Physiological Changes and Pathological Pain Associated with Sedentary Lifestyle-Induced Body Systems Fat Accumulation and Their Modulation by Physical Exercise



Abstract
:
1. Introduction
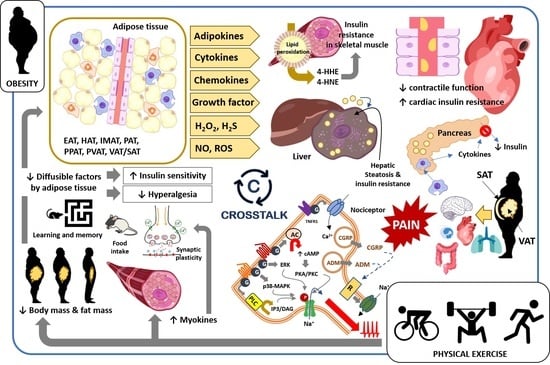
2. Obesity-Induced Changes in Tissues, Organs, and Body Systems: Crosstalk between Fat and Body Tissues
2.1. Skeletal Muscle
2.2. Cardiovascular System
2.3. Accumulation of Fat in Liver and Pancreas
2.4. Visceral, Abdominal, and Subcutaneous Adipose Tissues
2.5. Obesity: Fat Deposition, Dysfunctional Adipose Tissue, and Metabolic Complications
3. Obesity and Pain: Crosstalk between Adipose Tissue and Nociceptive Somatosensory Nervous System
3.1. Adipokines
3.2. Cytokines and Chemokines
3.3. Growth Factors and Other Diffusible Factors
4. Effect of Physical Exercise on Adipose Tissue and Other Body Tissues, Organs, and Systems in Obese Subjects and Its Impact on Pathological Pain
4.1. Skeletal Muscle
4.2. Bone Tissue
4.3. Immune System
4.4. Cardiovascular System
4.5. Respiratory System
4.6. Gastrointestinal System
4.7. Endocrine System
4.8. Nervous System
4.9. Pathological Pain: Relationship with Sedentary Behavior and Modulation by Physical Exercise
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hu, F.B.; Li, T.Y.; Colditz, G.A.; Willett, W.C.; Manson, J.E. Television watching and other sedentary behaviors in relation to risk of obesity and type 2 diabetes mellitus in women. JAMA 2003, 289, 1785–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kay, S.J.; Fiatarone Singh, M.A. The influence of physical activity on abdominal fat: A systematic review of the literature. Obes. Rev. 2006, 7, 183–200. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, S. Physical activity and abdominal obesity in youth. Appl. Physiol. Nutr. Metab. 2009, 34, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.; Edwardson, C.L.; Morgan, B.; Horsfield, M.A.; Bodicoat, D.H.; Biddle, S.J.; Gorely, T.; Nimmo, M.A.; McCann, G.P.; Khunti, K.; et al. Associations of Sedentary Time with Fat Distribution in a High-Risk Population. Med. Sci. Sports Exerc. 2015, 47, 1727–1734. [Google Scholar] [CrossRef] [Green Version]
- Galmes-Panades, A.M.; Konieczna, J.; Abete, I.; Colom, A.; Rosique-Esteban, N.; Zulet, M.A.; Vázquez, Z.; Estruch, R.; Vidal, J.; Toledo, E.; et al. PREDIMED-Plus investigators. Lifestyle factors and visceral adipose tissue: Results from the PREDIMED-PLUS study. PLoS ONE 2019, 14, e0210726. [Google Scholar] [CrossRef] [Green Version]
- Moschonis, G.; Kalliora, A.C.; Costarelli, V.; Papandreou, C.; Koutoukidis, D.; Lionis, C.; Chrousos, G.P.; Manios, Y.; Healthy Growth Study Group. Identification of lifestyle patterns associated with obesity and fat mass in children: The Healthy Growth Study. Public Health Nutr. 2014, 17, 614–624. [Google Scholar] [CrossRef] [Green Version]
- Hales, C.M.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Prevalence of Obesity and Severe Obesity among Adults: United States, 2017–2018; National Center for Health Statistics: Hyattsville, MD, USA, 2021. [Google Scholar]
- Eurostat. Overweight and Obesity—BMI Statistics; European Statistical System: Luxembourg, 2021. [Google Scholar]
- Spieker, E.A.; Pyzocha, N. Economic Impact of Obesity. Prim. Care 2016, 43, 83–95. [Google Scholar] [CrossRef]
- Hartman, M.; Martin, A.B.; Espinosa, N.; Catlin, A.; The National Health Expenditure Accounts Team. National Health Care Spending. In 2016: Spending and Enrollment Slow After Initial Growth Coverage Expansions. Health Aff. 2018, 37, 150–160. [Google Scholar] [CrossRef] [Green Version]
- OECD. The Heavy Burden of Obesity: The Economics of Prevention, OECD Health Policy Studies; OECD Publishing: Paris, France, 2019. [Google Scholar] [CrossRef]
- World Health Organization. Obesity and Overweight; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Pi-Sunyer, F.X. The obesity epidemic: Pathophysiology and consequences of obesity. Obes Res. 2002, 10, 97S–104S. [Google Scholar] [CrossRef]
- Standl, E. Dysglycemia and abdominal obesity. Curr. Vasc. Pharmacol. 2012, 10, 678–679. [Google Scholar] [CrossRef]
- Feakins, R.M. Obesity and metabolic syndrome: Pathological effects on the gastrointestinal tract. Histopathology 2016, 68, 630–640. [Google Scholar] [CrossRef]
- Chang, J.W.; Chen, H.L.; Su, H.J.; Lee, C.C. Abdominal Obesity and Insulin Resistance in People Exposed to Moderate-to-High Levels of Dioxin. PLoS ONE 2016, 11, e0145818. [Google Scholar] [CrossRef] [Green Version]
- Landin, K.; Stigendal, L.; Eriksson, E.; Krotkiewski, M.; Risberg, B.; Tengborn, L.; Smith, U. Abdominal obesity is associated with an impaired fibrinolytic activity and elevated plasminogen activator inhibitor-1. Metabolism 1990, 39, 1044–1048. [Google Scholar] [CrossRef]
- Katzel, L.I.; Busby-Whitehead, M.J.; Goldberg, A.P. Adverse effects of abdominal obesity on lipoprotein lipids in healthy older men. Exp. Gerontol. 1993, 28, 411–420. [Google Scholar] [CrossRef]
- Strasser, B.; Arvandi, M.; Pasha, E.P.; Haley, A.P.; Stanforth, P.; Tanaka, H. Abdominal obesity is associated with arterial stiffness in middle-aged adults. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 495–502. [Google Scholar] [CrossRef]
- Zhao, Y.; Qin, P.; Sun, H.; Liu, Y.; Liu, D.; Zhou, Q.; Guo, C.; Li, Q.; Tian, G.; Wu, X.; et al. Metabolically healthy general and abdominal obesity are associated with increased risk of hypertension. Br. J. Nutr. 2021, 123, 583–591. [Google Scholar] [CrossRef]
- Krzesiński, P.; Stańczyk, A.; Piotrowicz, K.; Gielerak, G.; Uziębło-Zyczkowska, B.; Skrobowski, A. Abdominal obesity and hypertension: A double burden to the heart. Hypertens. Res. 2016, 39, 349–355. [Google Scholar] [CrossRef]
- Thumann, B.F.; Michels, N.; Felső, R.; Hunsberger, M.; Kaprio, J.; Moreno, L.A.; Siani, A.; Tornaritis, M.; Veidebaum, T.; De Henauw, S.; et al. Associations between sleep duration and insulin resistance in European children and adolescents considering the mediating role of abdominal obesity. PLoS ONE 2021, 15, e0235049. [Google Scholar] [CrossRef]
- Ross, R.; Després, J.P. Abdominal obesity, insulin resistance, and the metabolic syndrome: Contribution of physical activity/exercise. Obesity 2009, 17, S1–S2. [Google Scholar] [CrossRef]
- Velásquez-Rodríguez, C.M.; Velásquez-Villa, M.; Gómez-Ocampo, L.; Bermúdez-Cardona, J. Abdominal obesity and low physical activity are associated with insulin resistance in overweight adolescents: A cross-sectional study. BMC Pediatr. 2014, 14, 258. [Google Scholar] [CrossRef] [Green Version]
- Marchand, N.E.; Sparks, J.A.; Tedeschi, S.K.; Malspeis, S.; Costenbader, K.H.; Karlson, E.W.; Lu, B. Abdominal Obesity in Comparison with General Obesity and Risk of Developing Rheumatoid Arthritis in Women. J. Rheumatol. 2021, 48, 165–173. [Google Scholar] [CrossRef]
- Chan, J.M.; Rimm, E.B.; Colditz, G.A.; Stampfer, M.J.; Willett, W.C. Obesity, fat distribution, and weight gain as risk factors for clinical diabetes in men. Diabetes Care 1994, 17, 961–969. [Google Scholar] [CrossRef] [Green Version]
- Souza, F.A.; Dias, R.; Fernandes, C.E.; Pimentel, F.; Dias, D. Menstrual irregularity: A possible clinical marker of metabolic dysfunction in women with class III obesity. Gynecol. Endocrinol. 2010, 10, 768–772. [Google Scholar] [CrossRef]
- Aune, D.; Norat, T.; Vatten, L.J. Body mass index, abdominal fatness and the risk of gallbladder disease. Eur. J. Epidemiol. 2015, 9, 1009–1019. [Google Scholar] [CrossRef]
- Shojaee-Moradie, F.; Baynes, K.C.; Pentecost, C.; Bell, J.D.; Thomas, E.L.; Jackson, N.C.; Stolinski, M.; Whyte, M.; Lovell, D.; Bowes, S.B.; et al. Exercise training reduces fatty acid availability and improves the insulin sensitivity of glucose metabolism. Diabetologia 2007, 50, 404–413. [Google Scholar] [CrossRef] [Green Version]
- Keating, S.E.; Coombes, J.S.; Stowasser, M.; Bailey, T.G. The Role of Exercise in Patients with Obesity and Hypertension. Curr. Hypertens. Rep. 2021, 22, 77. [Google Scholar] [CrossRef]
- Collier, S.R.; Sandberg, K.; Moody, A.M.; Frechette, V.; Curry, C.D.; Ji, H.; Gowdar, R.; Chaudhuri, D.; Meucci, M. Reduction of plasma aldosterone and arterial stiffness in obese pre- and stage1 hypertensive subjects after aerobic exercise. J. Hum. Hypertens. 2015, 29, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Aziz, C.B.; Omar, N.; Abdullah, W.Z.; Jalil, R.A.; Nik, W.S.; Zakaria, R. Reduced fibrinogen, fibrinolytic biomarkers, and physical parameters after a weight-loss program in obese subjects. N. Am. J. Med. Sci. 2014, 6, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Lemmey, A.B.; Williams, S.L.; Marcora, S.M.; Jones, J.; Maddison, P.J. Are the benefits of a high-intensity progressive resistance training program sustained in rheumatoid arthritis patients? A 3-year follow up study. Arthritis Care Res. 2012, 64, 71–75. [Google Scholar] [CrossRef]
- Huang, M.H.; Chen, C.H.; Chen, T.W.; Weng, M.C.; Wang, W.T.; Wang, Y.L. The effects of weight reduction on the rehabilitation of patients with knee osteoarthritis and obesity. Arthritis Care Res. 2000, 13, 398–405. [Google Scholar] [CrossRef]
- Mena, G.P.; Mielke, G.I.; Brown, W.J. Prospective associations between physical activity and BMI with irregular periods and heavy menstrual bleeding in a large cohort of Australian women. Hum. Reprod. 2021, 36, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Martín-Moraleda, E.; Delisle, C.; Collado Mateo, D.; Aznar-Lain, S. Weight loss and body composition changes through ketogenic diet and physical activity: A methodological and systematic review. Nutr. Hosp. 2019, 36, 1196–1204. [Google Scholar] [PubMed] [Green Version]
- Farias Edos, S.; Gonçalves, E.M.; Morcillo, A.M.; Guerra-Júnior, G.; Amancio, O.M. Effects of programmed physical activity on body composition in post-pubertal schoolchildren. J. Pediatr. 2015, 91, 122–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, L.H.; Slentz, C.A.; Bateman, L.A.; Shields, A.T.; Piner, L.W.; Bales, C.W.; Houmard, J.A.; Kraus, W.E. Effects of aerobic and/or resistance training on body mass and fat mass in overweight or obese adults. J. Appl. Physiol. 2012, 113, 1831–1837. [Google Scholar] [CrossRef]
- Westerterp, K.R. Exercise, energy balance and body composition. Eur. J. Clin. Nutr. 2018, 72, 1246–1250. [Google Scholar] [CrossRef]
- Yarizadeh, H.; Eftekhar, R.; Anjom-Shoae, J.; Speakman, J.R.; Djafarian, K. The Effect of Aerobic and Resistance Training and Combined Exercise Modalities on Subcutaneous Abdominal Fat: A Systematic Review and Meta-analysis of Randomized Clinical Trials. Adv. Nutr. 2021, 12, 179–196. [Google Scholar] [CrossRef]
- Schwingshackl, L.; Dias, S.; Strasser, B.; Hoffmann, G. Impact of different training modalities on anthropometric and metabolic characteristics in overweight/obese subjects: A systematic review and network meta-analysis. PLoS ONE 2013, 8, e82853. [Google Scholar] [CrossRef] [Green Version]
- Coker, R.H.; William, R.H.; Kortebein, P.M.; Sullivan, D.H.; Evans, W.J. Influence of exercise intensity on abdominal fat and adiponectin in elderly adults. Metab. Syndr. Relat. Disord. 2009, 7, 363–368. [Google Scholar] [CrossRef]
- Ohkawara, K.; Tanaka, S.; Miyachi, M.; Ishikawa-Takata, K.; Tabata, I. A dose-response relation between aerobic exercise and visceral fat reduction: Systematic review of clinical trials. Int. J. Obes. 2007, 31, 1786–1797. [Google Scholar] [CrossRef] [Green Version]
- Okura, T.; Nakata, Y.; Lee, D.J.; Ohkawara, K.; Tanaka, K. Effects of aerobic exercise and obesity phenotype on abdominal fat reduction in response to weight loss. Int. J. Obes. 2005, 29, 1259–1266. [Google Scholar] [CrossRef] [Green Version]
- Vissers, D.; Hens, W.; Taeymans, J.; Baeyens, J.P.; Poortmans, J.; Van Gaal, L. The effect of exercise on visceral adipose tissue in overweight adults: A systematic review and meta-analysis. PLoS ONE 2013, 8, e56415. [Google Scholar] [CrossRef] [Green Version]
- Ibrahimi-Kaçuri, D.; Murtezani, A.; Rrecaj, S.; Martinaj, M.; Haxhiu, B. Low back pain and obesity. Med. Arch. 2015, 69, 114–116. [Google Scholar] [CrossRef] [Green Version]
- Melissas, J.; Volakakis, E.; Hadjipavlou, A. Low-back pain in morbidly obese patients and the effect of weight loss following surgery. Obes. Surg. 2003, 13, 389–393. [Google Scholar] [CrossRef]
- Vismara, L.; Menegoni, F.; Zaina, F.; Galli, M.; Negrini, S.; Capodaglio, P. Effect of obesity and low back pain on spinal mobility: A cross sectional study in women. J. Neuroeng. Rehabil. 2010, 18, 73. [Google Scholar] [CrossRef] [Green Version]
- Shiri, R.; Solovieva, S.; Husgafvel-Pursiainen, K.; Telama, R.; Yang, X.; Viikari, J.; Raitakari, O.T.; Viikari-Juntura, E. The role of obesity and physical activity in non-specific and radiating low back pain: The Young Finns study. Semin. Arthritis Rheum. 2013, 42, 640–650. [Google Scholar] [CrossRef]
- Leboeuf-Yde, C.; Kyvik, K.O.; Bruun, N.H. Low back pain and lifestyle. Part II—Obesity. Information from a population-based sample of 29,424 twin subjects. Spine 1999, 24, 779–783. [Google Scholar] [CrossRef]
- Cimolin, V.; Vismara, L.; Galli, M.; Zaina, F.; Negrini, S.; Capodaglio, P. Effects of obesity and chronic low back pain on gait. J. Neuroeng. Rehabil. 2011, 8, 55. [Google Scholar] [CrossRef] [Green Version]
- Bener, A.; Alwash, R.; Gaber, T.; Lovasz, G. Obesity and low back pain. Coll. Antropol. 2003, 27, 95–104. [Google Scholar]
- Mangwani, J.; Giles, C.; Mullins, M.; Salih, T.; Natali, C. Obesity and recovery from low back pain: A prospective study to investigate the effect of body mass index on recovery from low back pain. Ann. R Coll. Surg. Engl. 2010, 92, 23–26. [Google Scholar] [CrossRef]
- Deyo, R.A.; Bass, J.E. Lifestyle and low-back pain. The influence of smoking and obesity. Spine 1989, 14, 501–506. [Google Scholar] [CrossRef]
- Han, T.S.; Schouten, J.S.; Lean, M.E.; Seidell, J.C. The prevalence of low back pain and associations with body fatness, fat distribution and height. Int. J. Obes. Relat. Metab. Disord. 1997, 21, 600–607. [Google Scholar] [CrossRef] [Green Version]
- Häuser, W.; Schmutzer, G.; Brähler, E.; Schiltenwolf, M.; Hilbert, A. The impact of body weight and depression on low back pain in a representative population sample. Pain Med. 2014, 15, 1316–1327. [Google Scholar] [CrossRef] [Green Version]
- Jannini, S.N.; Dória-Filh, U.; Damiani, D.; Silva, C.A. Musculoskeletal pain in obese adolescents. J. Pediatr 2011, 87, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Caberlon, C.F.; Padoin, A.V.; Mottin, C.C. Importance of musculoskeletal pain in work activities in obese individuals. Obes. Surg. 2013, 23, 2092–2095. [Google Scholar] [CrossRef]
- Stovitz, S.D.; Pardee, P.E.; Vazquez, G.; Duval, S.; Schwimmer, J.B. Musculoskeletal pain in obese children and adolescents. Acta Paediatr. 2008, 97, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Peltonen, M.; Lindroos, A.K.; Torgerson, J.S. Musculoskeletal pain in the obese: A comparison with a general population and long-term changes after conventional and surgical obesity treatment. Pain 2003, 104, 549–557. [Google Scholar] [CrossRef]
- Tamin, T.Z.; Murdana, N.; Pitoyo, Y.; Safitri, E.D. Exercise Intervention for Chronic Pain Management, Muscle Strengthening, and Functional Score in Obese Patients with Chronic Musculoskeletal Pain: A Systematic Review and Meta-analysis. Acta Med. Indones. 2018, 50, 299–308. [Google Scholar] [PubMed]
- Sperotto, F.; Balzarin, M.; Parolin, M.; Monteforte, N.; Vittadello, F.; Zulian, F. Joint hypermobility, growing pain and obesity are mutually exclusive as causes of musculoskeletal pain in schoolchildren. Clin. Exp. Rheumatol. 2014, 32, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Tsuritani, I.; Honda, R.; Noborisaka, Y.; Ishida, M.; Ishizaki, M.; Yamada, Y. Impact of obesity on musculoskeletal pain and difficulty of daily movements in Japanese middle-aged women. Maturitas 2002, 42, 23–30. [Google Scholar] [CrossRef]
- Widhalm, H.K.; Seemann, R.; Hamboeck, M.; Mittlboeck, M.; Neuhold, A.; Friedrich, K.; Hajdu, S.; Widhalm, K. Osteoarthritis in morbidly obese children and adolescents, an age-matched controlled study. Knee Surg. Sports Traumatol. Arthrosc. 2016, 24, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.J.; Fischer, M.E.; Bril, G.; Kelber, S.; Rupley, D., Jr.; Oken, B.; Rimm, A.A. The association of obesity with joint pain and osteoarthritis in the HANES data. J. Chronic. Dis. 1986, 39, 311–319. [Google Scholar] [CrossRef]
- Alfieri, F.M.; Silva, N.C.O.V.E.; Battistella, L.R. Study of the relation between body weight and functional limitations and pain in patients with knee osteoarthritis. Einstein 2017, 15, 307–312. [Google Scholar] [CrossRef] [Green Version]
- Goulston, L.M.; Kiran, A.; Javaid, M.K.; Soni, A.; White, K.M.; Hart, D.J.; Spector, T.D.; Arden, N.K. Does obesity predict knee pain over fourteen years in women, independently of radiographic changes? Arthritis Care Res. 2011, 63, 1398–1406. [Google Scholar] [CrossRef]
- Brandt, K.D.; Heilman, D.K.; Slemenda, C.; Katz, B.P.; Mazzuca, S.; Braunstein, E.M.; Byrd, D. A comparison of lower extremity muscle strength, obesity, and depression scores in elderly subjects with knee pain with and without radiographic evidence of knee osteoarthritis. J. Rheumatol. 2000, 27, 1937–1946. [Google Scholar]
- Miller, G.D.; Nicklas, B.J.; Loeser, R.F. Inflammatory biomarkers and physical function in older, obese adults with knee pain and self-reported osteoarthritis after intensive weight-loss therapy. J. Am. Geriatr. Soc. 2008, 56, 644–651. [Google Scholar] [CrossRef]
- Tanamas, S.K.; Wluka, A.E.; Davies-Tuck, M.; Wang, Y.; Strauss, B.J.; Proietto, J.; Dixon, J.B.; Jones, G.; Forbes, A.; Cicuttini, F.M. Association of weight gain with incident knee pain, stiffness, and functional difficulties: A longitudinal study. Arthritis Care Res. 2013, 65, 34–43. [Google Scholar] [CrossRef]
- Schwarze, M.; Häuser, W.; Schmutzer, G.; Brähler, E.; Beckmann, N.A.; Schiltenwolf, M. Obesity, depression and hip pain. Musculoskelet. Care 2019, 17, 126–132. [Google Scholar] [CrossRef] [Green Version]
- Tanamas, S.K.; Wluka, A.E.; Berry, P.; Menz, H.B.; Strauss, B.J.; Davies-Tuck, M.; Proietto, J.; Dixon, J.B.; Jones, G.; Cicuttini, F.M. Relationship between obesity and foot pain and its association with fat mass, fat distribution, and muscle mass. Arthritis Care Res. 2012, 64, 262–268. [Google Scholar] [CrossRef]
- Mekhail, N.; Mehanny, D.; Armanyous, S.; Saweris, Y.; Costandi, S. The impact of obesity on the effectiveness of spinal cord stimulation in chronic spine-related pain patients. Spine J. 2019, 19, 476–486. [Google Scholar] [CrossRef]
- Bigal, M.E.; Liberman, J.N.; Lipton, R.B. Obesity and migraine: A population study. Neurology 2006, 66, 545–550. [Google Scholar] [CrossRef]
- Bigal, M.E.; Gironda, M.; Tepper, S.J.; Feleppa, M.; Rapoport, A.M.; Sheftell, F.D.; Lipton, R.B. Headache prevention outcome and body mass index. Cephalalgia 2006, 26, 445–450. [Google Scholar] [CrossRef]
- Kristoffersen, E.S.; Børte, S.; Hagen, K.; Zwart, J.A.; Winsvold, B.S. Migraine, obesity and body fat distribution—A population-based study. J. Headache Pain 2021, 21, 97. [Google Scholar] [CrossRef]
- Trovato, G.; Brischetto, D.; Pace, P.; Fabio Martines, G. Perceived body weight status of youngsters interferes with headache in obese and non-obese subjects. Headache 2014, 54, 1062–1063. [Google Scholar] [CrossRef] [Green Version]
- Cha, N.C.; Scher, A.I.; Moghekar, A.; Bond, D.S.; Peterlin, B.L. Perceived body weight status of youngsters interferes with headache in obese and non-obese subjects: A response. Headache 2014, 54, 1063–1065. [Google Scholar] [CrossRef] [Green Version]
- Farris, S.G.; Thomas, J.G.; Abrantes, A.M.; Lipton, R.B.; Pavlovic, J.; Smitherman, T.A.; Irby, M.B.; Penzien, D.B.; Roth, J.; O’Leary, K.C.; et al. Pain worsening with physical activity during migraine attacks in women with overweight/obesity: A prospective evaluation of frequency, consistency, and correlates. Cephalalgia 2018, 38, 1707–1715. [Google Scholar] [CrossRef]
- Afshinmajd, S.; Davati, A.; Akbari, F. The effects of body mass index on the treatment of the patients with migraine headaches. Iran. J. Neurol. 2011, 10, 35–38. [Google Scholar]
- Saloom, H.F.; Papageorgiou, S.N.; Carpenter, G.H.; Cobourne, M.T. The effect of obesity on orofacial pain during early orthodontic treatment with fixed appliances: A prospective cohort study. Eur. J. Orthod. 2018, 40, 343–349. [Google Scholar] [CrossRef]
- Bonato, R.C.S.; Mapengo, M.A.A.; de Azevedo-Silva, L.J.; Janson, G.; de Carvalho Sales-Peres, S.H. Tooth movement, orofacial pain, and leptin, interleukin-1beta, and tumor necrosis factor-alpha levels in obese adolescents. Angle Orthod. 2021, 1–6. [Google Scholar] [CrossRef]
- Balderas-Peña, L.M.; Macías-López, G.G.; Zepeda-González, A.; González-Hernández, I.; Herrera-Rodríguez, R.; Fafutis-Morris, M. Association between obesity, gender and preoperative inflammatory markers with postsurgical pain in live kidney donors. Cir. Cir. 2011, 79, 526–533. [Google Scholar] [PubMed]
- Paley, C.A.; Johnson, M.I. Physical Activity to Reduce Systemic Inflammation Associated with Chronic Pain and Obesity: A Narrative Review. Clin. J. Pain. 2016, 32, 365–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zdziarski, L.A.; Wasser, J.G.; Vincent, H.K. Chronic pain management in the obese patient: A focused review of key challenges and potential exercise solutions. J. Pain Res. 2015, 8, 63–77. [Google Scholar]
- Wasser, J.G.; Vasilopoulos, T.; Zdziarski, L.A.; Vincent, H.K. Exercise Benefits for Chronic Low Back Pain in Overweight and Obese Individuals. PM&R 2017, 9, 181–192. [Google Scholar]
