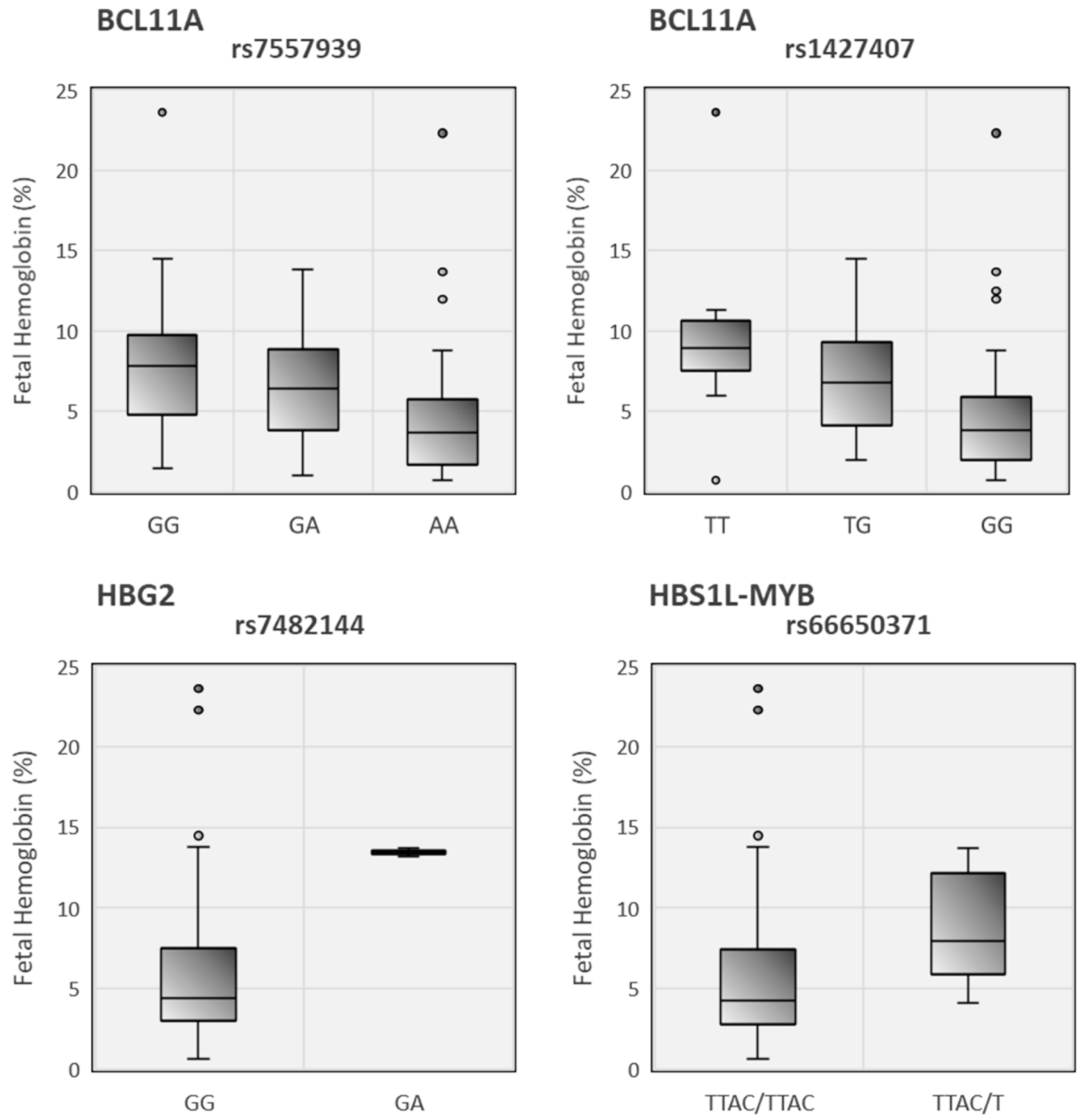
| p-value | 0.005 | <0.001 | 0.333 | 0.586 | 0.508 | 0.541 | 0.021 | 0.298 | 0.531 | 0.006 | ||
| rs11886868 | CC (n = 17) | 8.05 ± 1.00 | 9.37 ± 6.12 | 3.21 ± 0.71 | 75.69 ± 8.17 | 25.54 ± 3.06 | 13.92 ± 4.63 | 6.18 ± 3.16 | 465.26 ± 126.58 | 8.24 ± 3.09 | 0.65 ± 0.20 | |
| CT (n = 78) | 7.43 ± 0.90 | 6.69 ±3.44 | 2.95 ± 0.59 | 77.96 ± 8.26 | 25.68 ± 2.82 | 14.52 ± 5.43 | 6.45 ± 2.52 | 457.68 ± 193.02 | 10.55 ± 4.54 | 0.59 ± 0.23 | ||
| TT (n = 96) | 7.15 ± 0.94 | 4.21 ± 3.19 | 2.91 ± 0.62 | 76.37 ± 9.07 | 25.12 ± 3.10 | 13.58 ± 3.99 | 5.41 ± 1.91 | 415.50 ± 156.13 | 10.34 ± 4.73 | 0.49 ± 0.25 | ||
| p-value | 0.001 | <0.001 | 0.166 | 0.399 | 0.451 | 0.426 | 0.015 | 0.209 | 0.159 | 0.004 | ||
| rs1427407 | TT (n = 12) | 8.04 ± 1.15 | 10.51 ± 6.74 | 3.11 ± 0.73 | 78.12 ± 9.04 | 26.41 ± 3.67 | 13.19 ± 4.86 | 5.57 ± 3.14 | 474.51 ± 168.18 | 8.11 ± 3.02 | 0.65 ± 0.29 | |
| TG (n = 66) | 7.49 ± 0.81 | 7.01 ± 3.39 | 3.00 ± 0.58 | 76.98 ± 8.41 | 25.41 ± 2.70 | 14.86 ± 5.66 | 6.47 ± 2.53 | 465.25 ± 198.56 | 9.94 ± 4.35 | 0.57 ± 0.20 | ||
| GG (n = 114) | 7.16 ± 1.00 | 4.639 ± 3.22 | 2.90 ± 4.91 | 76.98 ± 8.96 | 25.28 ± 3.94 | 13.60 ± 3.94 | 5.63 ± 2.09 | 414.81 ± 151.47 | 10.76 ± 4.91 | 0.51 ± 0.25 | ||
| p-value | 0.003 | <0.001 | 0.336 | 0.909 | 0.457 | 0.183 | 0.071 | 0.117 | 0.124 | 0.081 | ||