- Perez-Huerta, B.D.; Díaz-Pulido, B.; Pecos-Martin, D.; Beckwee, D.; Lluch-Girbes, E.; Fernandez-Matias, R.; Rubio, M.J.B.; Gallego-Izquierdo, T. Effectiveness of a Program Combining Strengthening, Stretching, and Aerobic Training Exercises in a Standing versus a Sitting Position in Overweight Subjects with Knee Osteoarthritis: A Randomized Controlled Trial. J. Clin. Med. 2021, 9, 4113. [Google Scholar] [CrossRef]
- White, D.K.; Neogi, T.; Rejeski, W.J.; Walkup, M.P.; Lewis, C.E.; Nevitt, M.C.; Foy, C.G.; Felson, D.T.; Look Ahead Research Group. Can an intensive diet and exercise program prevent knee pain among overweight adults at high risk? Arthritis Care Res. 2015, 67, 965–971. [Google Scholar] [CrossRef] [Green Version]
- Irandoust, K.; Taheri, M. The effects of aquatic exercise on body composition and nonspecific low back pain in elderly males. J. Phys. Ther. Sci. 2015, 27, 433–435. [Google Scholar] [CrossRef] [Green Version]
- Goodpaster, B.H.; Theriault, R.; Watkins, S.C.; Kelley, D.E. Intramuscular lipid content is increased in obesity and decreased by weight loss. Metabolism 2000, 49, 467–472. [Google Scholar] [CrossRef]
- Malenfant, P.; Joanisse, D.R.; The’riault, R.; Goodpaster, B.H.; Kelley, D.E.; Simoneau, J.A. Fat content in individual muscle fibers of lean and obese subjects. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 1316–1321. [Google Scholar] [CrossRef] [Green Version]
- Velan, S.S.; Said, N.; Durst, C.; Frisbee, S.; Frisbee, J.; Raylman, R.R.; Thomas, M.A.; Rajendran, V.M.; Spencer, R.G.; Alway, S.E. Distinct patterns of fat metabolism in skeletal muscle of normal-weight, overweight, and obese humans. Am. J. Physiol Regul. Integr. Comp. Physiol. 2008, 295, R1060–R1065. [Google Scholar] [CrossRef] [Green Version]
- Morris, R.T.; Laye, M.J.; Lees, S.J.; Rector, R.S.; Thyfault, J.P.; Booth, F.W. Exercise-induced attenuation of obesity, hyperinsulinemia, and skeletal muscle lipid peroxidation in the OLETF rat. J. Appl. Physiol. 2008, 104, 708–715. [Google Scholar] [CrossRef] [Green Version]
- Ingram, K.H.; Hill, H.; Moellering, D.R.; Hill, B.G.; Lara-Castro, C.; Newcomer, B.; Brandon, L.J.; Ingalls, C.P.; Penumetcha, M.; Rupp, J.C.; et al. Skeletal muscle lipid peroxidation and insulin resistance in humans. J. Clin. Endocrinol. Metab. 2012, 97, E1182–E1186. [Google Scholar] [CrossRef] [Green Version]
- Di Meo, S.; Iossa, S.; Venditti, P. Skeletal muscle insulin resistance: Role of mitochondria and other ROS sources. J. Endocrinol. 2017, 233, R15–R42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiebaud, D.; Jacot, E.; DeFronzo, R.A.; Maeder, E.; Jequier, E.; Felber, J.P. The effect of graded doses of insulin on total glucose uptake, glucose oxidation, and glucose storage in man. Diabetes 1982, 31, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, G. Insulin and insulin resistance. Clin. Biochem. Rev. 2005, 26, 19–39. [Google Scholar] [PubMed]
- Soulage, C.O.; Sardón Puig, L.; Soulère, L.; Zarrouki, B.; Guichardant, M.; Lagarde, M.; Pillon, N.J. Skeletal muscle insulin resistance is induced by 4-hydroxy-2-hexenal, a by-product of n-3 fatty acid peroxidation. Diabetologia 2018, 61, 688–699. [Google Scholar] [CrossRef] [Green Version]
- Pillon, N.J.; Croze, M.L.; Vella, R.E.; Soulère, L.; Lagarde, M.; Soulage, C.O. The lipid peroxidation by-product 4-hydroxy-2-nonenal (4-HNE) induces insulin resistance in skeletal muscle through both carbonyl and oxidative stress. Endocrinology 2012, 153, 2099–2111. [Google Scholar] [CrossRef]
- Funai, K.; Song, H.; Yin, L.; Lodhi, I.J.; Wei, X.; Yoshino, J.; Coleman, T.; Semenkovich, C.F. Muscle lipogenesis balances insulin sensitivity and strength through calcium signaling. J. Clin. Invest. 2013, 123, 1229–1240. [Google Scholar] [CrossRef] [Green Version]
- Funai, K.; Lodhi, I.J.; Spears, L.D.; Yin, L.; Song, H.; Klein, S.; Semenkovich, C.F. Skeletal Muscle Phospholipid Metabolism Regulates Insulin Sensitivity and Contractile Function. Diabetes 2016, 65, 358–370. [Google Scholar] [CrossRef] [Green Version]
- Abdul-Ghani, M.A.; DeFronzo, R.A. Pathogenesis of insulin resistance in skeletal muscle. J. Biomed. Biotechnol. 2010, 2010, 476279. [Google Scholar] [CrossRef] [Green Version]
- Seebacher, F.; Tallis, J.; McShea, K.; James, R.S. Obesity-induced decreases in muscle performance are not reversed by weight loss. Int J. Obes. 2017, 41, 1271–1278. [Google Scholar] [CrossRef]
- Hilton, T.N.; Tuttle, L.J.; Bohnert, K.L.; Mueller, M.J.; Sinacore, D.R. Excessive adipose tissue infiltration in skeletal muscle in individuals with obesity, diabetes mellitus, and peripheral neuropathy: Association with performance and function. Phys. Ther. 2008, 88, 1336–1344. [Google Scholar] [CrossRef] [Green Version]
- Bittel, D.C.; Bittel, A.J.; Tuttle, L.J.; Hastings, M.K.; Commean, P.K.; Mueller, M.J.; Cade, W.T.; Sinacore, D.R. Adipose tissue content, muscle performance and physical function in obese adults with type 2 diabetes mellitus and peripheral neuropathy. J. Diabetes Complicat. 2015, 29, 250–257. [Google Scholar] [CrossRef] [Green Version]
- Sachs, S.; Zarini, S.; Kahn, D.E.; Harrison, K.A.; Perreault, L.; Phang, T.; Newsom, S.A.; Strauss, A.; Kerege, A.; Schoen, J.A.; et al. Intermuscular adipose tissue directly modulates skeletal muscle insulin sensitivity in humans. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E866–E879. [Google Scholar] [CrossRef] [Green Version]
- Griffin, M.E.; Marcucci, M.J.; Cline, G.W.; Bell, K.; Barucci, N.; Lee, D.; Goodyear, L.J.; Kraegen, E.W.; White, M.F.; Shulman, G.I. Free fatty acid-induced insulin resistance is associated with activation of protein kinase C theta and alterations in the insulin signaling cascade. Diabetes 1999, 48, 1270–1274. [Google Scholar] [CrossRef]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zong, H.; Wang, Y.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J.; et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef] [Green Version]
- Itani, S.I.; Ruderman, N.B.; Schmieder, F.; Boden, G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 2002, 51, 2005–2011. [Google Scholar] [CrossRef] [Green Version]
- Szendroedi, J.; Yoshimura, T.; Phielix, E.; Koliaki, C.; Marcucci, M.; Zhang, D.; Jelenik, T.; Müller, J.; Herder, C.; Nowotny, P.; et al. Role of diacylglycerol activation of PKCteta in lipid-induced muscle insulin resistance in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 9597–9602. [Google Scholar] [CrossRef] [Green Version]
- Timmers, S.; Schrauwen, P.; de Vogel, J. Muscular diacylglycerol metabolism and insulin resistance. Physiol. Behav. 2008, 94, 242–251. [Google Scholar] [CrossRef]
- Haam, J.H.; Kim, Y.S.; Koo, H.S.; Haam, J.; Seo, N.K.; Kim, H.Y.; Park, K.C.; Park, K.S.; Kim, M.J. Intermuscular adipose tissue is associated with monocyte chemoattractant protein-1, independent of visceral adipose tissue. Clin. Biochem. 2016, 49, 439–443. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.M.; Perrard, X.Y.; Brunner, G.; Lui, H.; Sparks, L.M.; Smith, S.R.; Wang, X.; Shi, Z.Z.; Lewis, D.E.; Wu, H.; et al. Intermuscular and perimuscular fat expansion in obesity correlates with skeletal muscle T cell and macrophage infiltration and insulin resistance. Int J. Obes. 2015, 39, 1607–1618. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Yu, S.; Wang, F.; Bai, L.; Xiao, J.; Jiang, Y.; Chen, L.; Wang, J.; Jiang, A.; Li, M.; et al. MicroRNA transcriptomes relate intermuscular adipose tissue to metabolic risk. Int J. Mol. Sci. 2013, 14, 8611–8624. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.J.; Lu, C.; Su, L.Y.; Sharma, A.M.; Lee, R.M.K.W. Modulation of vascular function by perivascular adipose tissue: The role of endothelium and hydrogen peroxide. Br. J. Pharmacol. 2007, 151, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Brown, N.K.; Zhou, Z.; Zhang, J.; Zeng, R.; Wu, J.; Eitzman, D.T.; Chen, Y.E.; Chang, L. Perivascular adipose tissue in vascular function and disease: A review of current research and animal models. Arter. Thromb. Vasc. Biol. 2014, 34, 1621–1630. [Google Scholar] [CrossRef] [Green Version]
- Gil-Ortega, M.; Somoza, B.; Huang, Y.; Gollasch, M.; Fernández-Alfonso, M.S. Regional differences in perivascular adipose tissue impacting vascular homeostasis. Trends Endocrinol. Metab. 2015, 26, 367–375. [Google Scholar] [CrossRef]
- Xia, N.; Li, H. The role of perivascular adipose tissue in obesity-induced vascular dysfunction. Br. J. Pharmacol. 2017, 174, 3425–3442. [Google Scholar] [CrossRef] [Green Version]
- Grigoras, A.; Amalinei, C.; Balan, R.A.; Giusca, S.E.; Caruntu, I.D. Perivascular adipose tissue in cardiovascular diseases—An update. Anatol. J. Cardiol. 2019, 22, 219–231. [Google Scholar] [CrossRef]
- Saxton, S.N.; Clark, B.J.; Withers, S.B.; Eringa, E.C.; Heagerty, A.M. Mechanistic Links Between Obesity, Diabetes, and Blood Pressure: Role of Perivascular Adipose Tissue. Physiol. Rev. 2019, 99, 1701–1763. [Google Scholar] [CrossRef]
- Ramirez, J.G.; O’Malley, E.J.; Ho, W.S.V. Pro-contractile effects of perivascular fat in health and disease. Br. J. Pharmacol. 2017, 174, 3482–3495. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Garcia-Barrio, M.T.; Chen, Y.E. Perivascular Adipose Tissue Regulates Vascular Function by Targeting Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2021, 40, 1094–1109. [Google Scholar] [CrossRef]
- Nosalski, R.; Guzik, T.J. Perivascular adipose tissue inflammation in vascular disease. Br. J. Pharmacol. 2017, 174, 3496–3513. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.K.; Bakar, H.A.; Gollasch, M.; Huang, Y. Perivascular Adipose Tissue: The Sixth Man of the Cardiovascular System. Cardiovasc. Drugs Ther. 2018, 32, 481–502. [Google Scholar] [CrossRef]
- Kangawa, K.; Kitamura, K.; Minamino, N.; Matsuo, H. Adrenomedullin: A new modulator of vascular tone. J. Card. Fail. 1996, 2, S135–S140. [Google Scholar] [CrossRef]
- Schroeter, M.R.; Eschholz, N.; Herzberg, S.; Jerchel, I.; Leifheit-Nestler, M.; Czepluch, F.S.; Chalikia, G.; Konstantinides, S.; Schafer, K. Leptin-dependent and leptin-independent paracrine effects of perivascular adipose tissue on neointima formation. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 980–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reidy, M.A.; Jackson, C.L. Factors controlling growth of arterial cells following injury. Toxicol. Pathol. 1990, 18, 547–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Ballantyne, L.L.; Yu, Y.; Funk, C.D. Perivascular adipose tissue-derived extracellular vesicle miR-221–3p mediates vascular remodeling. FASEB J. 2019, 33, 12704–12722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weston, A.H.; Egner, I.; Dong, Y.; Porter, E.L.; Heagerty, A.M.; Edwards, G. Stimulated release of a hyperpolarizing factor (ADHF) from mesenteric artery perivascular adipose tissue: Involvement of myocyte BKCa channels and adiponectin. Br. J. Pharmacol. 2013, 169, 1500–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uemura, Y.; Shibata, R.; Ohashi, K.; Enomoto, T.; Kambara, T.; Yamamoto, T.; Ogura, Y.; Yuasa, D.; Joki, Y.; Matsuo, K.; et al. Adipose-derived factor CTRP9 attenuates vascular smooth muscle cell proliferation and neointimal formation. FASEB J. 2013, 27, 25–33. [Google Scholar] [CrossRef]
- Butcher, M.J.; Waseem, T.C.; Galkina, E.V. Smooth muscle cell derived interleukin-17C plays an atherogenic role via the recruitment of proinflammatory interleukin-17A+ T cells to the aorta. Arter. Thromb. Vasc. Biol. 2016, 36, 1496–1506. [Google Scholar] [CrossRef] [Green Version]
- Xia, N.; Horke, S.; Habermeier, A.; Closs, E.I.; Reifenberg, G.; Gericke, A.; Mikhed, Y.; Münzel, T.; Daiber, A.; Förstermann, U.; et al. Uncoupling of Endothelial Nitric Oxide Synthase in Perivascular Adipose Tissue of Diet-Induced Obese Mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 78–85. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.J.; Wu, J.; Guo, W.; Zhu, Y.Z. Atherosclerosis and the Hydrogen Sulfide Signaling Pathway—Therapeutic Approaches to Disease Prevention. Cell. Physiol. Biochem. 2017, 42, 859–875. [Google Scholar] [CrossRef]
- Bussey, C.E.; Withers, S.B.; Aldous, R.G.; Edwards, G.; Heagerty, A.M. Obesity-Related Perivascular Adipose Tissue Damage Is Reversed by Sustained Weight Loss in the Rat. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1377–1385. [Google Scholar] [CrossRef] [Green Version]
- Henrichot, E.; Juge-Aubry, C.E.; Pernin, A.; Pache, J.C.; Velebit, V.; Dayer, J.M.; Meda, P.; Chizzolini, C.; Meier, C.A. Production of chemokines by perivascular adipose tissue: A role in the pathogenesis of atherosclerosis? Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2594–2599. [Google Scholar] [CrossRef] [Green Version]
- Xia, N.; Weisenburger, S.; Koch, E.; Burkart, M.; Reifenberg, G.; Förstermann, U.; Li, H. Restoration of perivascular adipose tissue function in diet-induced obese mice without changing bodyweight. Br. J. Pharmacol. 2017, 174, 3443–3453. [Google Scholar] [CrossRef] [Green Version]
- Mazzotta, C.; Basu, S.; Gower, A.C.; Karki, S.; Farb, M.G.; Sroczynski, E.; Zizza, E.; Sarhan, A.; Pande, A.N.; Walsh, K.; et al. Perivascular Adipose Tissue Inflammation in Ischemic Heart Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1239–1250. [Google Scholar] [CrossRef]
- Munjal, A.; Khandia, R. Atherosclerosis: Orchestrating cells and biomolecules involved in its activation and inhibition. Adv. Protein Chem. Struct. Biol. 2021, 120, 85–122. [Google Scholar]
- Xu, H.; Jiang, J.; Chen, W.; Li, W.; Chen, Z. Vascular Macrophages in Atherosclerosis. J. Immunol. Res. 2019, 2019, 4354786. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Gharaee-Kermani, M.; McGarry, B.; Remick, D.; Phan, S.H. TNF-alpha-mediated lung cytokine networking and eosinophil recruitment in pulmonary fibrosis. J. Immunol. 1997, 158, 954–959. [Google Scholar]
- Lukacs, N.W.; Strieter, R.M.; Chensue, S.W.; Widmer, M.; Kunkel, S.L. TNF-alpha mediates recruitment of neutrophils and eosinophils during airway inflammation. J. Immunol. 1995, 154, 5411–5417. [Google Scholar]
- Lampinen, M.; Carlson, M.; Sangfelt, P.; Taha, Y.; Thörn, M.; Lööf, L.; Raab, Y.; Venge, P. IL-5 and TNF-alpha participate in recruitment of eosinophils to intestinal mucosa in ulcerative colitis. Dig. Dis. Sci. 2001, 46, 2004–2009. [Google Scholar] [CrossRef]
- Marx, C.; Novotny, J.; Salbeck, D.; Zellner, K.R.; Nicolai, L.; Pekayvaz, K.; Kilani, B.; Stockhausen, S.; Bürgener, N.; Kupka, D.; et al. Eosinophil-platelet interactions promote atherosclerosis and stabilize thrombosis with eosinophil extracellular traps. Blood 2019, 134, 1859–1872. [Google Scholar] [CrossRef] [Green Version]
- Ouwens, D.M.; Sell, H.; Greulich, S.; Eckel, J. The role of epicardial and perivascular adipose tissue in the pathophysiology of cardiovascular disease. J. Cell. Mol. Med. 2010, 14, 2223–2234. [Google Scholar] [CrossRef] [Green Version]
- Mazurek, T.; Zhang, L.; Zalewski, A.; Mannion, J.D.; Diehl, J.T.; Arafat, H.; Sarov-Blat, L.; O’Brien, S.; Keiper, E.A.; Johnson, A.G.; et al. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation 2003, 108, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Baker, A.R.; da Silva, N.F.; Quinn, D.W.; Harte, A.L.; Pagano, D.; Bonser, R.S.; Kumar, S.; McTernan, P.G. Human epicardial adipose tissue expresses a pathogenic profile of adipocytokines in patients with cardiovascular disease. Cardiovasc. Diabetol. 2006, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Song, D.K.; Hong, Y.S.; Lee, H.; Oh, J.Y.; Sung, Y.A.; Kim, Y. Increased Epicardial Adipose Tissue Thickness in Type 2 Diabetes Mellitus and Obesity. Diabetes Metab. J. 2015, 39, 405–413. [Google Scholar] [CrossRef] [Green Version]
- Aitken-Buck, H.M.; Moharram, M.; Babakr, A.A.; Reijers, R.; Van Hout, I.; Fomison-Nurse, I.C.; Sugunesegran, R.; Bhagwat, K.; Davis, P.J.; Bunton, R.W.; et al. Relationship between epicardial adipose tissue thickness and epicardial adipocyte size with increasing body mass index. Adipocyte 2019, 8, 412–420. [Google Scholar] [CrossRef] [Green Version]
- Eren, E.; Koca, B.; Ture, M.; Guzel, B. Epicardial adiposity in children with obesity and metabolic syndrome. Iran. J. Pediatr. 2014, 24, 411–417. [Google Scholar]
- Boyraz, M.; Pirgon, O.; Akyol, B.; Dundar, B.; Cekmez, F.; Eren, N. Importance of epicardial adipose tissue thickness measurement in obese adolescents, its relationship with carotid intima-media thickness, and echocardiographic findings. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 3309–3317. [Google Scholar]
- Akyol, B.; Boyraz, M.; Aysoy, C. Relationship of epicardial adipose tissue thickness with early indicators of atherosclerosis and cardiac functional changes in obese adolescents with metabolic syndrome. J. Clin. Res. Pediatr. Endocrinol. 2013, 5, 156–163. [Google Scholar]
- Fu, C.P.; Sheu, W.H.; Lee, I.T.; Tsai, I.C.; Lee, W.J.; Liang, K.W.; Lee, W.L.; Lin, S.Y. Effects of weight loss on epicardial adipose tissue thickness and its relationship between serum soluble CD40 ligand levels in obese men. Clin. Chim. Acta 2013, 421, 98–103. [Google Scholar] [CrossRef]
- Chumakova, G.; Gritsenko, O.; Gruzdeva, O.; Dyleva, Y. Analysis of probable lipotoxic damage and myocardial fibrosis in epicardial obesity. Aging 2021, 13, 14806–14815. [Google Scholar] [CrossRef] [PubMed]
- Kankaanpää, M.; Lehto, H.R.; Pärkkä, J.P.; Komu, M.; Viljanen, A.; Ferrannini, E.; Knuuti, J.; Nuutila, P.; Parkkola, R.; Iozzo, P. Myocardial triglyceride content and epicardial fat mass in human obesity: Relationship to left ventricular function and serum free fatty acid levels. J. Clin. Endocrinol. Metab. 2006, 91, 4689–4695. [Google Scholar] [CrossRef] [PubMed]
- Scherer, M.; Montoliu, I.; Qanadli, S.D.; Collino, S.; Rezzi, S.; Kussmann, M.; Giusti, V.; Martin, F.P. Blood plasma lipidomic signature of epicardial fat in healthy obese women. Obesity 2015, 23, 130–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, U.; Heuer, S.; Meder, K.; Boehler, J.; Lange, V.; Quaschning, T.; Ertl, G.; Bonz, A. The proinflammatory cytokines TNF-alpha and IL-1 beta impair economy of contraction in human myocardium. Cytokine 2007, 39, 157–162. [Google Scholar] [CrossRef]
- Abe, I.; Teshima, Y.; Kondo, H.; Kaku, H.; Kira, S.; Ikebe, Y.; Saito, S.; Fukui, A.; Shinohara, T.; Yufu, K.; et al. Association of fibrotic remodeling and cytokines/chemokines content in epicardial adipose tissue with atrial myocardial fibrosis in patients with atrial fibrillation. Heart Rhythm. 2018, 15, 1717–1727. [Google Scholar] [CrossRef]
- Yu, M.; Wen, S.; Wang, M.; Liang, W.; Li, H.H.; Long, Q.; Guo, H.P.; Liao, Y.H.; Yuan, J. TNF-alpha-secreting B cells contribute to myocardial fibrosis in dilated cardiomyopathy. J. Clin. Immunol. 2013, 33, 1002–1008. [Google Scholar] [CrossRef]
- Venteclef, N.; Guglielmi, V.; Balse, E.; Gaborit, B.; Cotillard, A.; Atassi, F.; Amour, J.; Leprince, P.; Dutour, A.; Clément, K.; et al. Human epicardial adipose tissue induces fibrosis of the atrial myocardium through the secretion of adipo-fibrokines. Eur. Heart J. 2015, 36, 795–805a. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Liu, J.; Zhu, L.; Tan, F.; Qin, Y.; Huang, H.; Yu, Y. Free fatty acid can induce cardiac dysfunction and alter insulin signaling pathways in the heart. Lipids Health Dis. 2018, 17, 185. [Google Scholar] [CrossRef] [Green Version]
- Toemen, L.; Santos, S.; Roest, A.A.W.; Vernooij, M.W.; Helbing, W.A.; Gaillard, R.; Jaddoe, V.W.V. Pericardial adipose tissue, cardiac structures, and cardiovascular risk factors in school-age children. Eur. Heart J. Cardiovasc. Imaging 2021, 22, 307–313. [Google Scholar] [CrossRef]
- Gill, C.M.; Azevedo, D.C.; Oliveira, A.L.; Martinez-Salazar, E.L.; Torriani, M.; Bredella, M.A. Sex differences in pericardial adipose tissue assessed by PET/CT and association with cardiometabolic risk. Acta Radiol. 2018, 59, 1203–1209. [Google Scholar] [CrossRef]
- Wang, C.Y.; Li, S.J.; Wu, T.W.; Lin, H.J.; Chen, J.W.; Mersmann, H.J.; Ding, S.T.; Chen, C.Y. The role of pericardial adipose tissue in the heart of obese minipigs. Eur. J. Clin. Invest. 2018, 48, e12942. [Google Scholar] [CrossRef]
- Li, S.J.; Wu, T.W.; Chien, M.J.; Mersmann, H.J.; Chen, C.Y. Involvement of pericardial adipose tissue in cardiac fibrosis of dietary-induced obese minipigs—Role of mitochondrial function. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids. 2019, 1864, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Pucci, G.; Battista, F.; de Vuono, S.; Boni, M.; Scavizzi, M.; Ricci, M.A.; Lupattelli, G.; Schillaci, G. Pericardial fat, insulin resistance, and left ventricular structure and function in morbid obesity. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kim, S.W.; Lee, J.S.; Lee, S.K.; Abbott, R.; Lee, K.Y.; Lim, H.E.; Sung, K.C.; Cho, G.Y.; Koh, K.K.; et al. Association of pericardial adipose tissue with left ventricular structure and function: A region-specific effect? Cardiovasc. Diabetol. 2021, 20, 26. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Ten Dijke, P. TGF-beta Signaling in Control of Cardiovascular Function. Cold Spring Harb. Perspect. Biol. 2018, 10, a022210. [Google Scholar] [CrossRef] [Green Version]
- Gallini, R.; Lindblom, P.; Bondjers, C.; Betsholtz, C.; Andrae, J. PDGF-A and PDGF-B induces cardiac fibrosis in transgenic mice. Exp. Cell Res. 2016, 349, 282–290. [Google Scholar] [CrossRef]
- Kang, S.; Chemaly, E.R.; Hajjar, R.J.; Lebeche, D. Resistin promotes cardiac hypertrophy via the AMP-activated protein kinase/mammalian target of rapamycin (AMPK/mTOR) and c-Jun N-terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathways. J. Biol. Chem. 2011, 286, 18465–18473. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Oh, J.K.; Sakata, S.; Liang, I.; Park, W.; Hajjar, R.J.; Lebeche, D. Role of resistin in cardiac contractility and hypertrophy. J. Mol. Cell Cardiol. 2008, 45, 270–280. [Google Scholar] [CrossRef] [Green Version]
- Karmazyn, M.; Rajapurohitam, V. Leptin as a cardiac pro-hypertrophic factor and its potential role in the development of heart failure. Curr. Pharm. Des. 2014, 20, 646–651. [Google Scholar] [CrossRef]
- Ren, J. Leptin and hyperleptinemia—From friend to foe for cardiovascular function. J. Endocrinol. 2004, 181, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, Y.; Younossi, Z.M. Obesity-associated nonalcoholic fatty liver disease. Clin. Liver Dis. 2014, 18, 19–31. [Google Scholar] [CrossRef]
- Bellentani, S.; Saccoccio, G.; Masutti, F.; Crocè, L.S.; Brandi, G.; Sasso, F.; Cristanini, G.; Tiribelli, C. Prevalence of and risk factors for hepatic steatosis in Northern Italy. Ann. Intern. Med. 2000, 132, 112–117. [Google Scholar] [CrossRef]
- Santoro, N.; Feldstein, A.E.; Enoksson, E.; Pierpont, B.; Kursawe, R.; Kim, G.; Caprio, S. The association between hepatic fat content and liver injury in obese children and adolescents: Effects of ethnicity, insulin resistance, and common gene variants. Diabetes Care 2013, 36, 1353–1360. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, A.J.; Brunt, E.M.; Kleiner, D.E.; Kowdley, K.V.; Chalasani, N.; Lavine, J.E.; Ratziu, V.; Mccullough, A. Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatology 2011, 54, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Lian, C.Y.; Zhai, Z.Z.; Li, Z.F.; Wang, L. High fat diet-triggered non-alcoholic fatty liver disease: A review of proposed mechanisms. Chem. Biol. Interact. 2021, 330, 109199. [Google Scholar] [CrossRef]
- Al-Dayyat, H.M.; Rayyan, Y.M.; Tayyem, R.F. Non-alcoholic fatty liver disease and associated dietary and lifestyle risk factors. Diabetes Metab. Syndr. 2018, 12, 569–575. [Google Scholar] [CrossRef]
- Lu, F.B.; Hu, E.D.; Xu, L.M.; Chen, L.; Wu, J.L.; Li, H.; Chen, D.Z.; Chen, Y.P. The relationship between obesity and the severity of non-alcoholic fatty liver disease: Systematic review and meta-analysis. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 491–502. [Google Scholar] [CrossRef]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef]
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology 2021, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2021, 73, 202–209. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Mantzoros, C.S. Making progress in nonalcoholic fatty liver disease (NAFLD) as we are transitioning from the era of NAFLD to dys-metabolism associated fatty liver disease (DAFLD). Metabolism 2021, 111, 154318. [Google Scholar] [CrossRef]
- Makri, E.; Goulas, A.; Polyzos, S.A. Epidemiology, Pathogenesis, Diagnosis and Emerging Treatment of Nonalcoholic Fatty Liver Disease. Arch. Med. Res. 2021, 52, 25–37. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [Green Version]
- Koo, S.H. Nonalcoholic fatty liver disease: Molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 2013, 19, 210–215. [Google Scholar] [CrossRef]
- Wang, G.; Bonkovsky, H.L.; de Lemos, A.; Burczynski, F.J. Recent insights into the biological functions of liver fatty acid binding protein 1. J. Lipid Res. 2015, 56, 2238–2247. [Google Scholar] [CrossRef] [Green Version]
- Sanders, F.W.; Griffin, J.L. De novo lipogenesis in the liver in health and disease: More than just a shunting yard for glucose. Biol. Rev. Camb. Philos. Soc. 2016, 91, 452–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberle, D.; Hegarty, B.; Bossard, P.; Ferre, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, H.; Takenoshita, M.; Sakurai, M.; Bruick, R.K.; Henzel, W.J.; Shillinglaw, W.; Arnot, D.; Uyeda, K. A glucose-responsive transcription factor that regulates carbohydrate metabolism in the liver. Proc. Natl. Acad. Sci. USA 2001, 98, 9116–9121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohjima, M.; Enjoji, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2007, 20, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Kohjima, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; Enjoji, M.; et al. SREBP-1c. regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2008, 21, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Benhamed, F.; Denechaud, P.D.; Lemoine, M.; Robichon, C.; Moldes, M.; Bertrand-Michel, J.; Ratziu, V.; Serfaty, L.; Housset, C.; Capeau, J.; et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J. Clin. Invest. 2012, 122, 2176–2194. [Google Scholar] [CrossRef] [Green Version]
- Bril, F.; Barb, D.; Portillo-Sanchez, P.; Biernacki, D.; Lomonaco, R.; Suman, A.; Weber, M.H.; Budd, J.T.; Lupi, M.E.; Cusi, K. Metabolic and histological implications of intrahepatic triglyceride content in nonalcoholic fatty liver disease. Hepatology 2017, 65, 1132–1144. [Google Scholar] [CrossRef] [Green Version]
- Korenblat, K.M.; Fabbrini, E.; Mohammed, B.S.; Klein, S. Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology 2008, 134, 1369–1375. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Han, C.K.; Pan, L.L.; Zhang, H.J.; Ma, Z.M.; Huang, Z.F.; Chen, S.; Zhuang, X.J.; Li, Z.B.; Li, X.Y.; et al. Serum alanine aminotransferase independently correlates with intrahepatic triglyceride contents in obese subjects. Dig. Dis. Sci. 2014, 59, 2470–2476. [Google Scholar] [CrossRef]
- Zhang, H.J.; Zhang, X.F.; Ma, Z.M.; Pan, L.L.; Chen, Z.; Han, H.W.; Han, C.K.; Zhuang, X.J.; Lu, Y.; Li, X.J.; et al. Irisin is inversely associated with intrahepatic triglyceride contents in obese adults. J. Hepatol. 2013, 59, 557–562. [Google Scholar] [CrossRef]
- Flisiak-Jackiewicz, M.; Bobrus-Chociej, A.; Wasilewska, N.; Tarasow, E.; Wojtkowska, M.; Lebensztejn, D.M. Can hepatokines be regarded as novel non-invasive serum biomarkers of intrahepatic lipid content in obese children? Adv. Med. Sci. 2019, 64, 280–284. [Google Scholar] [CrossRef]
- Tang, H.; Yu, R.; Liu, S.; Huwatibieke, B.; Li, Z.; Zhang, W. Irisin Inhibits Hepatic Cholesterol Synthesis via AMPK-SREBP2 Signaling. EBioMedicine 2016, 6, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lei, T.; Huang, J.F.; Wang, S.B.; Zhou, L.L.; Yang, Z.Q.; Chen, X.D. The link between fibroblast growth factor 21 and sterol regulatory element binding protein 1c during lipogenesis in hepatocytes. Mol. Cell. Endocrinol. 2011, 342, 41–47. [Google Scholar] [CrossRef]
- Novotny, D.; Vaverkova, H.; Karasek, D.; Lukes, J.; Slavik, L.; Malina, P.; Orsag, J. Evaluation of total adiponectin, adipocyte fatty acid binding protein and fibroblast growth factor 21 levels in individuals with metabolic syndrome. Physiol. Res. 2014, 63, 219–228. [Google Scholar] [CrossRef]
- Maggio, A.B.; Mueller, P.; Wacker, J.; Viallon, M.; Belli, D.C.; Beghetti, M.; Farpour-Lambert, N.J.; McLin, V.A. Increased pancreatic fat fraction is present in obese adolescents with metabolic syndrome. J. Pediatr. Gastroenterol. Nutr. 2012, 54, 720–726. [Google Scholar] [CrossRef]
- Rossi, A.P.; Fantin, F.; Zamboni, G.A.; Mazzali, G.; Rinaldi, C.A.; Del Giglio, M.; Di Francesco, V.; Barillari, M.; Pozzi Mucelli, R.; Zamboni, M. Predictors of ectopic fat accumulation in liver and pancreas in obese men and women. Obesity 2011, 19, 1747–1754. [Google Scholar] [CrossRef]
- Targher, G.; Rossi, A.P.; Zamboni, G.A.; Fantin, F.; Antonioli, A.; Corzato, F.; Bambace, C.; Pozzi Mucelli, R.; Zamboni, M. Pancreatic fat accumulation and its relationship with liver fat content and other fat depots in obese individuals. J. Endocrinol. Investig. 2012, 35, 748–753. [Google Scholar]
- Lee, Y.; Lingvay, I.; Szczepaniak, L.S.; Ravazzola, M.; Orci, L.; Unger, R.H. Pancreatic steatosis: Harbinger of type 2 diabetes in obese rodents. Int. J. Obes. 2010, 34, 396–400. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.Y.; Ou, H.Y.; Chen, M.F.; Chang, T.C.; Chang, C.J. Enigmatic ectopic fat: Prevalence of nonalcoholic fatty pancreas disease and its associated factors in a Chinese population. J. Am. Heart Assoc. 2014, 3, e000297. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Li, M.L.; Zhang, D.D.; Lin, H.Y.; Dai, X.H.; Sun, X.L.; Li, J.T.; Song, L.Y.; Peng, H.; Wen, M.M. The correlation between pancreatic steatosis and metabolic syndrome in a Chinese population. Pancreatology 2016, 16, 578–583. [Google Scholar] [CrossRef]
- Smits, M.M.; van Geenen, E.J. The clinical significance of pancreatic steatosis. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 169–177. [Google Scholar] [CrossRef]
- Shah, N.; Rocha, J.P.; Bhutiani, N.; Endashaw, O. Nonalcoholic Fatty Pancreas Disease. Nutr. Clin. Pract. 2019, 34, S49–S56. [Google Scholar] [CrossRef] [Green Version]
- Silva, L.L.S.E.; Fernandes, M.S.S.; Lima, E.A.; Stefano, J.T.; Oliveira, C.P.; Jukemura, J. Fatty Pancreas: Disease or Finding? Clinics 2021, 76, e2439. [Google Scholar] [CrossRef]
- Rebuffat, S.A.; Sidot, E.; Guzman, C.; Azay-Milhau, J.; Jover, B.; Lajoix, A.D.; Peraldi-Roux, S. Adipose tissue derived-factors impaired pancreatic beta-cell function in diabetes. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3378–3387. [Google Scholar] [CrossRef]
- Watada, H. Role of VEGF-A in pancreatic beta cells. Endocr. J. 2010, 57, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; Schneider, D.; Kidszun, A.; Hauck-Schmalenberger, I.; Breier, G.; Brandhorst, D.; Brandhorst, H.; Iken, M.; Brendel, M.D.; Bretzel, R.G.; et al. Vascular endothelial growth factor increases functional beta-cell mass by improvement of angiogenesis of isolated human and murine pancreatic islets. Transplantation 2005, 79, 1530–1536. [Google Scholar] [CrossRef]
- De Leu, N.; Heremans, Y.; Coppens, V.; Van Gassen, N.; Cai, Y.; D’Hoker, J.; Magenheim, J.; Salpeter, S.; Swisa, A.; Khalaileh, A.; et al. Short-term overexpression of VEGF-A in mouse beta cells indirectly stimulates their proliferation and protects against diabetes. Diabetologia 2014, 57, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Agudo, J.; Ayuso, E.; Jimenez, V.; Casellas, A.; Mallol, C.; Salavert, A.; Tafuro, S.; Obach, M.; Ruzo, A.; Moya, M.; et al. Vascular endothelial growth factor-mediated islet hypervascularization and inflammation contribute to progressive reduction of beta-cell mass. Diabetes 2012, 61, 2851–2861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silha, J.V.; Krsek, M.; Sucharda, P.; Murphy, L.J. Angiogenic factors are elevated in overweight and obese individuals. Int. J. Obes. 2005, 29, 1308–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, S.; Imachi, H.; Lyu, J.; Miyai, Y.; Fukunaga, K.; Dong, T.; Ibata, T.; Kobayashi, T.; Yoshimoto, T.; Kikuchi, F.; et al. Effect of TNF-alpha on the expression of ABCA1 in pancreatic beta-cells. J. Mol. Endocrinol. 2018, 61, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Ellingsgaard, H.; Hauselmann, I.; Schuler, B.; Habib, A.M.; Baggio, L.L.; Meier, D.T.; Eppler, E.; Bouzakri, K.; Wueest, S.; Muller, Y.D.; et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nat. Med. 2011, 17, 1481–1489. [Google Scholar] [CrossRef] [Green Version]
- Azizian, M.; Mahdipour, E.; Mirhafez, S.R.; Shoeibi, S.; Nematy, M.; Esmaily, H.; Ferns, G.A.; Ghayour-Mobarhan, M. Cytokine profiles in overweight and obese subjects and normal weight individuals matched for age and gender. Ann. Clin. Biochem. 2016, 53, 663–668. [Google Scholar] [CrossRef]
- Dirice, E.; Kahraman, S.; Jiang, W.; El Ouaamari, A.; De Jesus, D.F.; Teo, A.K.; Hu, J.; Kawamori, D.; Gaglia, J.L.; Mathis, D.; et al. Soluble factors secreted by T cells promote beta-cell proliferation. Diabetes 2014, 63, 188–202. [Google Scholar] [CrossRef] [Green Version]
- Duan, L.F.; Xu, X.F.; Zhu, L.J.; Liu, F.; Zhang, X.Q.; Wu, N.; Fan, J.W.; Xin, J.Q.; Zhang, H. Dachaihu decoction ameliorates pancreatic fibrosis by inhibiting macrophage infiltration in chronic pancreatitis. World J. Gastroenterol. 2017, 23, 7242–7252. [Google Scholar] [CrossRef]
- Westerbacka, J.; Cornér, A.; Kolak, M.; Makkonen, J.; Turpeinen, U.; Hamsten, A.; Fisher, R.M.; Yki-Järvinen, H. Insulin regulation of MCP-1 in human adipose tissue of obese and lean women. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E841–E845. [Google Scholar] [CrossRef]
- Levelt, E.; Pavlides, M.; Banerjee, R.; Mahmod, M.; Kelly, C.; Sellwood, J.; Ariga, R.; Thomas, S.; Francis, J.; Rodgers, C.; et al. Ectopic and Visceral Fat Deposition in Lean and Obese Patients with Type 2 Diabetes. J. Am. Coll. Cardiol. 2016, 68, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.; Lee, A.; Yoo, H.J.; Kim, M.; Kim, M.; Shin, D.Y.; Lee, J.H. Association between increased visceral fat area and alterations in plasma fatty acid profile in overweight subjects: A cross-sectional study. Lipids Health Dis. 2017, 16, 248. [Google Scholar] [CrossRef] [Green Version]
- Walker, G.E.; Marzullo, P.; Prodam, F.; Bona, G.; Di Blasio, A.M. Obesity modifies expression profiles of metabolic markers in superficial and deep subcutaneous abdominal adipose tissue depots. Endocrine 2014, 46, 99–106. [Google Scholar] [CrossRef]
- Cancello, R.; Zulian, A.; Gentilini, D.; Maestrini, S.; Della Barba, A.; Invitti, C.; Corà, D.; Caselle, M.; Liuzzi, A.; Di Blasio, A.M. Molecular and morphologic characterization of superficial- and deep-subcutaneous adipose tissue subdivisions in human obesity. Obesity 2013, 21, 2562–2570. [Google Scholar] [CrossRef]
- Thomas, E.L.; Frost, G.; Taylor-Robinson, S.D.; Bell, J.D. Excess body fat in obese and normal-weight subjects. Nutr. Res. Rev. 2012, 25, 150–161. [Google Scholar] [CrossRef] [Green Version]
- Ezure, T.; Amano, S. Increased subcutaneous adipose tissue impairs dermal function in diet-induced obese mice. Exp. Dermatol. 2010, 19, 878–882. [Google Scholar] [CrossRef]
- Shen, W.; Wang, Z.; Punyanita, M.; Lei, J.; Sinav, A.; Kral, J.G.; Imielinska, C.; Ross, R.; Heymsfield, S.B. Adipose tissue quantification by imaging methods: A proposed classification. Obes. Res. 2003, 11, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Sbarbati, A.; Accorsi, D.; Benati, D.; Marchetti, L.; Orsini, G.; Rigotti, G.; Panettiere, P. Subcutaneous adipose tissue classification. Eur. J. Histochem. 2010, 54, e48. [Google Scholar] [CrossRef]
- Märin, P.; Andersson, B.; Ottosson, M.; Olbe, L.; Chowdhury, B.; Kvist, H.; Holm, G.; Sjöström, L.; Björntorp, P. The morphology and metabolism of intraabdominal adipose tissue in men. Metabolism 1992, 41, 1242–1248. [Google Scholar] [CrossRef]
- Abate, N.; Burns, D.; Peshock, R.M.; Garg, A.; Grundy, S.M. Estimation of adipose tissue mass by magnetic resonance imaging: Validation against dissection in human cadavers. J. Lipid Res. 1994, 35, 1490–1496. [Google Scholar] [CrossRef]
- Neeland, I.J.; Ross, R.; Després, J.P.; Matsuzawa, Y.; Yamashita, S.; Shai, I.; Seidell, J.; Magni, P.; Santos, R.D.; Arsenault, B.; et al. International Atherosclerosis Society; International Chair on Cardiometabolic Risk Working Group on Visceral Obesity. Visceral and ectopic fat, atherosclerosis, and cardiometabolic disease: A position statement. Lancet Diabetes Endocrinol. 2019, 7, 715–725. [Google Scholar] [CrossRef]
- Tchernof, A.; Després, J.P. Pathophysiology of human visceral obesity: An update. Physiol. Rev. 2013, 93, 359–404. [Google Scholar] [CrossRef]
- Walker, G.E.; Marzullo, P.; Ricotti, R.; Bona, G.; Prodam, F. The pathophysiology of abdominal adipose tissue depots in health and disease. Horm. Mol. Biol. Clin. Investig. 2014, 19, 57–74. [Google Scholar] [CrossRef]
- Cao, Y.L.; Hu, C.Z.; Meng, X.; Wang, D.F.; Zhang, J. Expression of TNF-alpha protein in omental and subcutaneous adipose tissue in obesity. Diabetes Res. Clin. Pract. 2008, 79, 214–219. [Google Scholar] [CrossRef]
- Terra, X.; Auguet, T.; Quesada, I.; Aguilar, C.; Luna, A.M.; Hernández, M.; Sabench, F.; Porras, J.A.; Martínez, S.; Lucas, A.; et al. Increased levels and adipose tissue expression of visfatin in morbidly obese women: The relationship with pro-inflammatory cytokines. Clin. Endocrinol. 2012, 77, 691–698. [Google Scholar] [CrossRef]
- Madani, R.; Karastergiou, K.; Ogston, N.C.; Miheisi, N.; Bhome, R.; Haloob, N.; Tan, G.D.; Karpe, F.; Malone-Lee, J.; Hashemi, M.; et al. RANTES release by human adipose tissue in vivo and evidence for depot-specific differences. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1262–E1268. [Google Scholar] [CrossRef]
- Jonas, M.I.; Kurylowicz, A.; Bartoszewicz, Z.; Lisik, W.; Jonas, M.; Wierzbicki, Z.; Chmura, A.; Pruszczyk, P.; Puzianowska-Kuznicka, M. Interleukins 6 and 15 Levels Are Higher in Subcutaneous Adipose Tissue, but Obesity Is Associated with Their Increased Content in Visceral Fat Depots. Int. J. Mol. Sci. 2015, 16, 25817–25830. [Google Scholar] [CrossRef] [Green Version]
- Chacón, M.R.; Richart, C.; Gómez, J.M.; Megía, A.; Vilarrasa, N.; Fernández-Real, J.M.; García-España, A.; Miranda, M.; Masdevall, C.; Ricard, W.; et al. Expression of TWEAK and its receptor Fn14 in human subcutaneous adipose tissue. Relationship with other inflammatory cytokines in obesity. Cytokine 2006, 33, 129–137. [Google Scholar] [CrossRef]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Ramírez, B.; Valentí, V.; Moncada, R.; Landecho, M.F.; Silva, C.; Salvador, J.; Frühbeck, G. Increased Interleukin-32 Levels in Obesity Promote Adipose Tissue Inflammation and Extracellular Matrix Remodeling: Effect of Weight Loss. Diabetes 2016, 65, 3636–3648. [Google Scholar] [CrossRef] [Green Version]
- Pierce, J.R.; Maples, J.M.; Hickner, R.C. IL-15 concentrations in skeletal muscle and subcutaneous adipose tissue in lean and obese humans: Local effects of IL-15 on adipose tissue lipolysis. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E1131–E1139. [Google Scholar] [CrossRef] [Green Version]
- Samaras, K.; Botelho, N.K.; Chisholm, D.J.; Lord, R.V. Subcutaneous and visceral adipose tissue gene expression of serum adipokines that predict type 2 diabetes. Obesity 2010, 18, 884–889. [Google Scholar] [CrossRef] [PubMed]
- Fried, S.K.; Bunkin, D.A.; Greenberg, A.S. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: Depot difference and regulation by glucocorticoid. J. Clin. Endocrinol. Metab. 1998, 83, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Good, M.; Newell, F.M.; Haupt, L.M.; Whitehead, J.P.; Hutley, L.J.; Prins, J.B. TNF and TNF receptor expression and insulin sensitivity in human omental and subcutaneous adipose tissue—Influence of BMI and adipose distribution. Diab. Vasc. Dis. Res. 2006, 3, 26–33. [Google Scholar] [CrossRef]
- Salas-Salvadó, J.; Bulló, M.; García-Lorda, P.; Figueredo, R.; Del Castillo, D.; Bonada, A.; Balanzà, R. Subcutaneous adipose tissue cytokine production is not responsible for the restoration of systemic inflammation markers during weight loss. Int. J. Obes. 2006, 30, 1714–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leick, L.; Lindegaard, B.; Stensvold, D.; Plomgaard, P.; Saltin, B.; Pilegaard, H. Adipose tissue interleukin-18 mRNA and plasma interleukin-18: Effect of obesity and exercise. Obesity 2007, 15, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Moro, C.; Klimcakova, E.; Lolmède, K.; Berlan, M.; Lafontan, M.; Stich, V.; Bouloumié, A.; Galitzky, J.; Arner, P.; Langin, D. Atrial natriuretic peptide inhibits the production of adipokines and cytokines linked to inflammation and insulin resistance in human subcutaneous adipose tissue. Diabetologia 2007, 50, 1038–1047. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, N.B.; Højbjerre, L.; Sonne, M.P.; Alibegovic, A.C.; Vaag, A.; Dela, F.; Stallknecht, B. Interstitial concentrations of adipokines in subcutaneous abdominal and femoral adipose tissue. Regul. Pept. 2009, 155, 39–45. [Google Scholar] [CrossRef]
- Vendrell, J.; Maymó-Masip, E.; Tinahones, F.; García-España, A.; Megia, A.; Caubet, E.; García-Fuentes, E.; Chacón, M.R. Tumor necrosis-like weak inducer of apoptosis as a proinflammatory cytokine in human adipocyte cells: Up-regulation in severe obesity is mediated by inflammation but not hypoxia. J. Clin. Endocrinol. Metab. 2010, 95, 2983–2992. [Google Scholar] [CrossRef] [Green Version]
- Murdolo, G.; Herder, C.; Wang, Z.; Rose, B.; Schmelz, M.; Jansson, P.A. In situ profiling of adipokines in subcutaneous microdialysates from lean and obese individuals. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1095–E1105. [Google Scholar] [CrossRef] [Green Version]
- Skopková, M.; Penesová, A.; Sell, H.; Rádiková, Z.; Vlcek, M.; Imrich, R.; Koska, J.; Ukropec, J.; Eckel, J.; Klimes, I.; et al. Protein array reveals differentially expressed proteins in subcutaneous adipose tissue in obesity. Obesity 2007, 15, 2396–2406. [Google Scholar] [CrossRef] [Green Version]
- Al-Attar, A.; Presnell, S.R.; Clasey, J.L.; Long, D.E.; Walton, R.G.; Sexton, M.; Starr, M.E.; Kern, P.A.; Peterson, C.A.; Lutz, C.T. Human Body Composition and Immunity: Visceral Adipose Tissue Produces IL-15 and Muscle Strength Inversely Correlates with NK Cell Function in Elderly Humans. Front. Immunol. 2018, 9, 440. [Google Scholar] [CrossRef] [Green Version]
- Jorge, A.S.B.; Andrade, J.M.O.; Paraíso, A.F.; Jorge, G.C.B.; Silveira, C.M.; de Souza, L.R.; Santos, E.P.; Guimaraes, A.L.S.; Santos, S.H.S.; De-Paula, A.M.B. Body mass index and the visceral adipose tissue expression of IL-6 and TNF-alpha are associated with the morphological severity of non-alcoholic fatty liver disease in individuals with class III obesity. Obes. Res. Clin. Pract. 2018, 12, 1–8. [Google Scholar] [CrossRef]
- Zeyda, M.; Wernly, B.; Demyanets, S.; Kaun, C.; Hämmerle, M.; Hantusch, B.; Schranz, M.; Neuhofer, A.; Itariu, B.K.; Keck, M.; et al. Severe obesity increases adipose tissue expression of interleukin-33 and its receptor ST2, both predominantly detectable in endothelial cells of human adipose tissue. Int. J. Obes. 2013, 37, 658–665. [Google Scholar] [CrossRef] [Green Version]
- Catalán, V.; Gómez-Ambrosi, J.; Ramirez, B.; Rotellar, F.; Pastor, C.; Silva, C.; Rodríguez, A.; Gil, M.J.; Cienfuegos, J.A.; Frühbeck, G. Proinflammatory cytokines in obesity: Impact of type 2 diabetes mellitus and gastric bypass. Obes. Surg. 2007, 17, 1464–1474. [Google Scholar] [CrossRef]