| rs7557939 | GG (n = 18) | 7.88 ± 1.20 | 8.93 ± 6.22 | 3.14 ± 0.76 | 76.73 ± 9.07 | 25.65 ± 3.01 | 14.07 ± 4.54 | 6.13 ± 3.07 | 450.05 ± 138.66 | 9.16 ± 4.90 | 0.64 ± 0.20 | |
| GA (n = 80) | 7.42 ± 0.90 | 6.64 ± 3.45 | 2.95 ± 0.58 | 77.85 ± 8.24 | 25.65 ± 2.81 | 14.43 ± 5.41 | 6.45 ± 2.51 | 455.53 ± 191.20 | 10.52 ± 4.49 | 0.58 ± 0.23 | ||
| AA (n = 94) | 7.15 ± 0.94 | 4.20 ± 3.18 | 2.91 ± 0.62 | 76.43 ± 9.12 | 25.13 ± 3.12 | 13.65 ± 3.98 | 5.40 ± 1.90 | 416.44 ± 157.50 | 10.36 ± 4.77 | 0.49 ± 0.25 | ||
| p-value | 0.007 | <0.001 | 0.349 | 0.563 | 0.481 | 0.549 | 0.013 | 0.304 | 0.531 | 0.007 | ||
| Chr6 HBSIL-MYB | rs66650371 | TTAC/TTAC (n = 182) | 7.29 ± 0.96 | 5.50 ± 03.94 | 2.91 ± 0.60 | 77.34 ± 8.60 | 25.48 ± 2.91 | 14.19 ± 4.71 | 5.99 ± 2.36 | 437.70 ± 174.01 | 10.53 ± 4.65 | 0.53 ± 0.23 |
| TTAC/T (n = 10) | 8.16 ± 0.72 | 8.66 ± 3.6 | 3.52 ± 0.82 | 71.79 ± 10.14 | 23.93 ± 3.90 | 10.79 ± 2.18 | 4.65 ± 1.60 | 402.75 ± 105.45 | 6.40 ± 2.73 | 0.68 ± 0.35 | ||
| p-value | 0.005 | 0.02 | 0.002 | 0.05 | 0.11 | 0.025 | 0.08 | 0.531 | 0.006 | 0.81 | ||
| Chr11 HBG2 | rs7482144 | GG (n = 190) | 7.33 ± 0.97 | 5.57 ± 3.91 | 2.94 ± 0.62 | 77.13 ± 8.68 | 25.42 ± 2.97 | 14.02 ± 4.69 | 5.93 ± 2.34 | 435.90 ± 171.86 | 10.33 ± 4.66 | 0.53 ± 0.23 |
| GA (n = 2) | 7.52 ± 1.19 | 13.45 ± 0.35 | 3.42 ± 1.11 | 69.43 ± 14.88 | 22.66 ± 3.91 | 12.83 ± 0.56 | 4.11 ± 2.31 | 433.58 ± 99.11 | 8.51 ± 5.81 | 1.38 ± 0.08 | ||