- Bruun, J.M.; Lihn, A.S.; Pedersen, S.B.; Richelsen, B. Monocyte chemoattractant protein-1 release is higher in visceral than subcutaneous human adipose tissue (AT): Implication of macrophages resident in the AT. J. Clin. Endocrinol. Metab. 2005, 90, 2282–2289. [Google Scholar] [CrossRef]
- Hueso, L.; Ortega, R.; Selles, F.; Wu-Xiong, N.Y.; Ortega, J.; Civera, M.; Ascaso, J.F.; Sanz, M.J.; Real, J.T.; Piqueras, L. Upregulation of angiostatic chemokines IP-10/CXCL10 and I-TAC/CXCL11 in human obesity and their implication for adipose tissue angiogenesis. Int J. Obes. 2018, 42, 1406–1417. [Google Scholar] [CrossRef]
- Rouault, C.; Pellegrinelli, V.; Schilch, R.; Cotillard, A.; Poitou, C.; Tordjman, J.; Sell, H.; Clément, K.; Lacasa, D. Roles of chemokine ligand-2 (CXCL2) and neutrophils in influencing endothelial cell function and inflammation of human adipose tissue. Endocrinology 2013, 154, 1069–1079. [Google Scholar] [CrossRef] [Green Version]
- Sbierski-Kind, J.; Mai, K.; Kath, J.; Jurisch, A.; Streitz, M.; Kuchenbecker, L.; Babel, N.; Nienen, M.; Jürchott, K.; Spranger, L.; et al. Association between Subcutaneous Adipose Tissue Inflammation, Insulin Resistance, and Calorie Restriction in Obese Females. J. Immunol. 2021, 205, 45–55. [Google Scholar] [CrossRef]
- Gómez-Ambrosi, J.; Catalán, V.; Ramírez, B.; Rodríguez, A.; Colina, I.; Silva, C.; Rotellar, F.; Mugueta, C.; Gil, M.J.; Cienfuegos, J.A.; et al. Plasma osteopontin levels and expression in adipose tissue are increased in obesity. J. Clin. Endocrinol. Metab. 2007, 92, 3719–3727. [Google Scholar] [CrossRef] [Green Version]
- Kiefer, F.W.; Zeyda, M.; Todoric, J.; Huber, J.; Geyeregger, R.; Weichhart, T.; Aszmann, O.; Ludvik, B.; Silberhumer, G.R.; Prager, G.; et al. Osteopontin expression in human and murine obesity: Extensive local up-regulation in adipose tissue but minimal systemic alterations. Endocrinology 2008, 149, 1350–1357. [Google Scholar] [CrossRef] [Green Version]
- Mogilenko, D.A.; Caiazzo, R.; L’homme, L.; Pineau, L.; Raverdy, V.; Noulette, J.; Derudas, B.; Pattou, F.; Staels, B.; Dombrowicz, D. IFNgamma-producing NK cells in adipose tissue are associated with hyperglycemia and insulin resistance in obese women. Int. J. Obes. 2021, 45, 1607–1617. [Google Scholar] [CrossRef]
- Berndt, J.; Klöting, N.; Kralisch, S.; Kovacs, P.; Fasshauer, M.; Schön, M.R.; Stumvoll, M.; Blüher, M. Plasma visfatin concentrations and fat depot-specific mRNA expression in humans. Diabetes 2005, 54, 2911–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulmyer-Lacroix, O.; Desbriere, R.; Poggi, M.; Achard, V.; Alessi, M.C.; Boudouresque, F.; Ouafik, L.; Vuaroqueaux, V.; Labuhn, M.; Dutourand, A.; et al. Expression of adrenomedullin in adipose tissue of lean and obese women. Eur. J. Endocrinol. 2006, 155, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, S.A.; Ciaraldi, T.P.; Oh, D.K.; Savu, M.K.; Henry, R.R. Adiponectin secretion and response to pioglitazone is depot dependent in cultured human adipose tissue. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E842–E850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbanová, M.; Dostálová, I.; Trachta, P.; Drápalová, J.; Kaválková, P.; Haluzíková, D.; Matoulek, M.; Lacinová, Z.; Mráz, M.; Kasalický, M.; et al. Serum concentrations and subcutaneous adipose tissue mRNA expression of omentin in morbid obesity and type 2 diabetes mellitus: The effect of very-low-calorie diet, physical activity and laparoscopic sleeve gastrectomy. Physiol. Res. 2014, 63, 207–218. [Google Scholar] [CrossRef]
- Reneau, J.; Goldblatt, M.; Gould, J.; Kindel, T.; Kastenmeier, A.; Higgins, R.; Rengel, L.R.; Schoyer, K.; James, R.; Obi, B.; et al. Effect of adiposity on tissue-specific adiponectin secretion. PLoS ONE 2018, 13, e0198889. [Google Scholar]
- Arner, E.; Mejhert, N.; Kulyté, A.; Balwierz, P.J.; Pachkov, M.; Cormont, M.; Lorente-Cebrián, S.; Ehrlund, A.; Laurencikiene, J.; Hedén, P.; et al. Adipose tissue microRNAs as regulators of CCL2 production in human obesity. Diabetes 2012, 61, 1986–1993. [Google Scholar] [CrossRef] [Green Version]
- Gu, N.; You, L.; Shi, C.; Yang, L.; Pang, L.; Cui, X.; Ji, C.; Zheng, W.; Guo, X. Expression of miR-199a-3p in human adipocytes is regulated by free fatty acids and adipokines. Mol. Med. Rep. 2016, 14, 1180–1186. [Google Scholar] [CrossRef] [Green Version]
- Lozano-Bartolomé, J.; Llauradó, G.; Portero-Otin, M.; Altuna-Coy, A.; Rojo-Martínez, G.; Vendrell, J.; Jorba, R.; Rodríguez-Gallego, E.; Chacón, M.R. Altered Expression of miR-181a-5p and miR-23a-3p Is Associated with Obesity and TNFalpha-Induced Insulin Resistance. J. Clin. Endocrinol. Metab. 2018, 103, 1447–1458. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, R.; Al-Mass, A.; Al-Ghawas, D.; Shareif, N.; Zghoul, N.; Melhem, M.; Hasan, A.; Al-Ghimlas, F.; Dermime, S.; Behbehani, K. Interaction of osteopontin with IL-18 in obese individuals: Implications for insulin resistance. PLoS ONE 2013, 8, e63944. [Google Scholar]
- Vianello, E.; Kalousová, M.; Dozio, E.; Tacchini, L.; Zima, T.; Corsi Romanelli, M.M. Osteopontin: The Molecular Bridge between Fat and Cardiac-Renal Disorders. Int. J. Mol. Sci. 2021, 21, 5568. [Google Scholar] [CrossRef]
- Okamoto, H. Osteopontin and cardiovascular system. Mol. Cell. Biochem. 2007, 300, 1–7. [Google Scholar] [CrossRef]
- Singh, M.; Ananthula, S.; Milhorn, D.M.; Krishnaswamy, G.; Singh, K. Osteopontin: A novel inflammatory mediator of cardiovascular disease. Front. Biosci. 2007, 12, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.M.; Wang, J.; Huang, H.R.; Jing, H. Osteopontin expression and its possible functions in the aortic disorders and coronary artery disease. Rev. Bras. Cir. Cardiovasc. 2011, 26, 173–182. [Google Scholar] [CrossRef]
- Kaleta, B. The role of osteopontin in kidney diseases. Inflamm. Res. 2019, 68, 93–102. [Google Scholar] [CrossRef]
- Si, J.; Wang, C.; Zhang, D.; Wang, B.; Zhou, Y. Osteopontin in Bone Metabolism and Bone Diseases. Med. Sci. Monit. 2021, 26, e919159. [Google Scholar] [CrossRef]
- Iida, T.; Wagatsuma, K.; Hirayama, D.; Nakase, H. Is Osteopontin a Friend or Foe of Cell Apoptosis in Inflammatory Gastrointestinal and Liver Diseases? Int. J. Mol. Sci. 2017, 19, 7. [Google Scholar] [CrossRef] [Green Version]
- O’Regan, A. The role of osteopontin in lung disease. Cytokine Growth Factor Rev. 2003, 14, 479–488. [Google Scholar] [CrossRef]
- Del Toro, R.; Cavallari, I.; Tramontana, F.; Park, K.; Strollo, R.; Valente, L.; De Pascalis, M.; Grigioni, F.; Pozzilli, P.; Buzzetti, R.; et al. Association of bone biomarkers with advanced atherosclerotic disease in people with overweight/obesity. Endocrine 2021, 73, 339–346. [Google Scholar] [CrossRef]
- Suliburska, J.; Bogdanski, P.; Gajewska, E.; Kalmus, G.; Sobieska, M.; Samborski, W. The association of insulin resistance with serum osteoprotegerin in obese adolescents. J. Physiol. Biochem. 2013, 69, 847–853. [Google Scholar] [CrossRef]
- Kotanidou, E.P.; Kotanidis, C.P.; Giza, S.; Serbis, A.; Tsinopoulou, V.R.; Karalazou, P.; Tzimagiorgis, G.; Galli-Tsinopoulou, A. Osteoprotegerin increases parallel to insulin resistance in obese adolescents. Endocr. Res. 2019, 44, 9–15. [Google Scholar] [CrossRef]
- Dimitri, P.; Wales, J.K.; Bishop, N. Adipokines, bone-derived factors and bone turnover in obese children; evidence for altered fat-bone signalling resulting in reduced bone mass. Bone 2011, 48, 189–196. [Google Scholar] [CrossRef]
- Makarović, S.; Makarović, Z.; Steiner, R.; Mihaljević, I.; Milas-Ahić, J. Osteoprotegerin and Vascular Calcification: Clinical and Prognostic Relevance. Coll. Antropol. 2015, 39, 461–468. [Google Scholar]
- Montagnana, M.; Lippi, G.; Danese, E.; Guidi, G.C. The role of osteoprotegerin in cardiovascular disease. Ann. Med. 2013, 45, 254–264. [Google Scholar] [CrossRef]
- Nacaroglu, H.T.; Büke, Ö.; Gayret, Ö.B.; Erol, M.; Zengi, O. Serum osteoprotegerin levels in school-aged children with asthma. Allergol. Immunopathol. 2021, 48, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Bilgin, E.; Yasasever, V.; Soydinc, H.O.; Yasasever, C.T.; Ozturk, N.; Duranyildiz, D. Markers of bone metastases in breast and lung cancers. Asian Pac. J. Cancer Prev. 2012, 13, 4331–4334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrie, A.; Waterman, E.; Southwood, M.; Evans, D.; Suntharalingam, J.; Francis, S.; Crossman, D.; Croucher, P.; Morrell, N.; Newman, C. Evidence of a role for osteoprotegerin in the pathogenesis of pulmonary arterial hypertension. Am. J. Pathol. 2008, 172, 256–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasiewicz, M.; Knapp, M.; Waszkiewicz, E.; Musiał, W.J.; Kamiński, K.A. Potential pathogenic role of soluble receptor activator of nuclear factor-qB ligand and osteoprotegerin in patients with pulmonary arterial hypertension. Pol. Arch. Med. Wewn. 2014, 124, 579–586. [Google Scholar] [CrossRef] [Green Version]
- De Voogd, F.A.; Gearry, R.B.; Mulder, C.J.; Day, A.S. Osteoprotegerin: A novel biomarker for inflammatory bowel disease and gastrointestinal carcinoma. J. Gastroenterol. Hepatol. 2016, 31, 1386–1392. [Google Scholar] [CrossRef] [Green Version]
- Krela-Kaźmierczak, I.; Szymczak-Tomczak, A.; Łykowska-Szuber, L.; Wysocka, E.; Michalak, M.; Stawczyk-Eder, K.; Waszak, K.; Linke, K.; Eder, P. Interleukin 6, osteoprotegerin, sRANKL and bone metabolism in inflammatory bowel diseases. Adv. Clin. Exp. Med. 2018, 27, 449–453. [Google Scholar] [CrossRef]
- Dufresne, S.S.; Dumont, N.A.; Bouchard, P.; Lavergne, É.; Penninger, J.M.; Frenette, J. Osteoprotegerin protects against muscular dystrophy. Am. J. Pathol. 2015, 185, 920–926. [Google Scholar] [CrossRef]
- Dufresne, S.S.; Boulanger-Piette, A.; Frenette, J. Osteoprotegerin and beta (2)-Agonists Mitigate Muscular Dystrophy in Slow- and Fast-Twitch Skeletal Muscles. Am. J. Pathol. 2017, 187, 498–504. [Google Scholar] [CrossRef] [Green Version]
- Pacifico, L.; Di Renzo, L.; Anania, C.; Osborn, J.F.; Ippoliti, F.; Schiavo, E.; Chiesa, C. Increased T-helper interferon-gamma-secreting cells in obese children. Eur. J. Endocrinol. 2006, 154, 691–697. [Google Scholar] [CrossRef] [Green Version]
- Asadikaram, G.; Ram, M.; Izadi, A.; Sheikh Fathollahi, M.; Nematollahi, M.H.; Najafipour, H.; Shahoozehi, B.; Mirhoseini, M.; Masoumi, M.; Shahrokhi, N.; et al. The study of the serum level of IL-4, TGF-beta, IFN-gamma, and IL-6 in overweight patients with and without diabetes mellitus and hypertension. J. Cell. Biochem. 2019, 120, 4147–4157. [Google Scholar] [CrossRef]
- Youssef, D.M.; Elbehidy, R.M.; Shokry, D.M.; Elbehidy, E.M. The influence of leptin on Th1/Th2 balance in obese children with asthma. J. Bras. Pneumol. 2013, 39, 562–568. [Google Scholar] [CrossRef]
- Lucas, R.; Parikh, S.J.; Sridhar, S.; Guo, D.H.; Bhagatwala, J.; Dong, Y.; Caldwell, R.; Mellor, A.; Caldwell, W.; Zhu, H.; et al. Cytokine profiling of young overweight and obese female African American adults with prediabetes. Cytokine 2013, 64, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Elyasi, A.; Voloshyna, I.; Ahmed, S.; Kasselman, L.J.; Behbodikhah, J.; De Leon, J.; Reiss, A.B. The role of interferon-gamma in cardiovascular disease: An update. Inflamm. Res. 2021, 69, 975–988. [Google Scholar] [CrossRef]
- Raundhal, M.; Morse, C.; Khare, A.; Oriss, T.B.; Milosevic, J.; Trudeau, J.; Huff, R.; Pilewski, J.; Holguin, F.; Kolls, J.; et al. High IFN-gamma and low SLPI mark severe asthma in mice and humans. J. Clin. Invest. 2015, 125, 3037–3050. [Google Scholar] [CrossRef] [Green Version]
- Nie, W.; Meng, L.; Wang, X.; Xiu, Q. Interferon-gamma +874A/T polymorphism is associated with asthma risk: A meta-analysis. J. Investig. Allergol. Clin. Immunol. 2014, 24, 324–330. [Google Scholar]
- Ten Hacken, N.H.; Oosterhoff, Y.; Kauffman, H.F.; Guevarra, L.; Satoh, T.; Tollerud, D.J.; Postma, D.S. Elevated serum interferon-gamma in atopic asthma correlates with increased airways responsiveness and circadian peak expiratory flow variation. Eur. Respir. J. 1998, 11, 312–316. [Google Scholar] [CrossRef] [Green Version]
- Cembrzynska-Nowak, M.; Szklarz, E.; Inglot, A.D.; Teodorczyk-Injeyan, J.A. Elevated release of tumor necrosis factor-alpha and interferon-gamma by bronchoalveolar leukocytes from patients with bronchial asthma. Am. Rev. Respir. Dis. 1993, 147, 291–295. [Google Scholar] [CrossRef]
- Bantulà, M.; Roca-Ferrer, J.; Arismendi, E.; Picado, C. Asthma and Obesity: Two Diseases on the Rise and Bridged by Inflammation. J. Clin. Med. 2021, 10, 169. [Google Scholar] [CrossRef]
- Yehuda-Shnaidman, E.; Schwartz, B. Mechanisms linking obesity, inflammation and altered metabolism to colon carcinogenesis. Obes. Rev. 2012, 13, 1083–1095. [Google Scholar] [CrossRef]
- Vazzana, N.; Riondino, S.; Toto, V.; Guadagni, F.; Roselli, M.; Davi, G.; Ferroni, P. Obesity-driven inflammation and colorectal cancer. Curr. Med. Chem. 2012, 19, 5837–5853. [Google Scholar] [CrossRef]
- Olszanecka-Glinianowicz, M.; Handzlik-Orlik, G.; Orlik, B.; Chudek, J. Adipokines in the pathogenesis of idiopathic inflammatory bowel disease. Endokrynol. Pol. 2013, 64, 226–231. [Google Scholar]
- Tewari, N.; Awad, S.; Macdonald, I.A.; Lobo, D.N. Obesity-related insulin resistance: Implications for the surgical patient. Int. J. Obes. 2015, 39, 1575–1588. [Google Scholar] [CrossRef]
- Janochova, K.; Haluzik, M.; Buzga, M. Visceral fat and insulin resistance—What we know? Biomed. Pap. Med Fac. Palacky Univ. Olomouc 2019, 163, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Alexopoulos, N.; Katritsis, D.; Raggi, P. Visceral adipose tissue as a source of inflammation and promoter of atherosclerosis. Atherosclerosis 2014, 233, 104–112. [Google Scholar] [CrossRef]
- Malavazos, A.E.; Corsi, M.M.; Ermetici, F.; Coman, C.; Sardanelli, F.; Rossi, A.; Morricone, L.; Ambrosi, B. Proinflammatory cytokines and cardiac abnormalities in uncomplicated obesity: Relationship with abdominal fat deposition. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 294–302. [Google Scholar] [CrossRef]
- Teplan, V., Jr.; Vyhnánek, F.; Gürlich, R.; Haluzík, M.; Racek, J.; Vyhnankova, I.; Stollová, M.; Teplan, V. Increased proinflammatory cytokine production in adipose tissue of obese patients with chronic kidney disease. Wien. Klin. Wochenschr. 2010, 122, 466–473. [Google Scholar] [CrossRef]
- Su, Y.; Yuan, J.; Zhang, F.; Lei, Q.; Zhang, T.; Li, K.; Guo, J.; Hong, Y.; Bu, G.; Lv, X.; et al. MicroRNA-181a-5p and microRNA-181a-3p cooperatively restrict vascular inflammation and atherosclerosis. Cell Death Dis. 2019, 10, 365. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Xue, Y.; Cai, H. Down-Regulation of miR-181a-5p Prevents Cerebral Ischemic Injury by Upregulating En2 and Activating Wnt/beta-catenin Pathway. J. Stroke Cerebrovasc. Dis. 2021, 30, 105485. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Long, M.Y.; Chen, Z.Q.; Huang, J.W.; Qin, Z.B.; Li, L. Downregulation of miR-181a-5p alleviates oxidative stress and inflammation in coronary microembolization-induced myocardial damage by directly targeting XIAP. J. Geriatr. Cardiol. 2021, 18, 426–439. [Google Scholar] [PubMed]
- Zhao, H.; Tao, Z.; Wang, R.; Liu, P.; Yan, F.; Li, J.; Zhang, C.; Ji, X.; Luo, Y. MicroRNA-23a-3p attenuates oxidative stress injury in a mouse model of focal cerebral ischemia-reperfusion. Brain Res. 2014, 1592, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Park, K.M.; Teoh, J.P.; Wang, Y.; Broskova, Z.; Bayoumi, A.S.; Tang, Y.; Su, H.; Weintraub, N.L.; Kim, I.M. Carvedilol-responsive microRNAs, miR-199a-3p and -214 protect cardiomyocytes from simulated ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H371–H383. [Google Scholar] [CrossRef]
- Joris, V.; Gomez, E.L.; Menchi, L.; Lobysheva, I.; Di Mauro, V.; Esfahani, H.; Condorelli, G.; Balligand, J.L.; Catalucci, D.; Dessy, C. MicroRNA-199a-3p and MicroRNA-199a-5p Take Part to a Redundant Network of Regulation of the NOS (NO Synthase)/NO Pathway in the Endothelium. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2345–2357. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Liu, W.X.; Huang, X.G. MicroRNA-199a-3p inhibits angiogenesis by targeting the VEGF/PI3K/AKT signalling pathway in an in vitro model of diabetic retinopathy. Exp. Mol. Pathol. 2021, 116, 104488. [Google Scholar] [CrossRef]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Ramírez, B.; Silva, C.; Rotellar, F.; Gil, M.J.; Cienfuegos, J.A.; Salvador, J.; Frühbeck, G. Increased adipose tissue expression of lipocalin-2 in obesity is related to inflammation and matrix metalloproteinase-2 and metalloproteinase-9 activities in humans. J. Mol. Med. 2009, 87, 803–813. [Google Scholar] [CrossRef]
- Auguet, T.; Quintero, Y.; Terra, X.; Martínez, S.; Lucas, A.; Pellitero, S.; Aguilar, C.; Hernández, M.; del Castillo, D.; Richart, C. Upregulation of lipocalin 2 in adipose tissues of severely obese women: Positive relationship with proinflammatory cytokines. Obesity 2011, 19, 2295–2300. [Google Scholar] [CrossRef]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Ramírez, B.; Rotellar, F.; Valentí, V.; Silva, C.; Gil, M.J.; Salvador, J.; Frühbeck, G. Increased tenascin C and Toll-like receptor 4 levels in visceral adipose tissue as a link between inflammation and extracellular matrix remodeling in obesity. J. Clin. Endocrinol. Metab. 2012, 97, E1880–E1889. [Google Scholar] [CrossRef] [Green Version]
- Osorio-Conles, O.; Guitart, M.; Chacón, M.R.; Maymo-Masip, E.; Moreno-Navarrete, J.M.; Montori-Grau, M.; Näf, S.; Fernandez-Real, J.M.; Vendrell, J.; Gómez-Foix, A.M. Plasma PTX3 protein levels inversely correlate with insulin secretion and obesity, whereas visceral adipose tissue PTX3 gene expression is increased in obesity. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E1254–E1261. [Google Scholar] [CrossRef]
- Wang, Y.; Lam, K.S.; Kraegen, E.W.; Sweeney, G.; Zhang, J.; Tso, A.W.; Chow, W.S.; Wat, N.M.; Xu, J.Y.; Hoo, R.L.; et al. Lipocalin-2 is an inflammatory marker closely associated with obesity, insulin resistance, and hyperglycemia in humans. Clin. Chem. 2007, 53, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Hoo, R.L.; Yeung, D.C.; Lam, K.S.; Xu, A. Inflammatory biomarkers associated with obesity and insulin resistance: A focus on lipocalin-2 and adipocyte fatty acid-binding protein. Expert Rev. Endocrinol. Metab. 2008, 3, 29–41. [Google Scholar] [CrossRef]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Ramírez, B.; Valentí, V.; Moncada, R.; Silva, C.; Salvador, J.; Frühbeck, G. Peripheral mononuclear blood cells contribute to the obesity-associated inflammatory state independently of glycemic status: Involvement of the novel proinflammatory adipokines chemerin, chitinase-3-like protein 1, lipocalin-2 and osteopontin. Genes Nutr. 2015, 10, 460. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wu, Y.; Zhang, Y.; Leroith, D.; Bernlohr, D.A.; Chen, X. The role of lipocalin 2 in the regulation of inflammation in adipocytes and macrophages. Mol. Endocrinol. 2008, 22, 1416–1426. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Huang, H.; Xia, T.; Liu, C.; Muhammad, S.; Sun, C. Homeobox a5 Promotes White Adipose Tissue Browning Through Inhibition of the Tenascin C/Toll-Like Receptor 4/Nuclear Factor Kappa B Inflammatory Signaling in Mice. Front. Immunol. 2018, 9, 647. [Google Scholar] [CrossRef] [Green Version]
- Renovato-Martins, M.; Moreira-Nunes, C.; Atella, G.C.; Barja-Fidalgo, C.; Moraes, J.A. Obese Adipose Tissue Secretion Induces Inflammation in Preadipocytes: Role of Toll-Like Receptor-4. Nutrients 2021, 12, 2828. [Google Scholar] [CrossRef]
- Kamble, P.G.; Pereira, M.J.; Sidibeh, C.O.; Amini, S.; Sundbom, M.; Börjesson, J.L.; Eriksson, J.W. Lipocalin 2 produces insulin resistance and can be upregulated by glucocorticoids in human adipose tissue. Mol. Cell. Endocrinol. 2016, 427, 124–132. [Google Scholar] [CrossRef]
- Tsukumo, D.M.; Carvalho-Filho, M.A.; Carvalheira, J.B.; Prada, P.O.; Hirabara, S.M.; Schenka, A.A.; Araújo, E.P.; Vassallo, J.; Curi, R.; Velloso, L.A.; et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 2007, 56, 1986–1998. [Google Scholar] [CrossRef] [Green Version]
- Yan, Q.W.; Yang, Q.; Mody, N.; Graham, T.E.; Hsu, C.H.; Xu, Z.; Houstis, N.E.; Kahn, B.B.; Rosen, E.D. The adipokine lipocalin 2 is regulated by obesity and promotes insulin resistance. Diabetes 2007, 56, 2533–2540. [Google Scholar] [CrossRef] [Green Version]
- Jun, L.S.; Siddall, C.P.; Rosen, E.D. A minor role for lipocalin 2 in high-fat diet-induced glucose intolerance. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E825–E835. [Google Scholar] [CrossRef] [Green Version]
- Song, E.; Fan, P.; Huang, B.; Deng, H.B.; Cheung, B.M.; Félétou, M.; Vilaine, J.P.; Villeneuve, N.; Xu, A.; Vanhoutte, P.M.; et al. Deamidated lipocalin-2 induces endothelial dysfunction and hypertension in dietary obese mice. J. Am. Heart Assoc. 2014, 3, e000837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alberti, L.; Gilardini, L.; Zulian, A.; Micheletto, G.; Peri, G.; Doni, A.; Mantovani, A.; Invitti, C. Expression of long pentraxin PTX3 in human adipose tissue and its relation with cardiovascular risk factors. Atherosclerosis 2009, 202, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, K.; Manabe, I. Toll-like receptor, lipotoxicity and chronic inflammation: The pathological link between obesity and cardiometabolic disease. J. Atheroscler. Thromb. 2014, 21, 629–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, J.D.; Surani, S.; Horseman, M.A. Endotoxin, Toll-like Receptor-4, and Atherosclerotic Heart Disease. Curr. Cardiol. Rev. 2017, 13, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Shibata, K.; Sato, K.; Shirai, R.; Seki, T.; Okano, T.; Yamashita, T.; Koide, A.; Mitsuboshi, M.; Mori, Y.; Hirano, T.; et al. Lipocalin-2 exerts pro-atherosclerotic effects as evidenced by in vitro and in vivo experiments. Heart Vessel. 2021, 35, 1012–1024. [Google Scholar] [CrossRef]
- Zwick, R.K.; Guerrero-Juarez, C.F.; Horsley, V.; Plikus, M.V. Anatomical, Physiological, and Functional Diversity of Adipose Tissue. Cell Metab. 2018, 27, 68–83. [Google Scholar] [CrossRef] [Green Version]
- Britton, K.A.; Fox, C.S. Ectopic fat depots and cardiovascular disease. Circulation 2011, 124, 837–841. [Google Scholar] [CrossRef]
- Lim, S.; Meigs, J.B. Links between ectopic fat and vascular disease in humans. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 820–826. [Google Scholar] [CrossRef] [Green Version]
- Smith, U. Abdominal obesity: A marker of ectopic fat accumulation. J. Clin. Invest. 2015, 125, 1790–1792. [Google Scholar] [CrossRef] [Green Version]
- Schneider, H.J.; Glaesmer, H.; Klotsche, J.; Böhler, S.; Lehnert, H.; Zeiher, A.M.; März, W.; Pittrow, D.; Stalla, G.K.; Wittchen, H.U. DETECT Study Group. Accuracy of anthropometric indicators of obesity to predict cardiovascular risk. J. Clin. Endocrinol. Metab. 2007, 92, 589–594. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.J.; Wu, Y.; Fried, S.K. Adipose tissue heterogeneity: Implication of depot differences in adipose tissue for obesity complications. Mol. Aspects Med. 2013, 34, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kwok, K.H.M.; Lam, K.S.L.; Xu, A. Heterogeneity of white adipose tissue: Molecular basis and clinical implications. Exp. Mol. Med. 2016, 48, 215. [Google Scholar] [CrossRef] [Green Version]
- Von Eyben, F.E.; Mouritsen, E.; Holm, J.; Montvilas, P.; Dimcevski, G.; Suciu, G.; Helleberg, I.; Kristensen, L.; von Eyben, R. Intra-abdominal obesity and metabolic risk factors: A study of young adults. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 941–949. [Google Scholar] [CrossRef] [Green Version]
- Vega, G.L.; Adams-Huet, B.; Peshock, R.; Willett, D.; Shah, B.; Grundy, S.M. Influence of body fat content and distribution on variation in metabolic risk. J. Clin. Endocrinol. Metab. 2006, 91, 4459–4466. [Google Scholar] [CrossRef] [Green Version]
- Reaven, G.M. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1600. [Google Scholar] [CrossRef]
- Grundy, S.M.; Brewer, H.B., Jr.; Cleeman, J.I.; Smith, S.C., Jr.; Lenfant, C.; American Heart Association; National Heart, Lung, and Blood Institute. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation 2004, 109, 433–438. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, E.J.; LeRoith, D.; Karnieli, E. The metabolic syndrome-from insulin resistance to obesity and diabetes. Endocrinol. Metab. Clin. N. Am. 2008, 37, 559–579. [Google Scholar] [CrossRef]
- Karelis, A.D.; St-Pierre, D.H.; Conus, F.; Rabasa-Lhoret, R.; Poehlman, E.T. Metabolic and body composition factors in subgroups of obesity: What do we know? J. Clin. Endocrinol. Metab. 2004, 89, 2569–2575. [Google Scholar] [CrossRef]
- Monteiro, R.; de Castro, P.M.S.T.; Calhau, C.; Azevedo, I. Adipocyte size and liability to cell death. Obes. Surg. 2006, 16, 804–806. [Google Scholar] [CrossRef]
- Monteiro, R.; Azevedo, I. Chronic inflammation in obesity and the metabolic syndrome. Mediat. Inflamm. 2010, 2010, 289645. [Google Scholar] [CrossRef]
- Fang, L.; Guo, F.; Zhou, L.; Stahl, R.; Grams, J. The cell size and distribution of adipocytes from subcutaneous and visceral fat is associated with type 2 diabetes mellitus in humans. Adipocyte 2015, 4, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laforest, S.; Labrecque, J.; Michaud, A.; Cianflone, K.; Tchernof, A. Adipocyte size as a determinant of metabolic disease and adipose tissue dysfunction. Crit. Rev. Clin. Lab. Sci. 2015, 52, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Cobb, W.S.; Burns, J.M.; Kercher, K.W.; Matthews, B.D.; James Norton, H.; Todd Heniford, B. Normal intraabdominal pressure in healthy adults. J. Surg. Res. 2005, 129, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.M.; Marceau, S.; Forse, R.A. Intra-abdominal pressure in the morbidly obese. Obes. Surg. 2005, 15, 1225–1232. [Google Scholar] [CrossRef]
- Monteiro, R.; Calhau, C.; Azevedo, I. Obstructive sleep apnea and adipocyte death. Eur. J. Heart Fail. 2007, 9, 103–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Invest. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Song, M.J.; Kim, K.H.; Yoon, J.M.; Kim, J.B. Activation of Toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochem. Biophys. Res. Commun. 2006, 346, 739–745. [Google Scholar] [CrossRef]
- Amar, J.; Chabo, C.; Waget, A.; Klopp, P.; Vachoux, C.; Bermúdez-Humarán, L.G.; Smirnova, N.; Bergé, M.; Sulpice, T.; Lahtinen, S.; et al. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: Molecular mechanisms and probiotic treatment. EMBO Mol. Med. 2011, 3, 559–572. [Google Scholar] [CrossRef]
- Yang, J.; Ran, Y.; Yang, Y.; Song, S.; Wu, Y.; Qi, Y.; Gao, Y.; Li, G. 4-Hydroxyisoleucine Alleviates Macrophage-Related Chronic Inflammation and Metabolic Syndrome in Mice Fed a High-Fat Diet. Front. Pharmacol. 2021, 11, 606514. [Google Scholar] [CrossRef]
- Primeau, V.; Coderre, L.; Karelis, A.D.; Brochu, M.; Lavoie, M.E.; Messier, V.; Sladek, R.; Rabasa-Lhoret, R. Characterizing the profile of obese patients who are metabolically healthy. Int. J. Obes. 2011, 35, 971–981. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.I.; Mittendorfer, B.; Klein, S. Metabolically healthy obesity: Facts and fantasies. J. Clin. Invest. 2019, 129, 3978–3989. [Google Scholar] [CrossRef] [Green Version]
- Phillips, C.M.; Perry, I.J. Does inflammation determine metabolic health status in obese and nonobese adults? J. Clin. Endocrinol. Metab. 2013, 98, E1610–E1619. [Google Scholar] [CrossRef]
- Muñoz-Garach, A.; Cornejo-Pareja, I.; Tinahones, F.J. Does Metabolically Healthy Obesity Exist? Nutrients 2016, 8, 320. [Google Scholar] [CrossRef] [Green Version]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Lionetti, L.; Mollica, M.P.; Lombardi, A.; Cavaliere, G.; Gifuni, G.; Barletta, A. From chronic overnutrition to insulin resistance: The role of fat-storing capacity and inflammation. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 146–152. [Google Scholar] [CrossRef]
- Klöting, N.; Fasshauer, M.; Dietrich, A.; Kovacs, P.; Schön, M.R.; Kern, M.; Stumvoll, M.; Blüher, M. Insulin-sensitive obesity. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E506–E515. [Google Scholar] [CrossRef]
- Blüher, M. Metabolically Healthy Obesity. Endocr. Rev. 2020, 41, 405–420. [Google Scholar] [CrossRef] [Green Version]
- Goossens, G.H. The metabolic phenotype in obesity: Fat mass, body fat distribution, and adipose tissue function. Obes. Facts. 2017, 10, 207–215. [Google Scholar] [CrossRef]
- Stefan, N.; Schick, F.; Häring, H.U. Causes, Characteristics, and Consequences of Metabolically Unhealthy Normal Weight in Humans. Cell Metab. 2017, 26, 292–300. [Google Scholar] [CrossRef]
- Gaiţă, D.; Moşteoru, S. Metabolically healthy versus unhealthy obesity and risk for diabetes mellitus and cardiovascular diseases. Cardiovasc. Endocrinol. 2017, 6, 23–26. [Google Scholar] [CrossRef]
- Ruggiero, A.D.; Key, C.C.; Kavanagh, K. Adipose Tissue Macrophage Polarization in Healthy and Unhealthy Obesity. Front. Nutr. 2021, 8, 625331. [Google Scholar] [CrossRef]
- Pasarica, M.; Sereda, O.R.; Redman, L.M.; Albarado, D.C.; Hymel, D.T.; Roan, L.E.; Rood, J.C.; Burk, D.H.; Smith, S.R. Reduced adipose tissue oxygenation in human obesity: Evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes 2009, 58, 718–725. [Google Scholar] [CrossRef] [Green Version]
- Dario, A.B.; Ferreira, M.L.; Refshauge, K.M.; Lima, T.S.; Ordoñana, J.R.; Ferreira, P.H. The relationship between obesity, low back pain, and lumbar disc degeneration when genetics and the environment are considered: A systematic review of twin studies. Spine J. 2015, 15, 1106–1117. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Matsudaira, K.; Sawada, S.S.; Gando, Y.; Kawakami, R.; Kinugawa, C.; Okamoto, T.; Tsukamoto, K.; Miyachi, M.; Naito, H. Obesity and low back pain: A retrospective cohort study of Japanese males. J. Phys. Ther. Sci. 2017, 29, 978–983. [Google Scholar] [CrossRef] [Green Version]
- Da Cruz Fernandes, I.M.; Pinto, R.Z.; Ferreira, P.; Lira, F.S. Low back pain, obesity, and inflammatory markers: Exercise as potential treatment. J. Exerc. Rehabil. 2018, 14, 168–174. [Google Scholar] [CrossRef] [Green Version]
- Puri, J.; Sharma, S.; Samuel, A.J.; Chahal, A. Investigate Correlation between Diastasis of Rectus Abdominis Muscle and Low Back Pain in Obese Women. J. Lifestyle Med. 2021, 11, 38–42. [Google Scholar] [CrossRef]
- Dufour, A.B.; Losina, E.; Menz, H.B.; LaValley, M.P.; Hannan, M.T. Obesity, foot pain and foot disorders in older men and women. Obes. Res. Clin. Pract. 2017, 11, 445–453. [Google Scholar] [CrossRef]
- Moon, J.L.; Moon, K.M.; Carlisle, D.M. Obesity-Related Foot Pain: Diagnosis and Surgical Planning. Clin. Podiatr. Med. Surg. 2019, 36, 141–151. [Google Scholar] [CrossRef]
- Li, J.S.; Tsai, T.Y.; Clancy, M.M.; Li, G.; Lewis, C.L.; Felson, D.T. Weight loss changed gait kinematics in individuals with obesity and knee pain. Gait Posture 2019, 68, 461–465. [Google Scholar] [CrossRef]
- Landsmeer, M.L.A.; Runhaar, J.; van Middelkoop, M.; Oei, E.H.G.; Schiphof, D.; Bindels, P.J.E.; Bierma-Zeinstra, S.M.A. Predicting Knee Pain and Knee Osteoarthritis Among Overweight Women. J. Am. Board Fam. Med. 2019, 32, 575–584. [Google Scholar] [CrossRef]
- Haebich, S.J.; Mark, P.; Khan, R.J.K.; Fick, D.P.; Brownlie, C.; Wimhurst, J.A. The Influence of Obesity on Hip Pain, Function, and Satisfaction 10 Years Following Total Hip Arthroplasty. J. Arthroplasty. 2021, 35, 818–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özkuk, K.; Ateş, Z. The effect of obesity on pain and disability in chronic shoulder pain patients. J. Back Musculoskelet. Rehabil. 2021, 33, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Vincent, H.K.; Adams, M.C.; Vincent, K.R.; Hurley, R.W. Musculoskeletal pain, fear avoidance behaviors, and functional decline in obesity: Potential interventions to manage pain and maintain function. Reg. Anesth. Pain Med. 2013, 38, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.; Ells, L.; Ryan, C.; Martin, D. Perceptions of adults with overweight/obesity and chronic musculoskeletal pain: An interpretative phenomenological analysis. J. Clin. Nurs. 2018, 27, e776–e786. [Google Scholar] [CrossRef] [Green Version]
- Hooper, M.M.; Stellato, T.A.; Hallowell, P.T.; Seitz, B.A.; Moskowitz, R.W. Musculoskeletal findings in obese subjects before and after weight loss following bariatric surgery. Int J. Obes. 2007, 31, 114–120. [Google Scholar] [CrossRef] [Green Version]
- Di Lorenzo, C.; Pinto, A.; Ienca, R.; Coppola, G.; Sirianni, G.; Di Lorenzo, G.; Parisi, V.; Serrao, M.; Spagnoli, A.; Vestri, A.; et al. A Randomized Double-Blind, Cross-Over Trial of very Low-Calorie Diet in Overweight Migraine Patients: A Possible Role for Ketones? Nutrients 2019, 11, 1742. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Davis-Ajami, M.L.; Lu, Z.K. Impact of migraine on health care utilization and expenses in obese adults: A US population-based study. Clinicoecon. Outcomes Res. 2018, 11, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Miri, A.; Nasiri, M.; Zonoori, S.; Yarahmad, F.; Dabbagh-Moghadam, A.; Askari, G.; Sadeghi, O.; Asadi, M. The association between obesity and migraine in a population of Iranian adults: A case-control study. Diabetes Metab. Syndr. 2018, 12, 733–736. [Google Scholar] [CrossRef]
- Verrotti, A.; Di Fonzo, A.; Penta, L.; Agostinelli, S.; Parisi, P. Obesity and headache/migraine: The importance of weight reduction through lifestyle modifications. Biomed. Res. Int. 2014, 2014, 420858. [Google Scholar] [CrossRef]
- Verrotti, A.; Carotenuto, M.; Altieri, L.; Parisi, P.; Tozzi, E.; Belcastro, V.; Esposito, M.; Guastaferro, N.; Ciuti, A.; Mohn, A.; et al. Migraine and obesity: Metabolic parameters and response to a weight loss programme. Pediatr. Obes. 2015, 10, 220–225. [Google Scholar] [CrossRef]
- Hatami, M.; Soveid, N.; Lesani, A.; Djafarian, K.; Shab-Bidar, S. Migraine and Obesity: Is There a Relationship? A Systematic Review and Meta-Analysis of Observational Studies. CNS Neurol. Disord. Drug Targets 2021, 20, 863–870. [Google Scholar] [CrossRef]
- Miscio, G.; Guastamacchia, G.; Brunani, A.; Priano, L.; Baudo, S.; Mauro, A. Obesity and peripheral neuropathy risk: A dangerous liaison. J. Peripher. Nerv. Syst. 2005, 10, 354–358. [Google Scholar] [CrossRef]
- Hozumi, J.; Sumitani, M.; Matsubayashi, Y.; Abe, H.; Oshima, Y.; Chikuda, H.; Takeshita, K.; Yamada, Y. Relationship between Neuropathic Pain and Obesity. Pain Res. Manag. 2016, 2016, 2487924. [Google Scholar] [CrossRef] [Green Version]
- Herman, R.M.; Brower, J.B.; Stoddard, D.G.; Casano, A.R.; Targovnik, J.H.; Herman, J.H.; Tearse, P. Prevalence of somatic small fiber neuropathy in obesity. Int. J. Obes. 2007, 31, 226–235. [Google Scholar] [CrossRef] [Green Version]
- De Araújo, T.A.; Mota, M.C.; Crispim, C.A. Obesity and sleepiness in women with fibromyalgia. Rheumatol. Int. 2015, 35, 281–287. [Google Scholar] [CrossRef]
- Paiva, E.S.; Andretta, A.; Batista, E.D.; Lobo, M.M.M.T.; Miranda, R.C.; Nisihara, R.; Schieferdecker, M.E.M.; Boguszewski, C.L. Serum levels of leptin and adiponectin and clinical parameters in women with fibromyalgia and overweight/obesity. Arch. Endocrinol. Metab. 2017, 61, 249–256. [Google Scholar] [CrossRef] [Green Version]
- Callaghan, B.C.; Xia, R.; Banerjee, M.; de Rekeneire, N.; Harris, T.B.; Newman, A.B.; Satterfield, S.; Schwartz, A.V.; Vinik, A.I.; Feldman, E.L.; et al. Health ABC Study. Metabolic Syndrome Components Are Associated with Symptomatic Polyneuropathy Independent of Glycemic Status. Diabetes Care 2016, 39, 801–807. [Google Scholar] [CrossRef] [Green Version]
- Stino, A.M.; Smith, A.G. Peripheral neuropathy in prediabetes and the metabolic syndrome. J. Diabetes Investig. 2017, 8, 646–655. [Google Scholar] [CrossRef]
- Truini, A.; Biasiotta, A.; Cesa, S.; Stefano, D.G.; Galeotti, F.; Petrucci, M.T.; Inghilleri, M.; Cartoni, C.; Pergolini, M.; Cruccu, G. Mechanisms of pain in distal symmetric polyneuropathy: A combined clinical and neurophysiological study. Pain 2010, 150, 516–521. [Google Scholar] [CrossRef]
- Truini, A.; Biasiotta, A.; Di Stefano, G.; La Cesa, S.; Leone, C.; Cartoni, C.; Leonetti, F.; Casato, M.; Pergolini, M.; Petrucci, M.T.; et al. Peripheral nociceptor sensitization mediates allodynia in patients with distal symmetric polyneuropathy. J. Neurol. 2013, 260, 761–766. [Google Scholar] [CrossRef]
- Mondelli, M.; Aretini, A.; Baldasseroni, A. Distal symmetric polyneuropathy in diabetes. Differences between patients with and without neuropathic pain. Exp. Clin. Endocrinol. Diabetes 2012, 120, 45–50. [Google Scholar] [CrossRef]
- Tesfaye, S.; Boulton, A.J.; Dickenson, A.H. Mechanisms and management of diabetic painful distal symmetrical polyneuropathy. Diabetes Care 2013, 36, 2456–2465. [Google Scholar] [CrossRef] [Green Version]
- Callaghan, B.; Kerber, K.; Langa, K.M.; Banerjee, B.; Rodgers, A.; McCammon, R.; Burke, J.; Feldman, E. Longitudinal patient-oriented outcomes in neuropathy: Importance of early detection and falls. Neurology 2015, 85, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Callaghan, B.C.; Price, R.S.; Feldman, E.L. Distal Symmetric Polyneuropathy: A Review. JAMA 2015, 314, 2172–2181. [Google Scholar] [CrossRef] [PubMed]
- Veresiu, A.I.; Bondor, C.I.; Florea, B.; Vinik, E.J.; Vinik, A.I.; Gâvan, N.A. Detection of undisclosed neuropathy and assessment of its impact on quality of life: A survey in 25,000 Romanian patients with diabetes. J. Diabetes Complicat. 2015, 29, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, P.; Hincker, A.M.; Jensen, T.S.; Freeman, R.; Haroutounian, S. Structural, functional, and symptom relations in painful distal symmetric polyneuropathies: A systematic review. Pain 2019, 160, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Shillo, P.; Sloan, G.; Greig, M.; Hunt, L.; Selvarajah, D.; Elliott, J.; Gandhi, R.; Wilkinson, I.D.; Tesfaye, S. Painful and Painless Diabetic Neuropathies: What Is the Difference? Curr. Diab. Rep. 2019, 19, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galosi, E.; Hu, X.; Michael, N.; Nyengaard, J.R.; Truini, A.; Karlsson, P. Redefining distal symmetrical polyneuropathy features in type 1 diabetes: A systematic review. Acta Diabetol. 2021, 1–19. [Google Scholar] [CrossRef]
- Kazamel, M.; Stino, A.M.; Smith, A.G. Metabolic syndrome and peripheral neuropathy. Muscle Nerve 2021, 63, 285–293. [Google Scholar] [CrossRef]
- Wolfe, G.I.; Baker, N.S.; Amato, A.A.; Jackson, C.E.; Nations, S.P.; Saperstein, D.S.; Cha, C.H.; Katz, J.S.; Bryan, W.W.; Barohn, R.J. Chronic cryptogenic sensory polyneuropathy: Clinical and laboratory characteristics. Arch. Neurol. 1999, 56, 540–547. [Google Scholar] [CrossRef] [Green Version]
- Pasnoor, M.; Dimachkie, M.M.; Barohn, R.J. Cryptogenic sensory polyneuropathy. Neurol. Clin. 2013, 31, 463–476. [Google Scholar] [CrossRef] [Green Version]
- Emerson, N.M.; Nahman-Averbuch, H.; Peugh, J.L.; Coghill, R.C. Pain sensitivity does not differ between obese and healthy weight individuals. Pain Rep. 2021, 6, e942. [Google Scholar] [CrossRef]
- Garzillo, M.J.; Garzillo, T.A. Does obesity cause low back pain? J. Manip. Physiol. Ther. 1994, 17, 601–604. [Google Scholar]
- Janke, E.A.; Collins, A.; Kozak, A.T. Overview of the relationship between pain and obesity: What do we know? Where do we go next? J. Rehabil. Res. Dev. 2007, 44, 245–262. [Google Scholar] [CrossRef]
- Ursini, F.; Naty, S.; Grembiale, R.D. Fibromyalgia and obesity: The hidden link. Rheumatol. Int. 2011, 31, 1403–1408. [Google Scholar] [CrossRef]
- Arranz, L.I.; Rafecas, M.; Alegre, C. Effects of obesity on function and quality of life in chronic pain conditions. Curr. Rheumatol. Rep. 2014, 16, 390. [Google Scholar] [CrossRef]
- Chai, N.C.; Scher, A.I.; Moghekar, A.; Bond, D.S.; Peterlin, B.L. Obesity and headache: Part I—A systematic review of the epidemiology of obesity and headache. Headache 2014, 54, 219–234. [Google Scholar] [CrossRef]
- Smith, S.M.; Sumar, B.; Dixon, K.A. Musculoskeletal pain in overweight and obese children. Int. J. Obes. 2014, 38, 11–15. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R., Jr.; Pergolizzi, J.V.; Raffa, R.B.; Nalamachu, S.; Balestrieri, P.J. Pain and obesity in the older adult. Curr. Pharm. Des. 2014, 20, 6037–6041. [Google Scholar]
- Narouze, S.; Souzdalnitski, D. Obesity and chronic pain: Systematic review of prevalence and implications for pain practice. Reg. Anesth. Pain Med. 2015, 40, 91–111. [Google Scholar] [CrossRef]
- Okifuji, A.; Hare, B.D. The association between chronic pain and obesity. J. Pain Res. 2015, 8, 399–408. [Google Scholar] [CrossRef] [Green Version]
- Laino, D.; Vitaliti, G.; Parisi, P.; Pavone, P.; Verrotti, A.; Lubrano, R.; Matin, N.; Falsaperla, R. Headache, migraine and obesity: An overview on plausible links. J. Biol. Regul. Homeost. Agents 2016, 30, 333–338. [Google Scholar]
- Torensma, B.; Thomassen, I.; van Velzen, M.; In‘t Veld, B.A. Pain Experience and Perception in the Obese Subject Systematic Review (Revised Version). Obes. Surg. 2016, 26, 631–639. [Google Scholar] [CrossRef]
- Pavlovic, J.M.; Vieira, J.R.; Lipton, R.B.; Bond, D.S. Association Between Obesity and Migraine in Women. Curr. Pain Headache Rep. 2017, 21, 41. [Google Scholar] [CrossRef]
- Chin, S.H.; Huang, W.L.; Akter, S.; Binks, M. Obesity and pain: A systematic review. Int. J. Obes. 2020, 44, 969–979. [Google Scholar] [CrossRef]
- D’Onghia, M.; Ciaffi, J.; Lisi, L.; Mancarella, L.; Ricci, S.; Stefanelli, N.; Meliconi, R.; Ursini, F. Fibromyalgia and obesity: A comprehensive systematic review and meta-analysis. Semin. Arthritis Rheum. 2021, 51, 409–424. [Google Scholar] [CrossRef]
- Qian, M.; Shi, Y.; Yu, M. The association between obesity and chronic pain among community-dwelling older adults: A systematic review and meta-analysis. Geriatr. Nurs. 2021, 42, 8–15. [Google Scholar] [CrossRef]
- Mäntyselkä, P.; Kautiainen, H.; Vanhala, M. Prevalence of neck pain in subjects with metabolic syndrome--a cross-sectional population-based study. BMC Musculoskelet. Disord. 2010, 11, 171. [Google Scholar] [CrossRef] [Green Version]
- Ono, R.; Yamazaki, S.; Takegami, M.; Otani, K.; Sekiguchi, M.; Onishi, Y.; Hayashino, Y.; Kikuchi, S.; Konno, S.; Fukuhara, S. Gender difference in association between low back pain and metabolic syndrome: Locomotive syndrome and health outcome in Aizu cohort study (LOHAS). Spine 2012, 37, 1130–1137. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, T.; Ochiai, H.; Shirasawa, T.; Nagahama, S.; Uehara, A.; Sai, S.; Kokaze, A. Sex differences in the association of metabolic syndrome with low back pain among middle-aged Japanese adults: A large-scale cross-sectional study. Biol. Sex Differ. 2019, 10, 33. [Google Scholar] [CrossRef]
- Kalita, J.; Sonkar, K.K.; Misra, U.K.; Bhoi, S.K. Does Metabolic Syndrome Determine Severity and Disability of Chronic Low Backache? J. Neurosci. Rural. Pract. 2018, 9, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Guh, D.P.; Zhang, W.; Bansback, N.; Amarsi, Z.; Birmingham, C.L.; Anis, A.H. The incidence of co-morbidities related to obesity and overweight: A systematic review and meta-analysis. BMC Public Health 2009, 9, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, J.A.; Sabia, S.; Singh-Manoux, A.; Hamer, M.; Kivimäki, M. Healthy obesity and risk of accelerated functional decline and disability. Int J. Obes 2017, 41, 866–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IASP—International Association for the Study of Pain. Terminology. Updated August 2021. Available online: https://www.iasp-pain.org/resources/terminology (accessed on 9 August 2021).