| p-value | 0.782 | 0.005 | 0.284 | 0.216 | 0.193 | 0.721 | 0.275 | 0.985 | 0.584 | <0.001 | ||
| Chr11 BGLT3 | rs7924684 | CC (n = 188) | 7.34 ± 0.97 | 5.74 ± 3.97 | 2.95 ± 0.63 | 77.04 ± 8.81 | 25.38 ± 2.99 | 14.03 ± 4.69 | 5.91 ± 2.36 | 437.22 ± 171.35 | 10.32 ± 4.70 | 0.55 ± 0.24 |
| CT (n = 4) | 7.10 ± 0.82 | 1.85 ± 2.04 | 2.73 ± 0.33 | 77.60 ± 4.59 | 26.15 ± 2.38 | 13.30 ± 4.38 | 5.97 ± 1.55 | 372.50 ± 168.56 | 9.84 ± 2.62 | 0.23 ± 0.26 | ||
| p-value | 0.644 | 0.053 | 0.488 | 0.899 | 0.608 | 0.759 | 0.961 | 0.456 | 0.837 | 0.007 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delgadinho, M.; Ginete, C.; Santos, B.; Miranda, A.; Brito, M. Genotypic Diversity among Angolan Children with Sickle Cell Anemia. Int. J. Environ. Res. Public Health 2021, 18, 5417. https://doi.org/10.3390/ijerph18105417
Delgadinho M, Ginete C, Santos B, Miranda A, Brito M. Genotypic Diversity among Angolan Children with Sickle Cell Anemia. International Journal of Environmental Research and Public Health. 2021; 18(10):5417. https://doi.org/10.3390/ijerph18105417
Chicago/Turabian StyleDelgadinho, Mariana, Catarina Ginete, Brígida Santos, Armandina Miranda, and Miguel Brito. 2021. "Genotypic Diversity among Angolan Children with Sickle Cell Anemia" International Journal of Environmental Research and Public Health 18, no. 10: 5417. https://doi.org/10.3390/ijerph18105417