- Scholz, J.; Finnerup, N.B.; Attal, N.; Aziz, Q.; Baron, R.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Cruccu, G.; Davis, K.D.; et al. Classification Committee of the Neuropathic Pain Special Interest Group (NeuPSIG). The IASP classification of chronic pain for ICD-11: Chronic neuropathic pain. Pain 2019, 160, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Trouvin, A.P.; Perrot, S. New concepts of pain. Best Pract. Res. Clin. Rheumatol. 2019, 33, 101415. [Google Scholar] [CrossRef]
- Freynhagen, R.; Parada, H.A.; Calderon-Ospina, C.A.; Chen, J.; Rakhmawati Emril, D.; Fernández-Villacorta, F.J.; Franco, H.; Ho, K.Y.; Lara-Solares, A.; Li, C.C.; et al. Current understanding of the mixed pain concept: A brief narrative review. Curr. Med. Res. Opin. 2019, 35, 1011–1018. [Google Scholar] [CrossRef] [Green Version]
- Nijs, J.; Lahousse, A.; Kapreli, E.; Bilika, P.; Saraçoğlu, İ.; Malfliet, A.; Coppieters, I.; De Baets, L.; Leysen, L.; Roose, E.; et al. Nociplastic Pain Criteria or Recognition of Central Sensitization? Pain Phenotyping in the Past, Present and Future. J. Clin. Med. 2021, 10, 3203. [Google Scholar] [CrossRef]
- Fitzcharles, M.A.; Cohen, S.P.; Clauw, D.J.; Littlejohn, G.; Usui, C.; Häuser, W. Nociplastic pain: Towards an understanding of prevalent pain conditions. Lancet 2021, 397, 2098–2110. [Google Scholar] [CrossRef]
- Li, L.; Hatcher, J.T.; Hoover, D.B.; Gu, H.; Wurster, R.D.; Cheng, Z.J. Distribution and morphology of calcitonin gene-related peptide and substance P immunoreactive axons in the whole-mount atria of mice. Auton. Neurosci. 2014, 181, 37–48. [Google Scholar] [CrossRef]
- Staszewska-Woolley, J.; Luk, D.E.; Nolan, P.N. Cardiovascular reflexes mediated by capsaicin sensitive cardiac afferent neurones in the dog. Cardiovasc. Res. 1986, 20, 897–906. [Google Scholar] [CrossRef]
- Baker, D.G.; Coleridge, H.M.; Coleridge, J.C.; Nerdrum, T. Search for a cardiac nociceptor: Stimulation by bradykinin of sympathetic afferent nerve endings in the heart of the cat. J. Physiol. 1980, 306, 519–536. [Google Scholar] [CrossRef]
- Marron, K.; Wharton, J.; Sheppard, M.N.; Fagan, D.; Royston, D.; Kuhn, D.M.; de Leval, M.R.; Whitehead, B.F.; Anderson, R.H.; Polak, J.M. Distribution, morphology, and neurochemistry of endocardial and epicardial nerve terminal arborizations in the human heart. Circulation 1995, 92, 2343–2351. [Google Scholar] [CrossRef]
- Corvetti, G.; Andreotti, L.; Sisto Daneo, L. Chick heart peptidergic innervation: Localization and development. Basic Appl. Histochem. 1988, 32, 485–493. [Google Scholar]
- Gordon, L.; Polak, J.M.; Moscoso, G.J.; Smith, A.; Kuhn, D.M.; Wharton, J. Development of the peptidergic innervation of human heart. J. Anat. 1993, 183, 131–140. [Google Scholar]
- Saricaoglu, Ö.C.; Teller, S.; Wang, X.; Wang, S.; Stupakov, P.; Heinrich, T.; Istvanffy, R.; Friess, H.; Ceyhan, G.O.; Demir, I.E. Localisation analysis of nerves in the mouse pancreas reveals the sites of highest nerve density and nociceptive innervation. Neurogastroenterol. Motil. 2021, 32, e13880. [Google Scholar] [CrossRef]
- Carobi, C.; Magni, F. Capsaicin-sensitive afferent vagal neurons innervating the rat liver. Neurosci. Lett. 1985, 62, 261–265. [Google Scholar] [CrossRef]
- Hockley, J.R.F.; Smith, E.S.J.; Bulmer, D.C. Human visceral nociception: Findings from translational studies in human tissue. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G464–G472. [Google Scholar] [CrossRef]
- Siri, S.; Maier, F.; Chen, L.; Santos, S.; Pierce, D.M.; Feng, B. Differential biomechanical properties of mouse distal colon and rectum innervated by the splanchnic and pelvic afferents. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G473–G481. [Google Scholar] [CrossRef]
- Kwong, K.; Carr, M.J.; Gibbard, A.; Savage, T.J.; Singh, K.; Jing, J.; Meeker, S.; Undem, B.J. Voltage-gated sodium channels in nociceptive versus non-nociceptive nodose vagal sensory neurons innervating guinea pig lungs. J. Physiol. 2008, 586, 1321–1336. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Sato, Y.; Taniguchi, K. Distribution of TRPV1- and TRPV2-immunoreactive afferent nerve endings in rat trachea. J. Anat. 2007, 211, 775–783. [Google Scholar] [CrossRef]
- Nassenstein, C.; Kwong, K.; Taylor-Clark, T.; Kollarik, M.; Macglashan, D.M.; Braun, A.; Undem, B.J. Expression and function of the ion channel TRPA1 in vagal afferent nerves innervating mouse lungs. J. Physiol. 2008, 586, 1595–1604. [Google Scholar] [CrossRef]
- Tanaka, K.; Hayakawa, T.; Maeda, S.; Kuwahara-Otani, S.; Seki, M. Distribution and ultrastructure of afferent fibers in the parietal peritoneum of the rat. Anat. Rec. 2011, 294, 1736–1742. [Google Scholar] [CrossRef]
- Bentley, G.A.; Newton, S.H.; Starr, J. Evidence for an action of morphine and the enkephalins on sensory nerve endings in the mouse peritoneum. Br. J. Pharmacol. 1981, 73, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Jankowski, M.P.; Rau, K.K.; Ekmann, K.M.; Anderson, C.E.; Koerber, H.R. Comprehensive phenotyping of group III and IV muscle afferents in mouse. J. Neurophysiol. 2013, 109, 2374–2381. [Google Scholar] [CrossRef]
- Taguchi, T.; Yasui, M.; Kubo, A.; Abe, M.; Kiyama, H.; Yamanaka, A.; Mizumura, K. Nociception originating from the crural fascia in rats. Pain 2013, 154, 1103–1114. [Google Scholar] [CrossRef]
- Alhilou, A.M.; Shimada, A.; Svensson, C.I.; Ernberg, M.; Cairns, B.E.; Christidis, N. Density of nerve fibres and expression of substance P, NR2B-receptors and nerve growth factor in healthy human masseter muscle: An immunohistochemical study. J. Oral Rehabil. 2021, 48, 35–44. [Google Scholar] [CrossRef]
- Marchettini, P.; Simone, D.A.; Caputi, G.; Ochoa, J.L. Pain from excitation of identified muscle nociceptors in humans. Brain Res. 1996, 740, 109–116. [Google Scholar] [CrossRef]
- Dauch, J.R.; Lindblad, C.N.; Hayes, J.M.; Lentz, S.I.; Cheng, H.T. Three-dimensional imaging of nociceptive intraepidermal nerve fibers in human skin biopsies. J. Vis. Exp. 2013, 74, e50331. [Google Scholar] [CrossRef] [Green Version]
- Mouraux, A.; Ragé, M.; Bragard, D.; Plaghki, L. Estimation of intraepidermal fiber density by the detection rate of nociceptive laser stimuli in normal and pathological conditions. Neurophysiol. Clin. 2012, 42, 281–291. [Google Scholar] [CrossRef]
- Lauria, G.; Morbin, M.; Lombardi, R.; Capobianco, R.; Camozzi, F.; Pareyson, D.; Manconi, M.; Geppetti, P. Expression of capsaicin receptor immunoreactivity in human peripheral nervous system and in painful neuropathies. J. Peripher. Nerv. Syst. 2006, 11, 262–271. [Google Scholar] [CrossRef]
- Simone, D.A.; Nolano, M.; Johnson, T.; Wendelschafer-Crabb, G.; Kennedy, W.R. Intradermal injection of capsaicin in humans produces degeneration and subsequent reinnervation of epidermal nerve fibers: Correlation with sensory function. J. Neurosci. 1998, 18, 8947–8959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, T. Intraepidermal free nerve fiber endings in the hairless skin of the rat as revealed by the zinc iodide-osmium tetroxide technique. Histol. Histopathol. 2000, 15, 493–498. [Google Scholar] [PubMed]
- Kruger, L.; Sampogna, S.L.; Rodin, B.E.; Clague, J.; Brecha, N.; Yeh, Y. Thin-fiber cutaneous innervation and its intraepidermal contribution studied by labeling methods and neurotoxin treatment in rats. Somat. Res. 1985, 2, 335–356. [Google Scholar] [CrossRef] [PubMed]
- Navarro, X.; Verdú, E.; Wendelschafer-Crabb, G.; Kennedy, W.R. Immunohistochemical study of skin reinnervation by regenerative axons. J. Comp. Neurol. 1997, 380, 164–174. [Google Scholar] [CrossRef]
- Navarro, X.; Verdú, E.; Wendelscafer-Crabb, G.; Kennedy, W.R. Innervation of cutaneous structures in the mouse hind paw: A confocal microscopy immunohistochemical study. J. Neurosci. Res. 1995, 41, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Vijay, J.; Gauthier, M.F.; Biswell, R.L.; Louiselle, D.A.; Johnston, J.J.; Cheung, W.A.; Belden, B.; Pramatarova, A.; Biertho, L.; Gibson, M.; et al. Single-cell analysis of human adipose tissue identifies depot and disease specific cell types. Nat. Metab. 2021, 2, 97–109. [Google Scholar] [CrossRef]
- Esteve Ràfols, M. Adipose tissue: Cell heterogeneity and functional diversity. Endocrinol. Nutr. 2014, 61, 100–112. [Google Scholar] [CrossRef]
- Iannitti, T.; Graham, A.; Dolan, S. Adiponectin-Mediated Analgesia and Anti-Inflammatory Effects in Rat. PLoS ONE 2015, 10, e0136819. [Google Scholar]
- Luo, Y.; Liu, M. Adiponectin: A versatile player of innate immunity. J. Mol. Cell Biol. 2016, 8, 120–128. [Google Scholar] [CrossRef]
- Żelechowska, P.; Brzezińska-Błaszczyk, E.; Wiktorska, M.; Różalska, S.; Wawrocki, S.; Kozłowska, E.; Agier, J. Adipocytokines leptin and adiponectin function as mast cell activity modulators. Immunology 2019, 158, 3–18. [Google Scholar] [CrossRef]
- Ma, W.; Chabot, J.G.; Quirion, R. A role for adrenomedullin as a pain-related peptide in the rat. Proc. Natl. Acad. Sci. USA 2006, 103, 16027–16032. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, A.J.; Copits, B.A.; Mickle, A.D.; Karlsson, P.; Kadunganattil, S.; Haroutounian, S.; Tadinada, S.M.; de Kloet, A.D.; Valtcheva, M.V.; McIlvried, L.A.; et al. Angiotensin II Triggers Peripheral Macrophage-to-Sensory Neuron Redox Crosstalk to Elicit Pain. J. Neurosci. 2018, 38, 7032–7057. [Google Scholar] [CrossRef] [Green Version]
- Canpolat, S.; Ozcan, M.; Saral, S.; Kalkan, O.F.; Ayar, A. Effects of apelin-13 in mice model of experimental pain and peripheral nociceptive signaling in rat sensory neurons. J. Recept. Signal Transduct. Res. 2016, 36, 243–247. [Google Scholar] [CrossRef]
- Lv, S.; Zhang, X.; Zhou, Y.; Feng, Y.; Yang, Y.; Wang, X. Intrathecally Administered Apelin-13 Alleviated Complete Freund’s Adjuvant-Induced Inflammatory Pain in Mice. Front. Pharmacol. 2021, 11, 1335. [Google Scholar] [CrossRef]
- Lv, S.; Zhang, X.; Feng, Y.; Zhou, Y.; Cui, B.; Yang, Y.; Wang, X. Intravenous Administration of Pyroglutamyl Apelin-13 Alleviates Murine Inflammatory Pain via the Kappa Opioid Receptor. Front. Neurosci. 2021, 14, 929. [Google Scholar] [CrossRef]
- Xiong, Q.; He, W.; Wang, H.; Zhou, J.; Zhang, Y.; He, J.; Yang, C.; Zhang, B. Effect of the spinal apelin-APJ system on the pathogenesis of chronic constriction injury-induced neuropathic pain in rats. Mol. Med. Rep. 2017, 16, 1223–1231. [Google Scholar] [CrossRef] [Green Version]
- Doyle, J.R.; Krishnaji, S.T.; Zhu, G.; Xu, Z.Z.; Heller, D.; Ji, R.R.; Levy, B.D.; Kumar, K.; Kopin, A.S. Development of a membrane-anchored chemerin receptor agonist as a novel modulator of allergic airway inflammation and neuropathic pain. J. Biol. Chem. 2014, 289, 13385–13396. [Google Scholar] [CrossRef] [Green Version]
- Dickie, A.C.; Torsney, C. The chemerin receptor 23 agonist, chemerin, attenuates monosynaptic C-fibre input to lamina I neurokinin 1 receptor expressing rat spinal cord neurons in inflammatory pain. Mol. Pain 2014, 10, 24. [Google Scholar] [CrossRef] [Green Version]
- Oehler, B.; Mohammadi, M.; Perpina Viciano, C.; Hackel, D.; Hoffmann, C.; Brack, A.; Rittner, H.L. Peripheral Interaction of Resolvin D1 and E1 with Opioid Receptor Antagonists for Antinociception in Inflammatory Pain in Rats. Front. Mol. Neurosci. 2017, 10, 242. [Google Scholar] [CrossRef]
- Hu, Z.J.; Han, W.; Cao, C.Q.; Mao-Ying, Q.L.; Mi, W.L.; Wang, Y.Q. Peripheral Leptin Signaling Mediates Formalin-Induced Nociception. Neurosci. Bull. 2018, 34, 321–329. [Google Scholar] [CrossRef]
- Maeda, T.; Kiguchi, N.; Kobayashi, Y.; Ikuta, T.; Ozaki, M.; Kishioka, S. Leptin derived from adipocytes in injured peripheral nerves facilitates development of neuropathic pain via macrophage stimulation. Proc. Natl. Acad. Sci. USA 2009, 106, 13076–13081. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Kang, L.; Li, G.; Zeng, H.; Zhang, L.; Ling, X.; Dong, H.; Liang, S.; Chen, H. Intrathecal leptin inhibits expression of the P2 × 2/3 receptors and alleviates neuropathic pain induced by chronic constriction sciatic nerve injury. Mol. Pain 2013, 9, 65. [Google Scholar] [CrossRef] [Green Version]
- Harmon, J.B.; Sanders, A.E.; Wilder, R.S.; Essick, G.K.; Slade, G.D.; Hartung, J.E.; Nackley, A.G. Circulating Omentin-1 and Chronic Painful Temporomandibular Disorders. J. Oral Facial Pain Headache 2016, 30, 203–209. [Google Scholar] [CrossRef]
- Slovacek, H.; Khanna, R.; Poredos, P.; Jezovnik, M.; Hoppensteadt, D.; Fareed, J.; Hopkinson, W. Interrelationship of Osteopontin, MMP-9 and ADAMTS4 in Patients With Osteoarthritis Undergoing Total Joint Arthroplasty. Clin. Appl. Thromb. Hemost. 2021, 26, 1076029620964864. [Google Scholar] [CrossRef]
- Yamaga, M.; Tsuji, K.; Miyatake, K.; Yamada, J.; Abula, K.; Ju, Y.J.; Sekiya, I.; Muneta, T. Osteopontin level in synovial fluid is associated with the severity of joint pain and cartilage degradation after anterior cruciate ligament rupture. PLoS ONE 2012, 7, e49014. [Google Scholar] [CrossRef] [Green Version]
- Marsh, B.C.; Kerr, N.C.; Isles, N.; Denhardt, D.T.; Wynick, D. Osteopontin expression and function within the dorsal root ganglion. Neuroreport 2007, 18, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Sagar, D.R.; Ashraf, S.; Xu, L.; Burston, J.J.; Menhinick, M.R.; Poulter, C.L.; Bennett, A.J.; Walsh, D.A.; Chapman, V. Osteoprotegerin reduces the development of pain behaviour and joint pathology in a model of osteoarthritis. Ann. Rheum. Dis. 2014, 73, 1558–1565. [Google Scholar] [CrossRef] [Green Version]
- Roudier, M.P.; Bain, S.D.; Dougall, W.C. Effects of the RANKL inhibitor, osteoprotegerin, on the pain and histopathology of bone cancer in rats. Clin. Exp. Metastasis. 2006, 23, 167–175. [Google Scholar] [CrossRef]
- Luger, N.M.; Honore, P.; Sabino, M.A.; Schwei, M.J.; Rogers, S.D.; Mach, D.B.; Clohisy, D.R.; Mantyh, P.W. Osteoprotegerin diminishes advanced bone cancer pain. Cancer Res. 2001, 61, 4038–4047. [Google Scholar]
- Fioravanti, A.; Giannitti, C.; Cheleschi, S.; Simpatico, A.; Pascarelli, N.A.; Galeazzi, M. Circulating levels of adiponectin, resistin, and visfatin after mud-bath therapy in patients with bilateral knee osteoarthritis. Int. J. Biometeorol. 2015, 59, 1691–1700. [Google Scholar] [CrossRef]
- Bas, S.; Finckh, A.; Puskas, G.J.; Suva, D.; Hoffmeyer, P.; Gabay, C.; Lübbeke, A. Adipokines correlate with pain in lower limb osteoarthritis: Different associations in hip and knee. Int. Orthop. 2014, 38, 2577–2583. [Google Scholar] [CrossRef] [PubMed]
- Tarabeih, N.; Kalinkovich, A.; Shalata, A.; Livshits, G. Circulating Levels of Visceral Adipose Tissue-Derived Serine Protease Inhibitor (Vaspin) Appear as a Marker of Musculoskeletal Pain Disability. Diagnostics 2021, 10, 797. [Google Scholar] [CrossRef] [PubMed]
- Askari, A.; Arasteh, P.; Homayounfar, R.; Naghizadeh, M.M.; Ehrampoush, E.; Mousavi, S.M.; Alipoor, R. The role of adipose tissue secretion in the creation and pain level in osteoarthritis. Endocr. Regul. 2021, 54, 6–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorkin, L.S.; Xiao, W.H.; Wagner, R.; Myers, R.R. Tumour necrosis factor-alpha induces ectopic activity in nociceptive primary afferent fibres. Neuroscience 1997, 81, 255–262. [Google Scholar] [CrossRef]
- Hakim, A.W.; Dong, X.D.; Svensson, P.; Kumar, U.; Cairns, B.E. TNFalpha mechanically sensitizes masseter muscle afferent fibers of male rats. J. Neurophysiol. 2009, 102, 1551–1559. [Google Scholar] [CrossRef]
- Brenn, D.; Richter, F.; Schaible, H.G. Sensitization of unmyelinated sensory fibers of the joint nerve to mechanical stimuli by interleukin-6 in the rat: An inflammatory mechanism of joint pain. Arthritis Rheum. 2007, 56, 351–359. [Google Scholar] [CrossRef]
- Fu, L.W.; Longhurst, J.C. Interleukin-1beta sensitizes abdominal visceral afferents of cats to ischaemia and histamine. J. Physiol. 1999, 521, 249–260. [Google Scholar] [CrossRef]
- Ek, M.; Kurosawa, M.; Lundeberg, T.; Ericsson, A. Activation of vagal afferents after intravenous injection of interleukin-1beta: Role of endogenous prostaglandins. J. Neurosci. 1998, 18, 9471–9479. [Google Scholar] [CrossRef]
- Fukuoka, H.; Kawatani, M.; Hisamitsu, T.; Takeshige, C. Cutaneous hyperalgesia induced by peripheral injection of interleukin-1 beta in the rat. Brain Res. 1994, 657, 133–140. [Google Scholar] [CrossRef]
- Flatters, S.J.; Fox, A.J.; Dickenson, A.H. Nerve injury alters the effects of interleukin-6 on nociceptive transmission in peripheral afferents. Eur. J. Pharmacol. 2004, 484, 183–191. [Google Scholar] [CrossRef]
- Day, Y.J.; Liou, J.T.; Lee, C.M.; Lin, Y.C.; Mao, C.C.; Chou, A.H.; Liao, C.C.; Lee, H.C. Lack of interleukin-17 leads to a modulated micro-environment and amelioration of mechanical hypersensitivity after peripheral nerve injury in mice. Pain 2014, 155, 1293–1302. [Google Scholar] [CrossRef]
- Liou, J.T.; Mao, C.C.; Ching-Wah Sum, D.; Liu, F.C.; Lai, Y.S.; Li, J.C.; Day, Y.J. Peritoneal administration of Met-RANTES attenuates inflammatory and nociceptive responses in a murine neuropathic pain model. J. Pain 2013, 14, 24–35. [Google Scholar] [CrossRef]
- McNamee, K.E.; Alzabin, S.; Hughes, J.P.; Anand, P.; Feldmann, M.; Williams, R.O.; Inglis, J.J. IL-17 induces hyperalgesia via TNF-dependent neutrophil infiltration. Pain 2011, 152, 1838–1845. [Google Scholar] [CrossRef]
- Fattori, V.; Hohmann, M.S.N.; Rossaneis, A.C.; Manchope, M.F.; Alves-Filho, J.C.; Cunha, T.M.; Cunha, F.Q.; Verri, W.A. Targeting IL-33/ST2 signaling: Regulation of immune function and analgesia. Expert Opin. Ther. Targets 2017, 21, 1141–1152. [Google Scholar] [CrossRef]
- Richards, N.; Batty, T.; Dilley, A. CCL2 has similar excitatory effects to TNF-alpha in a subgroup of inflamed C-fiber axons. J. Neurophysiol. 2011, 106, 2838–2848. [Google Scholar] [CrossRef]
- Kao, D.J.; Li, A.H.; Chen, J.C.; Luo, R.S.; Chen, Y.L.; Lu, J.C.; Wang, H.L. CC chemokine ligand 2 upregulates the current density and expression of TRPV1 channels and Nav1.8 sodium channels in dorsal root ganglion neurons. J. Neuroinflammation 2012, 9, 189. [Google Scholar] [CrossRef] [Green Version]
- Bhangoo, S.; Ren, D.; Miller, R.J.; Henry, K.J.; Lineswala, J.; Hamdouchi, C.; Li, B.; Monahan, P.E.; Chan, D.M.; Ripsch, M.S.; et al. Delayed functional expression of neuronal chemokine receptors following focal nerve demyelination in the rat: A mechanism for the development of chronic sensitization of peripheral nociceptors. Mol. Pain 2007, 3, 38. [Google Scholar] [CrossRef] [Green Version]
- Liou, J.T.; Yuan, H.B.; Mao, C.C.; Lai, Y.S.; Day, Y.J. Absence of C-C motif chemokine ligand 5 in mice leads to decreased local macrophage recruitment and behavioral hypersensitivity in a murine neuropathic pain model. Pain 2012, 153, 1283–1291. [Google Scholar] [CrossRef]
- Silva, R.L.; Lopes, A.H.; Guimarães, R.M.; Cunha, T.M. CXCL1/CXCR2 signaling in pathological pain: Role in peripheral and central sensitization. Neurobiol. Dis. 2017, 105, 109–116. [Google Scholar] [CrossRef]
- Sun, Y.; Sahbaie, P.; Liang, D.; Li, W.; Clark, J.D. Opioids enhance CXCL1 expression and function after incision in mice. J. Pain 2014, 15, 856–866. [Google Scholar] [CrossRef] [Green Version]
- Souza, G.R.; Talbot, J.; Lotufo, C.M.; Cunha, F.Q.; Cunha, T.M.; Ferreira, S.H. Fractalkine mediates inflammatory pain through activation of satellite glial cells. Proc. Natl. Acad. Sci. USA 2013, 110, 11193–11198. [Google Scholar] [CrossRef] [Green Version]
- Andres, C.; Hasenauer, J.; Ahn, H.S.; Joseph, E.K.; Isensee, J.; Theis, F.J.; Allgöwer, F.; Levine, J.D.; Dib-Hajj, S.D.; Waxman, S.G.; et al. Wound-healing growth factor, basic FGF, induces Erk1/2-dependent mechanical hyperalgesia. Pain 2013, 154, 2216–2226. [Google Scholar] [CrossRef]
- Qiu, C.Y.; Liu, T.T.; Wei, S.; Zhou, Y.M.; Wu, L.; Jin, Y.; Hu, W.P. TGF-beta1 enhances the activity of acid-sensing ion channel in rat primary sensory neurons. J. Neurosci. Res. 2019, 97, 1298–1305. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, X.M.; Duan, K.Z.; Gu, X.Y.; Han, M.; Liu, B.L.; Zhao, Z.Q.; Zhang, Y.Q. Peripheral TGF-beta1 signaling is a critical event in bone cancer-induced hyperalgesia in rodents. J. Neurosci. 2013, 33, 19099–19111. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhu, Y.; Noë, M.; Li, Q.; Pasricha, P.J. Neuronal Transforming Growth Factor beta Signaling via SMAD3 Contributes to Pain in Animal Models of Chronic Pancreatitis. Gastroenterology 2018, 154, 2252–2265.e2. [Google Scholar] [CrossRef] [Green Version]
- Kiguchi, N.; Kobayashi, Y.; Kadowaki, Y.; Fukazawa, Y.; Saika, F.; Kishioka, S. Vascular endothelial growth factor signaling in injured nerves underlies peripheral sensitization in neuropathic pain. J. Neurochem. 2014, 129, 169–178. [Google Scholar] [CrossRef]
- Hulse, R.P.; Beazley-Long, N.; Hua, J.; Kennedy, H.; Prager, J.; Bevan, H.; Qiu, Y.; Fernandes, E.S.; Gammons, M.V.; Ballmer-Hofer, K.; et al. Regulation of alternative VEGF-A mRNA splicing is a therapeutic target for analgesia. Neurobiol. Dis. 2014, 71, 245–259. [Google Scholar] [CrossRef] [Green Version]
- Salvemini, D.; Little, J.W.; Doyle, T.; Neumann, W.L. Roles of reactive oxygen and nitrogen species in pain. Free Radic. Biol. Med. 2011, 51, 951–966. [Google Scholar] [CrossRef] [Green Version]
- Kallenborn-Gerhardt, W.; Schröder, K.; Del Turco, D.; Lu, R.; Kynast, K.; Kosowski, J.; Niederberger, E.; Shah, A.M.; Brandes, R.P.; Geisslinger, G.; et al. NADPH oxidase-4 maintains neuropathic pain after peripheral nerve injury. J. Neurosci. 2012, 32, 10136–10145. [Google Scholar] [CrossRef]
- Holthusen, H.; Arndt, J.O. Nitric oxide evokes pain in humans on intracutaneous injection. Neurosci. Lett. 1994, 165, 71–74. [Google Scholar] [CrossRef]
- Kawabata, A.; Ishiki, T.; Nagasawa, K.; Yoshida, S.; Maeda, Y.; Takahashi, T.; Sekiguchi, F.; Wada, T.; Ichida, S.; Nishikawa, H. Hydrogen sulfide as a novel nociceptive messenger. Pain 2007, 132, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Aoki, Y.; Sekiguchi, F.; Matsunami, M.; Takahashi, T.; Nishikawa, H.; Kawabata, A. Hyperalgesia induced by spinal and peripheral hydrogen sulfide: Evidence for involvement of Cav3.2 T-type calcium channels. Pain 2009, 142, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Jakicic, J.M.; Clark, K.; Coleman, E.; Donnelly, J.E.; Foreyt, J.; Melanson, E.; Volek, J.; Volpe, S.L. American College of Sports Medicine. American College of Sports Medicine position stand. Appropriate intervention strategies for weight loss and prevention of weight regain for adults. Med. Sci. Sports Exerc. 2001, 33, 2145–2156. [Google Scholar] [CrossRef] [PubMed]
- American College of Sports Medicine. ACSM’s Guidelines for Exercise Testing and Prescription, 10th ed.; Wolters Kluwer Health: Philadelphia, PA, USA, 2018. [Google Scholar]
- Yumuk, V.; Tsigos, C.; Fried, M.; Schindler, K.; Busetto, L.; Micic, D.; Toplak, H.; Obesity Management Task Force of the European Association for the Study of Obesity. European Guidelines for Obesity Management in Adults. Obes. Facts 2015, 8, 402–424. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, G.; Blake, C.; Cunningham, C.; Lennon, O.; Perrotta, C. What exercise prescription is optimal to improve body composition and cardiorespiratory fitness in adults living with obesity? A network meta-analysis. Obes. Rev. 2021, 22, e13137. [Google Scholar] [CrossRef]
- Sultana, R.N.; Sabag, A.; Keating, S.E.; Johnson, N.A. The Effect of Low-Volume High-Intensity Interval Training on Body Composition and Cardiorespiratory Fitness: A Systematic Review and Meta-Analysis. Sports Med. 2019, 49, 1687–1721. [Google Scholar] [CrossRef]
- Sabag, A.; Little, J.P.; Johnson, N.A. Low-volume high-intensity interval training for cardiometabolic health. J. Physiol. 2021, 1–14. [Google Scholar] [CrossRef]
- Ho, S.S.; Dhaliwal, S.S.; Hills, A.P.; Pal, S. The effect of 12 weeks of aerobic, resistance or combination exercise training on cardiovascular risk factors in the overweight and obese in a randomized trial. BMC Public Health. 2012, 12, 704. [Google Scholar] [CrossRef] [Green Version]
- Stinkens, R.; Brouwers, B.; Jocken, J.W.; Blaak, E.E.; Teunissen-Beekman, K.F.; Hesselink, M.K.; van Baak, M.A.; Schrauwen, P.; Goossens, G.H. Exercise training-induced effects on the abdominal subcutaneous adipose tissue phenotype in humans with obesity. J. Appl. Physiol. 2018, 125, 1585–1593. [Google Scholar] [CrossRef] [Green Version]
- Donges, C.E.; Duffield, R.; Drinkwater, E.J. Effects of resistance or aerobic exercise training on interleukin-6, C-reactive protein, and body composition. Med. Sci. Sports Exerc. 2010, 42, 304–313. [Google Scholar] [CrossRef]
- Amano, Y.; Nonaka, Y.; Takeda, R.; Kano, Y.; Hoshino, D. Effects of electrical stimulation-induced resistance exercise training on white and brown adipose tissues and plasma meteorin-like concentration in rats. Physiol. Rep. 2021, 8, e14540. [Google Scholar] [CrossRef]
- Walton, R.G.; Finlin, B.S.; Mula, J.; Long, D.E.; Zhu, B.; Fry, C.S.; Westgate, P.M.; Lee, J.D.; Bennett, T.; Kern, P.A.; et al. Insulin-resistant subjects have normal angiogenic response to aerobic exercise training in skeletal muscle, but not in adipose tissue. Physiol. Rep. 2015, 3, e12415. [Google Scholar] [CrossRef] [Green Version]
- Van Pelt, D.W.; Guth, L.M.; Horowitz, J.F. Aerobic exercise elevates markers of angiogenesis and macrophage IL-6 gene expression in the subcutaneous adipose tissue of overweight-to-obese adults. J. Appl. Physiol. 2017, 123, 1150–1159. [Google Scholar] [CrossRef] [Green Version]
- De Glisezinski, I.; Crampes, F.; Harant, I.; Berlan, M.; Hejnova, J.; Langin, D.; Rivière, D.; Stich, V. Endurance training changes in lipolytic responsiveness of obese adipose tissue. Am. J. Physiol. 1998, 275, E951–E956. [Google Scholar] [CrossRef]
- Lakhdar, N.; Denguezli, M.; Zaouali, M.; Zbidi, A.; Tabka, Z.; Bouassida, A. Six months training alone or combined with diet alters HOMA-AD, HOMA-IR and plasma and adipose tissue adiponectin in obese women. Neuro. Endocrinol. Lett. 2014, 35, 373–379. [Google Scholar]
- Oh, S.; So, R.; Shida, T.; Matsuo, T.; Kim, B.; Akiyama, K.; Isobe, T.; Okamoto, Y.; Tanaka, K.; Shoda, J. High-Intensity Aerobic Exercise Improves Both Hepatic Fat Content and Stiffness in Sedentary Obese Men with Nonalcoholic Fatty Liver Disease. Sci. Rep. 2017, 7, 43029. [Google Scholar] [CrossRef] [Green Version]
- Langleite, T.M.; Jensen, J.; Norheim, F.; Gulseth, H.L.; Tangen, D.S.; Kolnes, K.J.; Heck, A.; Storås, T.; Grøthe, G.; Dahl, M.A.; et al. Insulin sensitivity, body composition and adipose depots following 12 w combined endurance and strength training in dysglycemic and normoglycemic sedentary men. Arch. Physiol. Biochem. 2016, 122, 167–179. [Google Scholar] [CrossRef]
- Jamka, M.; Mądry, E.; Krzyżanowska-Jankowska, P.; Skrypnik, D.; Szulińska, M.; Mądry, R.; Lisowska, A.; Batyrova, G.; Duś-Żuchowska, M.; Gotz-Więckowska, A.; et al. The effect of endurance and endurance-strength training on body composition and cardiometabolic markers in abdominally obese women: A randomised trial. Sci. Rep. 2021, 11, 12339. [Google Scholar] [CrossRef]
- Costa, J.S.R.; Fonseca, G.F.A.C.; Ottone, N.C.D.S.; Silva, P.A.; Antonaccio, R.F.; Silva, G.; Rocha, M.D.S.A.; Coimbra, C.C.; Esteves, E.A.; Mang, Z.A.; et al. Strength training improves insulin resistance and differently affects mitochondria in skeletal muscle and visceral adipose tissue in high-fat fed mice. Life Sci. 2021, 278, 119639. [Google Scholar] [CrossRef]
- Lee, S.; Norheim, F.; Gulseth, H.L.; Langleite, T.M.; Kolnes, K.J.; Tangen, D.S.; Stadheim, H.K.; Gilfillan, G.D.; Holen, T.; Birkeland, K.I.; et al. Interaction between plasma fetuin-A and free fatty acids predicts changes in insulin sensitivity in response to long-term exercise. Physiol. Rep. 2017, 5, e13183. [Google Scholar] [CrossRef]
- Donges, C.E.; Duffield, R.; Guelfi, K.J.; Smith, G.C.; Adams, D.R.; Edge, J.A. Comparative effects of single-mode vs. duration-matched concurrent exercise training on body composition, low-grade inflammation, and glucose regulation in sedentary, overweight, middle-aged men. Appl. Physiol. Nutr. Metab. 2013, 38, 779–788. [Google Scholar] [CrossRef]
- Mauriège, P.; Prud’Homme, D.; Marcotte, M.; Yoshioka, M.; Tremblay, A.; Després, J.P. Regional differences in adipose tissue metabolism between sedentary and endurance-trained women. Am. J. Physiol. 1997, 273, E497–E506. [Google Scholar] [CrossRef]
- Lamarche, B.; Després, J.P.; Moorjani, S.; Nadeau, A.; Lupien, P.J.; Tremblay, A.; Thériault, G.; Bouchard, C. Evidence for a role of insulin in the regulation of abdominal adipose tissue lipoprotein lipase response to exercise training in obese women. Int. J. Obes. Relat. Metab. Disord. 1993, 17, 255–261. [Google Scholar]
- Nordby, P.; Auerbach, P.L.; Rosenkilde, M.; Kristiansen, L.; Thomasen, J.R.; Rygaard, L.; Groth, R.; Brandt, N.; Helge, J.W.; Richter, E.A.; et al. Endurance training per se increases metabolic health in young, moderately overweight men. Obesity 2012, 20, 2202–2212. [Google Scholar] [CrossRef] [PubMed]
- Després, J.P.; Bouchard, C.; Savard, R.; Tremblay, A.; Marcotte, M.; Thériault, G. The effect of a 20-week endurance training program on adipose-tissue morphology and lipolysis in men and women. Metabolism 1984, 33, 235–239. [Google Scholar] [CrossRef]
- Moghadasi, M.; Mohebbi, H.; Rahmani-Nia, F.; Hassan-Nia, S.; Noroozi, H.; Pirooznia, N. High-intensity endurance training improves adiponectin mRNA and plasma concentrations. Eur. J. Appl. Physiol. 2012, 112, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.; Eijnde, B.O.; Roelants, M.; Broekmans, T.; Rummens, J.L.; Hensen, K.; Daniels, A.; Van Erum, M.; Bonné, K.; Reyckers, I.; et al. Clinical benefits of the addition of lower extremity low-intensity resistance muscle training to early aerobic endurance training intervention in patients with coronary artery disease: A randomized controlled trial. J. Rehabil. Med. 2011, 43, 800–807. [Google Scholar] [CrossRef] [Green Version]
- Halverstadt, A.; Phares, D.A.; Wilund, K.R.; Goldberg, A.P.; Hagberg, J.M. Endurance exercise training raises high-density lipoprotein cholesterol and lowers small low-density lipoprotein and very low-density lipoprotein independent of body fat phenotypes in older men and women. Metabolism 2007, 56, 444–450. [Google Scholar] [CrossRef]
- Couillard, C.; Després, J.P.; Lamarche, B.; Bergeron, J.; Gagnon, J.; Leon, A.S.; Rao, D.C.; Skinner, J.S.; Wilmore, J.H.; Bouchard, C. Effects of endurance exercise training on plasma HDL cholesterol levels depend on levels of triglycerides: Evidence from men of the Health, Risk Factors, Exercise Training and Genetics (HERITAGE) Family Study. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1226–1232. [Google Scholar] [CrossRef] [Green Version]
- Bladbjerg, E.M.; Skov, J.; Nordby, P.; Stallknecht, B. Endurance exercise per se reduces the cardiovascular risk marker t-PA antigen in healthy, younger, overweight men. Thromb. Res. 2017, 152, 69–73. [Google Scholar] [CrossRef]
- Tjønna, A.E.; Leinan, I.M.; Bartnes, A.T.; Jenssen, B.M.; Gibala, M.J.; Winett, R.A.; Wisløff, U. Low- and high-volume of intensive endurance training significantly improves maximal oxygen uptake after 10-weeks of training in healthy men. PLoS ONE 2013, 8, e65382. [Google Scholar]
- Polak, J.; Moro, C.; Klimcakova, E.; Hejnova, J.; Majercik, M.; Viguerie, N.; Langin, D.; Lafontan, M.; Stich, V.; Berlan, M. Dynamic strength training improves insulin sensitivity and functional balance between adrenergic alpha 2A and beta pathways in subcutaneous adipose tissue of obese subjects. Diabetologia 2005, 48, 2631–2640. [Google Scholar] [CrossRef]
- Klimcakova, E.; Polak, J.; Moro, C.; Hejnova, J.; Majercik, M.; Viguerie, N.; Berlan, M.; Langin, D.; Stich, V. Dynamic strength training improves insulin sensitivity without altering plasma levels and gene expression of adipokines in subcutaneous adipose tissue in obese men. J. Clin. Endocrinol. Metab. 2006, 91, 5107–5112. [Google Scholar] [CrossRef] [Green Version]
- Avila, J.J.; Gutierres, J.A.; Sheehy, M.E.; Lofgren, I.E.; Delmonico, M.J. Effect of moderate intensity resistance training during weight loss on body composition and physical performance in overweight older adults. Eur. J. Appl. Physiol. 2010, 109, 517–525. [Google Scholar] [CrossRef]
- Cottell, K.E.; Dorfman, L.R.; Straight, C.R.; Delmonico, M.J.; Lofgren, I.E. The effects of diet education plus light resistance training on coronary heart disease risk factors in community-dwelling older adults. J. Nutr. Health Aging 2011, 15, 762–767. [Google Scholar] [CrossRef]
- Falk, B.; Sadres, E.; Constantini, N.; Zigel, L.; Lidor, R.; Eliakim, A. The association between adiposity and the response to resistance training among pre- and early-pubertal boys. J. Pediatr. Endocrinol. Metab. 2002, 15, 597–606. [Google Scholar] [CrossRef]
- Kordi, R.; Dehghani, S.; Noormohammadpour, P.; Rostami, M.; Mansournia, M.A. Effect of abdominal resistance exercise on abdominal subcutaneous fat of obese women: A randomized controlled trial using ultrasound imaging assessments. J. Manip. Physiol. Ther. 2015, 38, 203–209. [Google Scholar] [CrossRef]
- Larsen, S.; Danielsen, J.H.; Søndergård, S.D.; Søgaard, D.; Vigelsoe, A.; Dybboe, R.; Skaaby, S.; Dela, F.; Helge, J.W. The effect of high-intensity training on mitochondrial fat oxidation in skeletal muscle and subcutaneous adipose tissue. Scand. J. Med. Sci. Sports 2015, 25, e59–e69. [Google Scholar] [CrossRef]
- Dohlmann, T.L.; Hindsø, M.; Dela, F.; Helge, J.W.; Larsen, S. High-intensity interval training changes mitochondrial respiratory capacity differently in adipose tissue and skeletal muscle. Physiol. Rep. 2018, 6, e13857. [Google Scholar] [CrossRef]
- Devries, M.C.; Samjoo, I.A.; Hamadeh, M.J.; McCready, C.; Raha, S.; Watt, M.J.; Steinberg, G.R.; Tarnopolsky, M.A. Endurance training modulates intramyocellular lipid compartmentalization and morphology in skeletal muscle of lean and obese women. J. Clin. Endocrinol. Metab. 2013, 98, 4852–4862. [Google Scholar] [CrossRef] [Green Version]
- Booth, F.W.; Ruegsegger, G.N.; Toedebusch, R.G.; Yan, Z. Endurance Exercise and the Regulation of Skeletal Muscle Metabolism. Prog. Mol. Biol. Transl. Sci. 2015, 135, 129–151. [Google Scholar]
- Seip, R.L.; Semenkovich, C.F. Skeletal muscle lipoprotein lipase: Molecular regulation and physiological effects in relation to exercise. Exerc. Sport Sci. Rev. 1998, 26, 191–218. [Google Scholar] [CrossRef]
- Yoshida, T.; Delafontaine, P. Mechanisms of IGF-1-Mediated Regulation of Skeletal Muscle Hypertrophy and Atrophy. Cells 2021, 9, 1970. [Google Scholar] [CrossRef]
- Sullivan, B.P.; Weiss, J.A.; Nie, Y.; Garner, R.T.; Drohan, C.J.; Kuang, S.; Stout, J.; Gavin, T.P. Skeletal muscle IGF-1 is lower at rest and after resistance exercise in humans with obesity. Eur. J. Appl. Physiol. 2021, 120, 2835–2846. [Google Scholar] [CrossRef]
- Kras, K.A.; Hoffman, N.; Roust, L.R.; Benjamin, T.R.; De Filippis, E.A.; Katsanos, C.S. Adenosine Triphosphate Production of Muscle Mitochondria after Acute Exercise in Lean and Obese Humans. Med. Sci. Sports Exerc. 2019, 51, 445–453. [Google Scholar] [CrossRef]
- Berggren, J.R.; Boyle, K.E.; Chapman, W.H.; Houmard, J.A. Skeletal muscle lipid oxidation and obesity: Influence of weight loss and exercise. Am. J. Physiol. Endocrinol. Metab. 2008, 294, E726–E732. [Google Scholar] [CrossRef]
- Louche, K.; Badin, P.M.; Montastier, E.; Laurens, C.; Bourlier, V.; de Glisezinski, I.; Thalamas, C.; Viguerie, N.; Langin, D.; Moro, C. Endurance exercise training up-regulates lipolytic proteins and reduces triglyceride content in skeletal muscle of obese subjects. J. Clin. Endocrinol. Metab. 2013, 98, 4863–4871. [Google Scholar] [CrossRef] [Green Version]
- Hansen, D.; Meeusen, R.; Mullens, A.; Dendale, P. Effect of acute endurance and resistance exercise on endocrine hormones directly related to lipolysis and skeletal muscle protein synthesis in adult individuals with obesity. Sports Med. 2012, 42, 415–431. [Google Scholar] [CrossRef]
- Leon, B.; Jenkins, S.; Pepin, K.; Chaudhry, H.; Smith, K.; Zalos, G.; Miller, B.V., 3rd; Chen, K.Y.; Remaley, A.T.; Waclawiw, M.A.; et al. Insulin and extremity muscle mass in overweight and obese women. Int. J. Obes. 2013, 37, 1560–1564. [Google Scholar] [CrossRef] [Green Version]
- Jensen, J.; Tantiwong, P.; Stuenæs, J.T.; Molina-Carrion, M.; DeFronzo, R.A.; Sakamoto, K.; Musi, N. Effect of acute exercise on glycogen synthase in muscle from obese and diabetic subjects. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E82–E89. [Google Scholar] [CrossRef] [Green Version]
- Shojaa, M.; Von Stengel, S.; Schoene, D.; Kohl, M.; Barone, G.; Bragonzoni, L.; Dallolio, L.; Marini, S.; Murphy, M.H.; Stephenson, A.; et al. Effect of Exercise Training on Bone Mineral Density in Post-menopausal Women: A Systematic Review and Meta-Analysis of Intervention Studies. Front. Physiol. 2021, 11, 652. [Google Scholar] [CrossRef] [PubMed]
- Schwab, P.; Scalapino, K. Exercise for bone health: Rationale and prescription. Curr. Opin. Rheumatol. 2011, 23, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.P.; Sale, C.; Greeves, J.P.; Casey, A.; Dutton, J.; Fraser, W.D. The role of exercise intensity in the bone metabolic response to an acute bout of weight-bearing exercise. J. Appl. Physiol. 2011, 110, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, I.; Kemppainen, J.; Kaskinoro, K.; Langberg, H.; Knuuti, J.; Boushel, R.; Kjaer, M.; Kalliokoski, K.K. Bone blood flow and metabolism in humans: Effect of muscular exercise and other physiological perturbations. J. Bone Miner. Res. 2013, 28, 1068–1074. [Google Scholar] [CrossRef]
- Fujimura, R.; Ashizawa, N.; Watanabe, M.; Mukai, N.; Amagai, H.; Fukubayashi, T.; Hayashi, K.; Tokuyama, K.; Suzuki, M. Effect of resistance exercise training on bone formation and resorption in young male subjects assessed by biomarkers of bone metabolism. J. Bone Min. Res. 1997, 12, 656–662. [Google Scholar] [CrossRef]
- Li, K.C.; Zernicke, R.F.; Barnard, R.J.; Li, A.F. Differential response of rat limb bones to strenuous exercise. J. Appl. Physiol. 1991, 70, 554–560. [Google Scholar] [CrossRef]
- Suniaga, S.; Rolvien, T.; Vom Scheidt, A.; Fiedler, I.A.K.; Bale, H.A.; Huysseune, A.; Witten, P.E.; Amling, M.; Busse, B. Increased mechanical loading through controlled swimming exercise induces bone formation and mineralization in adult zebrafish. Sci. Rep. 2018, 8, 3646. [Google Scholar] [CrossRef]
- Gleeson, M. Immune function in sport and exercise. J. Appl. Physiol. 2007, 103, 693–699. [Google Scholar] [CrossRef] [Green Version]
- Brolinson, P.G.; Elliott, D. Exercise and the immune system. Clin. Sports Med. 2007, 26, 311–319. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Hoffman-Goetz, L. Exercise and the immune system: Regulation, integration, and adaptation. Physiol. Rev. 2000, 80, 1055–1081. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.R.; Ha, G.C.; Ko, K.J.; Kang, S.J. Effects of exercise type on estrogen, tumor markers, immune function, antioxidant function, and physical fitness in postmenopausal obese women. J. Exerc. Rehabil. 2018, 14, 1032–1040. [Google Scholar] [CrossRef]
- Nieman, D.C.; Nehlsen-Cannarella, S.L.; Henson, D.A.; Butterworth, D.E.; Fagoaga, O.R.; Warren, B.J.; Rainwater, M.K. Immune response to obesity and moderate weight loss. Int. J. Obes. Relat. Metab. Disord. 1996, 20, 353–360. [Google Scholar]
- Tanaka, S.; Inoue, S.; Isoda, F.; Waseda, M.; Ishihara, M.; Yamakawa, T.; Sugiyama, A.; Takamura, Y.; Okuda, K. Impaired immunity in obesity: Suppressed but reversible lymphocyte responsiveness. Int. J. Obes. Relat. Metab. Disord. 1993, 17, 631–636. [Google Scholar]
- Kimura, M.; Tanaka, S.; Isoda, F.; Sekigawa, K.; Yamakawa, T.; Sekihara, H. T lymphopenia in obese diabetic (db/db) mice is nonselective and thymus independent. Life Sci. 1998, 62, 1243–1250. [Google Scholar] [CrossRef]
- Plotkin, B.J.; Paulson, D.; Chelich, A.; Jurak, D.; Cole, J.; Kasimos, J.; Burdick, J.R.; Casteel, N. Immune responsiveness in a rat model for type II diabetes (Zucker rat, fa/fa): Susceptibility to Candida albicans infection and leucocyte function. J. Med. Microbiol. 1996, 44, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.S.; Kim, K.A.; Kim, J.K.; Nho, H. Augmented Hemodynamic Responses in Obese Young Men during Dynamic Exercise: Role of the Muscle Metaboreflex. Int. J. Environ. Res. Public Health 2021, 17, 7321. [Google Scholar] [CrossRef]
- Hambrecht, R.; Adams, V.; Erbs, S.; Linke, A.; Kränkel, N.; Shu, Y.; Baither, Y.; Gielen, S.; Thiele, H.; Gummert, J.F.; et al. Regular physical activity improves endothelial function in patients with coronary artery disease by increasing phosphorylation of endothelial nitric oxide synthase. Circulation 2003, 107, 3152–3158. [Google Scholar] [CrossRef] [Green Version]
- Thijssen, D.H.; Maiorana, A.J.; O’Driscoll, G.; Cable, N.T.; Hopman, M.T.; Green, D.J. Impact of inactivity and exercise on the vasculature in humans. Eur. J. Appl. Physiol. 2010, 108, 845–875. [Google Scholar] [CrossRef] [Green Version]
- Evans, D.L. Cardiovascular adaptations to exercise and training. Vet. Clin. N. Am. Equine Pract. 1985, 1, 513–531. [Google Scholar] [CrossRef]
- Lavie, C.J.; Arena, R.; Swift, D.L.; Johannsen, N.M.; Sui, X.; Lee, D.; Earnest, C.P.; Church, T.S.; O’Keefe, J.H.; Milani, R.V.; et al. Exercise and the Cardiovascular System. Clinical Science and Cardiovascular Outcomes. Circ. Res. 2015, 117, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Weiner, R.B.; Baggish, A.L. Exercise-induced cardiac remodeling. Prog. Cardiovasc. Dis. 2012, 54, 380–386. [Google Scholar] [CrossRef]
- Delp, M.D. Differential effects of training on the control of skeletal muscle perfusion. Med. Sci. Sports Exerc. 1998, 30, 361–374. [Google Scholar] [CrossRef]
- Hogan, T.S. Exercise-induced reduction in systemic vascular resistance: A covert killer and an unrecognised resuscitation challenge? Med. Hypotheses 2009, 73, 479–484. [Google Scholar] [CrossRef]
- Breisch, E.A.; White, F.C.; Nimmo, L.E.; McKirnan, M.D.; Bloor, C.M. Exercise-induced cardiac hypertrophy: A correlation of blood flow and microvasculature. J. Appl. Physiol. 1986, 60, 1259–1267. [Google Scholar] [CrossRef]
- Wisloff, U.; Loennechen, J.P.; Currie, S.; Smith, G.L.; Ellingsen, O. Aerobic exercise reduces cardiomyocyte hypertrophy and increases contractility, Ca2+ sensitivity and SERCA-2 in rat after myocardial infarction. Cardiovasc. Res. 2002, 54, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Natali, A.J.; Wilson, L.A.; Peckham, M.; Turner, D.L.; Harrison, S.M.; White, E. Different regional effects of voluntary exercise on the mechanical and electrical properties of rat ventricular myocytes. J. Physiol. 2002, 541, 863–875. [Google Scholar] [CrossRef]
- Kemi, O.J.; Wisloff, U. Mechanisms of exercise-induced improvements in the contractile apparatus of the mammalian myocardium. Acta Physiol. 2010, 199, 425–439. [Google Scholar] [CrossRef]
- Hallen, J. K+ balance in humans during exercise. Acta Physiol. Scand. 1996, 156, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Radegran, G.; Calbet, J.A. Role of adenosine in exercise-induced human skeletal muscle vasodilatation. Acta Physiol. Scand. 2001, 171, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Sarelius, I.; Pohl, U. Control of muscle blood flow during exercise: Local factors and integrative mechanisms. Acta Physiol. 2010, 199, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Haverkamp, H.C.; Dempsey, J.A.; Miller, J.D.; Romer, L.M.; Eldridge, M.W. Physiologic response to exercise. In Physiological Basis of Respiratory Disease, 1st ed.; Hamid, Q., Shannon, J., Martin, J., Eds.; BC Decker, Inc.: Hamilton, ON, Canada, 2005; pp. 525–540. [Google Scholar]
- Chlif, M.; Chaouachi, A.; Ahmaidi, S. Effect of Aerobic Exercise Training on Ventilatory Efficiency and Respiratory Drive in Obese Subjects. Respir. Care 2017, 62, 936–946. [Google Scholar] [CrossRef] [Green Version]
- Chlif, M.; Temfemo, A.; Keochkerian, D.; Choquet, D.; Chaouachi, A.; Ahmaidi, S. Advanced Mechanical Ventilatory Constraints During Incremental Exercise in Class III Obese Male Subjects. Respir. Care 2015, 60, 549–560. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.Y.; Cerny, F.J. Ventilatory response to exercise in simulated obesity by chest loading. Med. Sci. Sports Exerc. 2004, 36, 780–786. [Google Scholar] [CrossRef]
- Li, A.M.; Chan, D.; Wong, E.; Yin, J.; Nelson, E.A.; Fok, T.F. The effects of obesity on pulmonary function. Arch. Dis. Child. 2003, 88, 361–363. [Google Scholar] [CrossRef] [Green Version]
- Koenig, S.M. Pulmonary complications of obesity. Am. J. Med. Sci. 2001, 321, 249–279. [Google Scholar] [CrossRef]
- Faria, A.G.; Ribeiro, M.A.; Marson, F.A.; Schivinski, C.I.; Severino, S.D.; Ribeiro, J.D.; Barros Filho, A.A. Effect of exercise test on pulmonary function of obese adolescents. J. Pediatr. 2014, 90, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Torchio, R.; Gobb, A.; Gulotta, C.; Dellacà, R.; Tinivella, M.; Hyatt, R.E.; Brusasco, V.; Pellegrino, R. Mechanical effects of obesity on airway responsiveness in otherwise healthy humans. J. Appl. Physiol. 2009, 107, 408–416. [Google Scholar] [CrossRef]
- Moses, F.M. The effect of exercise on the gastrointestinal tract. Sports Med. 1990, 9, 159–172. [Google Scholar] [CrossRef]
- De Oliveira, E.P.; Burini, R.C.; Jeukendrup, A. Gastrointestinal complaints during exercise: Prevalence, etiology, and nutritional recommendations. Sports Med. 2014, 44, S79–S85. [Google Scholar] [CrossRef] [Green Version]
- Sari, R.; Balci, N.; Balci, M.K. Effects of exercise on gallbladder volume and motility in obese women. J. Clin. Ultrasound 2005, 33, 218–222. [Google Scholar] [CrossRef]
- Wilund, K.R.; Feeney, L.A.; Tomayko, E.J.; Weiss, E.P.; Hagberg, J.M. Effects of endurance exercise training on markers of cholesterol absorption and synthesis. Physiol. Res. 2009, 58, 545–552. [Google Scholar] [CrossRef]
- Devries, M.C.; Samjoo, I.A.; Hamadeh, M.J.; Tarnopolsky, M.A. Effect of endurance exercise on hepatic lipid content, enzymes, and adiposity in men and women. Obesity 2008, 16, 2281–2288. [Google Scholar] [CrossRef]
- Skrypnik, D.; Ratajczak, M.; Karolkiewicz, J.; Mądry, E.; Pupek-Musialik, D.; Hansdorfer-Korzon, R.; Walkowiak, J.; Jakubowski, H.; Bogdański, P. Effects of endurance and endurance-strength exercise on biochemical parameters of liver function in women with abdominal obesity. Biomed. Pharmacother. 2016, 80, 1–7. [Google Scholar] [CrossRef]
- Hackney, A.C.; Lane, A.R. Exercise and the Regulation of Endocrine Hormones. Prog. Mol. Biol. Transl. Sci. 2015, 135, 293–311. [Google Scholar]
- Cano Sokoloff, N.; Misra, M.; Ackerman, K.E. Exercise, Training, and the Hypothalamic-Pituitary-Gonadal Axis in Men and Women. Front. Horm. Res. 2016, 47, 27–43. [Google Scholar] [PubMed]
- Mastorakos, G.; Pavlatou, M.; Diamanti-Kandarakis, E.; Chrousos, G.P. Exercise and the stress system. Hormones 2005, 4, 73–89. [Google Scholar] [PubMed]
- Slentz, C.A.; Tanner, C.J.; Bateman, L.A.; Durheim, M.T.; Huffman, K.M.; Houmard, J.A.; Kraus, W.E. Effects of exercise training intensity on pancreatic beta-cell function. Diabetes Care 2009, 32, 1807–1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, S.M.; Thorup, A.C.; Overgaard, K.; Jeppesen, P.B. High Intensity Interval Training Improves Glycaemic Control and Pancreatic beta Cell Function of Type 2 Diabetes Patients. PLoS ONE 2015, 10, e0133286. [Google Scholar] [CrossRef]
- Heiskanen, M.A.; Motiani, K.K.; Mari, A.; Saunavaara, V.; Eskelinen, J.J.; Virtanen, K.A.; Koivumäki, M.; Löyttyniemi, E.; Nuutila, P.; Kalliokoski, K.K.; et al. Exercise training decreases pancreatic fat content and improves beta cell function regardless of baseline glucose tolerance: A randomised controlled trial. Diabetologia 2018, 61, 1817–1828. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.H.; Farmer, K.; Windscheffel, J.; Yost, K.; Power, M.; Wright, D.E.; Stehno-Bittel, L. Exercise increases insulin content and basal secretion in pancreatic islets in type 1 diabetic mice. Exp. Diabetes Res. 2011, 2011, 481427. [Google Scholar] [CrossRef]
- Malin, S.K.; Francois, M.E.; Eichner, N.Z.M.; Gilbertson, N.M.; Heiston, E.M.; Fabris, C.; Breton, M. Impact of short-term exercise training intensity on beta-cell function in older obese adults with prediabetes. J. Appl. Physiol. 2018, 125, 1979–1986. [Google Scholar] [CrossRef] [Green Version]
- Morgan, J.A.; Corrigan, F.; Baune, B.T. Effects of physical exercise on central nervous system functions: A review of brain region specific adaptations. J. Mol. Psychiatry 2015, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Dishman, R.K.; Berthoud, H.R.; Booth, F.W.; Cotman, C.W.; Edgerton, V.R.; Fleshner, M.R.; Gandevia, S.C.; Gomez-Pinilla, F.; Greenwood, B.N.; Hillman, C.H.; et al. Neurobiology of exercise. Obesity 2006, 14, 345–356. [Google Scholar] [CrossRef]
- Di Liegro, C.M.; Schiera, G.; Proia, P.; Di Liegro, I. Physical Activity and Brain Health. Genes 2019, 10, 720. [Google Scholar] [CrossRef] [Green Version]
- Díaz, B.B.; González, D.A.; Gannar, F.; Pérez, M.C.R.; de León, A.C. Myokines, physical activity, insulin resistance and autoimmune diseases. Immunol. Lett. 2018, 203, 1–5. [Google Scholar] [CrossRef]
- Sholl-Franco, A.; da Silva, A.G.; Adão-Novaes, J. Interleukin-4 as a neuromodulatory cytokine: Roles and signaling in the nervous system. Ann. N. Y. Acad. Sci. 2009, 1153, 65–75. [Google Scholar] [CrossRef]
- Plata-Salamán, C.R.; Borkoski, J.P. Interleukin-8 modulates feeding by direct action in the central nervous system. Am. J. Physiol. 1993, 265, R877–R882. [Google Scholar] [CrossRef]
- Pan, W.; Wu, X.; He, Y.; Hsuchou, H.; Huang, E.Y.K.; Mishra, P.K.; Kastin, A.J. Brain interleukin-15 in neuroinflammation and behavior. Neurosci. Biobehav. Rev. 2013, 37, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Dyer, A.H.; Vahdatpour, C.; Sanfeliu, A.; Tropea, D. The role of Insulin-Like Growth Factor 1 (IGF-1) in brain development, maturation and neuroplasticity. Neuroscience 2016, 325, 89–99. [Google Scholar] [CrossRef]
- Von Holstein-Rathlou, S.; Gillum, M.P. Fibroblast growth factor 21: An endocrine inhibitor of sugar and alcohol appetite. J. Physiol. 2019, 597, 3539–3548. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, W. Can irisin be a linker between physical activity and brain function? Biomol. Concepts 2016, 7, 253–258. [Google Scholar] [CrossRef]
- Genc, S.; Koroglu, T.F.; Genc, K. Erythropoietin and the nervous system. Brain Res. 2004, 1000, 19–31. [Google Scholar] [CrossRef]
- Buemi, M.; Cavallaro, E.; Floccari, F.; Sturiale, A.; Aloisi, C.; Trimarchi, M.; Corica, F.; Frisina, N. The pleiotropic effects of erythropoietin in the central nervous system. J. Neuropathol. Exp. Neurol. 2003, 62, 228–236. [Google Scholar] [CrossRef] [Green Version]
- Nagai, N.; Moritani, T. Effect of physical activity on autonomic nervous system function in lean and obese children. Int. J. Obes. Relat. Metab. Disord. 2004, 28, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Amano, M.; Kanda, T.; Ue, H.; Moritani, T. Exercise training and autonomic nervous system activity in obese individuals. Med. Sci. Sports Exerc. 2001, 33, 1287–1291. [Google Scholar] [CrossRef]
- Cornier, M.A.; Melanson, E.L.; Salzberg, A.K.; Bechtell, J.L.; Tregellas, J.R. The effects of exercise on the neuronal response to food cues. Physiol. Behav. 2012, 105, 1028–1034. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.Y.; Roh, H.T. Effects of aerobic exercise training on peripheral brain-derived neurotrophic factor and eotaxin-1 levels in obese young men. J. Phys. Ther. Sci. 2016, 28, 1355–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araya, A.V.; Orellana, X.; Godoy, D.; Soto, L.; Fiedler, J. Effect of exercise on circulating levels of brain-derived neurotrophic factor (BDNF) in overweight and obese subjects. Horm. Metab. Res. 2013, 45, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.L.; Whitehurst, M.; Fico, B.G.; Dodge, K.M.; Ferrandi, P.J.; Pena, G.; Adelman, A.; Huang, C. Acute high-intensity interval exercise induces greater levels of serum brain-derived neurotrophic factor in obese individuals. Exp. Biol. Med. 2018, 243, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Pate, R.R.; O’Neill, J.R.; Lobelo, F. The evolving definition of “sedentary”. Exerc. Sport Sci. Rev. 2008, 36, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Sedentary Behaviour and Research Network. Standardized use of the terms “sedentary” and “sedentary behaviours”. Appl. Physiol. Nutr. Metab. 2012, 37, 540–542. [Google Scholar] [CrossRef] [Green Version]
- Chau, J.Y.; Grunseit, A.C.; Chey, T.; Stamatakis, E.; Brown, W.J.; Matthews, C.E.; Bauman, A.E.; van der Ploeg, H.P. Daily sitting time and all-cause mortality: A meta-analysis. PLoS ONE 2013, 8, e80000. [Google Scholar]
- Ching, P.L.; Willett, W.C.; Rimm, E.B.; Colditz, G.A.; Gortmaker, S.L.; Stampfer, M.J. Activity level and risk of overweight in male health professionals. Am. J. Public Health 1996, 86, 25–30. [Google Scholar] [CrossRef] [Green Version]
- Bowman, S.A. Television-viewing characteristics of adults: Correlations to eating practices and overweight and health status. Prev. Chronic. Dis. 2006, 3, A38. [Google Scholar]
- Thomson, M.; Spence, J.C.; Raine, K.; Laing, L. The association of television viewing with snacking behavior and body weight of young adults. Am. J. Health Promot. 2008, 22, 329–335. [Google Scholar] [CrossRef]
- Frank, L.D.; Andresen, M.A.; Schmid, T.L. Obesity relationships with community design, physical activity, and time spent in cars. Am. J. Prev. Med. 2004, 27, 87–96. [Google Scholar] [CrossRef]
- Lopez-Zetina, J.; Lee, H.; Friis, R. The link between obesity and the built environment. Evidence from an ecological analysis of obesity and vehicle miles of travel in California. Health Place 2006, 12, 656–664. [Google Scholar] [CrossRef]
- Dunton, G.F.; Berrigan, D.; Ballard-Barbash, R.; Graubard, B.; Atienza, A.A. Joint associations of physical activity and sedentary behaviors with body mass index: Results from a time use survey of US adults. Int. J. Obes. 2009, 33, 1427–1436. [Google Scholar] [CrossRef] [Green Version]
- Stefansdottir, R.; Gudmundsdottir, S.L. Sedentary behavior and musculoskeletal pain: A five-year longitudinal Icelandic study. Public Health 2017, 149, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Santos, M.C.S.; de Andrade, S.M.; González, A.D.; Dias, D.F.; Mesas, A.E. Association Between Chronic Pain and Leisure Time Physical Activity and Sedentary Behavior in Schoolteachers. Behav. Med. 2018, 44, 335–343. [Google Scholar] [CrossRef]
- Nijs, J.; D’Hondt, E.; Clarys, P.; Deliens, T.; Polli, A.; Malfliet, A.; Coppieters, I.; Willaert, W.; Tumkaya Yilmaz, S.; Elma, Ö.; et al. Lifestyle and Chronic Pain across the Lifespan: An Inconvenient Truth? PM&R 2020, 12, 410–419. [Google Scholar]
- Zhaoyang, R.; Martire, L.M.; Darnall, B.D. Daily pain catastrophizing predicts less physical activity and more sedentary behavior in older adults with osteoarthritis. Pain 2020, 161, 2603–2610. [Google Scholar] [CrossRef]
- Lee, S.H.; Son, C.; Yeo, S.; Ha, I.H. Cross-sectional analysis of self-reported sedentary behaviors and chronic knee pain among South Korean adults over 50 years of age in KNHANES 2013–2015. BMC Public Health 2019, 19, 1375. [Google Scholar] [CrossRef]
- Mendonça, C.R.; Noll, M.; Rodrigues, A.P.D.S.; Vitorino, P.V.O.; Mendes, M.A.; Silveira, E.A. Association of Pain, Severe Pain, and Multisite Pain with the Level of Physical Activity and Sedentary Behavior in Severely Obese Adults: Baseline Data from the DieTBra Trial. Int. J. Environ. Res. Public Health 2020, 17, 4478. [Google Scholar] [CrossRef]
- O’Leary, H.; Larkin, L.; Murphy, G.M.; Quinn, K. Relationship Between Pain and Sedentary Behavior in Rheumatoid Arthritis Patients: A Cross-Sectional Study. Arthritis Care Res. 2021, 73, 990–997. [Google Scholar] [CrossRef]
- Segura-Jiménez, V.; Borges-Cosic, M.; Soriano-Maldonado, A.; Estévez-López, F.; Álvarez-Gallardo, I.C.; Herrador-Colmenero, M.; Delgado-Fernández, M.; Ruiz, J.R. Association of sedentary time and physical activity with pain, fatigue, and impact of fibromyalgia: The al-Andalus study. Scand. J. Med. Sci. Sports 2017, 27, 83–92. [Google Scholar] [CrossRef]
- Baena-Beato, P.Á.; Artero, E.G.; Arroyo-Morales, M.; Robles-Fuentes, A.; Gatto-Cardia, M.C.; Delgado-Fernández, M. Aquatic therapy improves pain, disability, quality of life, body composition and fitness in sedentary adults with chronic low back pain. A controlled clinical trial. Clin. Rehabil. 2014, 28, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, L.; Tu, Y.; Chen, X.; Hu, K.; Tu, X.; Lin, M.; Xie, G.; Chen, S.; Huang, J.; et al. Different exercise modalities relieve pain syndrome in patients with knee osteoarthritis and modulate the dorsolateral prefrontal cortex: A multiple mode MRI study. Brain Behav. Immun. 2019, 82, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Knauf, M.T.; Koltyn, K.F. Exercise-induced modulation of pain in adults with and without painful diabetic neuropathy. J. Pain 2014, 15, 656–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowans, S.E.; deHueck, A.; Voss, S.; Richardson, M. A randomized, controlled trial of exercise and education for individuals with fibromyalgia. Arthritis Care Res. 1999, 12, 120–128. [Google Scholar] [CrossRef]
- Jentoft, E.S.; Kvalvik, A.G.; Mengshoel, A.M. Effects of pool-based and land-based aerobic exercise on women with fibromyalgia/chronic widespread muscle pain. Arthritis Rheum. 2001, 45, 42–47. [Google Scholar] [CrossRef]
- Dönmez, A.; Karagülle, M.Z.; Tercan, N.; Dinler, M.; Işsever, H.; Karagülle, M.; Turan, M. SPA therapy in fibromyalgia: A randomised controlled clinic study. Rheumatol. Int. 2005, 26, 168–172. [Google Scholar] [CrossRef]
- Jones, K.D.; Adams, D.; Winters-Stone, K.; Burckhardt, C.S. A comprehensive review of 46 exercise treatment studies in fibromyalgia (1988–2005). Health Qual. Life Outcomes 2006, 4, 67. [Google Scholar] [CrossRef] [Green Version]
- Palandi, J.; Bobinski, F.; de Oliveira, G.M.; Ilha, J. Neuropathic pain after spinal cord injury and physical exercise in animal models: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2021, 108, 781–795. [Google Scholar] [CrossRef]
- Li, X.; Wang, Q.; Ding, J.; Wang, S.; Dong, C.; Wu, Q. Exercise training modulates glutamic acid decarboxylase-65/67 expression through TrkB signaling to ameliorate neuropathic pain in rats with spinal cord injury. Mol. Pain 2021, 16, 1744806920924511. [Google Scholar] [CrossRef]
- Detloff, M.R.; Smith, E.J.; Quiros Molina, D.; Ganzer, P.D.; Houlé, J.D. Acute exercise prevents the development of neuropathic pain and the sprouting of non-peptidergic (GDNF- and artemin-responsive) c-fibers after spinal cord injury. Exp. Neurol. 2014, 255, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Dugan, E.A.; Sagen, J. An Intensive Locomotor Training Paradigm Improves Neuropathic Pain following Spinal Cord Compression Injury in Rats. J. Neurotrauma 2015, 32, 622–632. [Google Scholar] [CrossRef]
- Nees, T.A.; Tappe-Theodor, A.; Sliwinski, C.; Motsch, M.; Rupp, R.; Kuner, R.; Weidner, N.; Blesch, A. Early-onset treadmill training reduces mechanical allodynia and modulates calcitonin gene-related peptide fiber density in lamina III/IV in a mouse model of spinal cord contusion injury. Pain 2016, 157, 687–697. [Google Scholar] [CrossRef]
- Dugan, E.A.; Schachner, B.; Jergova, S.; Sagen, J. Intensive Locomotor Training Provides Sustained Alleviation of Chronic Spinal Cord Injury-Associated Neuropathic Pain: A Two-Year Pre-Clinical Study. J. Neurotrauma 2021, 38, 789–802. [Google Scholar] [CrossRef]
- Chen, Y.W.; Li, Y.T.; Chen, Y.C.; Li, Z.Y.; Hung, C.H. Exercise training attenuates neuropathic pain and cytokine expression after chronic constriction injury of rat sciatic nerve. Anesth. Analg. 2012, 114, 1330–1337. [Google Scholar] [CrossRef]
- Lopes, B.C.; Medeiros, L.F.; Silva de Souza, V.; Cioato, S.G.; Medeiros, H.R.; Regner, G.G.; Lino de Oliveira, C.; Fregni, F.; Caumo, W.; Torres, I.L.S. Transcranial direct current stimulation combined with exercise modulates the inflammatory profile and hyperalgesic response in rats subjected to a neuropathic pain model: Long-term effects. Brain Stimul. 2021, 13, 774–782. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.C.; Tsai, K.L.; Chen, Y.W.; Lin, H.T.; Hung, C.H. Exercise Combined With Ultrasound Attenuates Neuropathic Pain in Rats Associated With Downregulation of IL-6 and TNF-alpha, but With Upregulation of IL-10. Anesth. Analg. 2017, 124, 2038–2044. [Google Scholar] [CrossRef]
- Farzad, B.; Rajabi, H.; Gharakhanlou, R.; Allison, D.J.; Hayat, P.; Jameie, S.B. Swimming Training Attenuates Allodynia and Hyperalgesia Induced by Peripheral Nerve Injury in an Adult Male Rat Neuropathic Model: Effects on Irisin and GAD65. Pain Med. 2018, 19, 2236–2245. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.H.; Kim, J.E.; Seo, T.B. Effect of treadmill exercise on pain-related Wnt/beta-catenin signaling pathway in dorsal root ganglion neurons at the early phase regeneration of the injured sciatic nerve. J. Exerc. Rehabil. 2021, 17, 96–102. [Google Scholar] [CrossRef]
- Almeida, C.; DeMaman, A.; Kusuda, R.; Cadetti, F.; Ravanelli, M.I.; Queiroz, A.L.; Sousa, T.A.; Zanon, S.; Silveira, L.R.; Lucas, G. Exercise therapy normalizes BDNF upregulation and glial hyperactivity in a mouse model of neuropathic pain. Pain 2015, 156, 504–513. [Google Scholar] [CrossRef]
- Kami, K.; Taguchi Ms, S.; Tajima, F.; Senba, E. Improvements in impaired GABA and GAD65/67 production in the spinal dorsal horn contribute to exercise-induced hypoalgesia in a mouse model of neuropathic pain. Mol. Pain 2016, 12, 1744806916629059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, A.; Palstam, A.; Löfgren, M.; Ernberg, M.; Bjersing, J.; Bileviciute-Ljungar, I.; Gerdle, B.; Kosek, E.; Mannerkorpi, K. Resistance exercise improves muscle strength.; health status and pain intensity in fibromyalgia—A randomized controlled trial. Arthritis Res. Ther. 2015, 17, 161. [Google Scholar] [CrossRef] [Green Version]
- Toprak Celenay, S.; Mete, O.; Akan, S.; Un Yildirim, N.; Erten, S. Comparison of the effects of stabilization exercise plus kinesio taping and stabilization exercise alone on pain and well-being in fibromyalgia. Complement. Ther. Clin. Pract. 2021, 38, 101076. [Google Scholar] [CrossRef] [PubMed]
- Munguía-Izquierdo, D.; Legaz-Arrese, A. Exercise in warm water decreases pain and improves cognitive function in middle-aged women with fibromyalgia. Clin. Exp. Rheumatol. 2007, 25, 823–830. [Google Scholar] [PubMed]
- Bjersing, J.L.; Dehlin, M.; Erlandsson, M.; Bokarewa, M.I.; Mannerkorpi, K. Changes in pain and insulin-like growth factor 1 in fibromyalgia during exercise: The involvement of cerebrospinal inflammatory factors and neuropeptides. Arthritis Res. Ther. 2012, 14, R162. [Google Scholar] [CrossRef] [Green Version]
- Ferro Moura Franco, K.; Lenoir, D.; Dos Santos Franco, Y.R.; Jandre Reis, F.J.; Nunes Cabral, C.M.; Meeus, M. Prescription of exercises for the treatment of chronic pain along the continuum of nociplastic pain: A systematic review with meta-analysis. Eur. J. Pain 2021, 25, 51–70. [Google Scholar] [CrossRef]
- Zhang, J.M.; An, J. Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Obata, K.; Noguchi, K. BDNF in sensory neurons and chronic pain. Neurosci. Res. 2006, 55, 1–10. [Google Scholar] [CrossRef]
- Zhang, Y.; Qin, W.; Qian, Z.; Liu, X.; Wang, H.; Gong, S.; Sun, Y.G.; Snutch, T.P.; Jiang, X.; Tao, J. Peripheral pain is enhanced by insulin-like growth factor 1 through a G protein-mediated stimulation of T-type calcium channels. Sci. Signal 2014, 7, ra94. [Google Scholar] [CrossRef]
- Mazur-Bialy, A.I.; Pocheć, E.; Zarawski, M. Anti-Inflammatory Properties of Irisin, Mediator of Physical Activity, Are Connected with TLR4/MyD88 Signaling Pathway Activation. Int. J. Mol. Sci. 2017, 18, 701. [Google Scholar] [CrossRef]
- Dameni, S.; Janzadeh, A.; Yousefifard, M.; Nasirinezhad, F. The effect of intrathecal injection of irisin on pain threshold and expression rate of GABAB receptors in peripheral neuropathic pain model. J. Chem. Neuroanat. 2018, 91, 17–26. [Google Scholar] [CrossRef]
- Huang, L.; Yan, S.; Luo, L.; Yang, L. Irisin regulates the expression of BDNF and glycometabolism in diabetic rats. Mol. Med. Rep. 2019, 19, 1074–1082. [Google Scholar] [CrossRef]
- Groover, A.L.; Ryals, J.M.; Guilford, B.L.; Wilson, N.M.; Christianson, J.A.; Wright, D.E. Exercise-mediated improvements in painful neuropathy associated with prediabetes in mice. Pain 2013, 154, 2658–2667. [Google Scholar] [CrossRef] [Green Version]
- Coradini, J.G.; Kunz, R.I.; Kakihata, C.M.; Errero, T.K.; Bonfleur, M.L.; Ribeiro Lde, F.; Brancalhão, R.M.; Bertolini, G.R. Swimming does not alter nociception threshold in obese rats submitted to median nerve compression. Neurol. Res. 2015, 37, 1118–1124. [Google Scholar] [CrossRef]
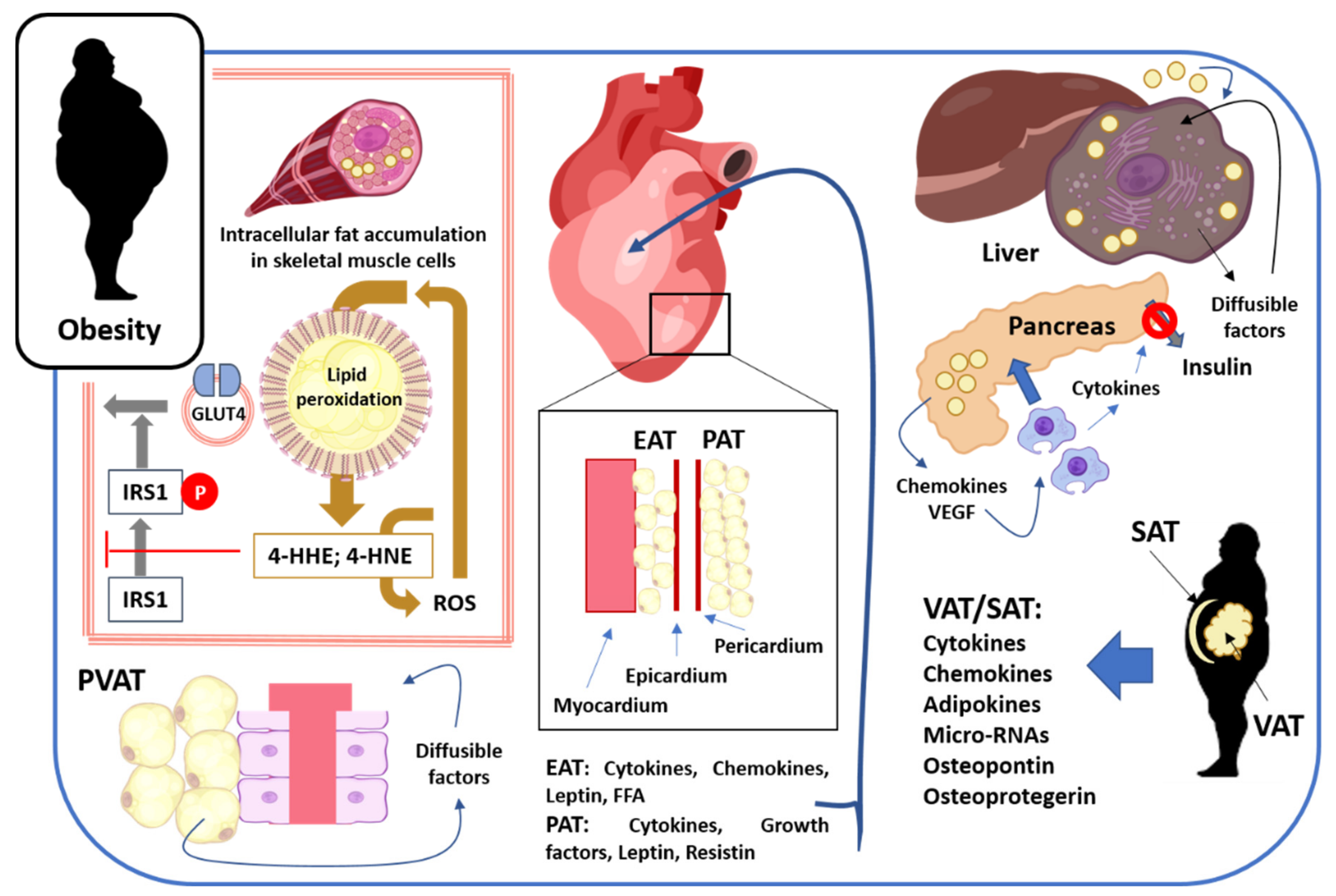




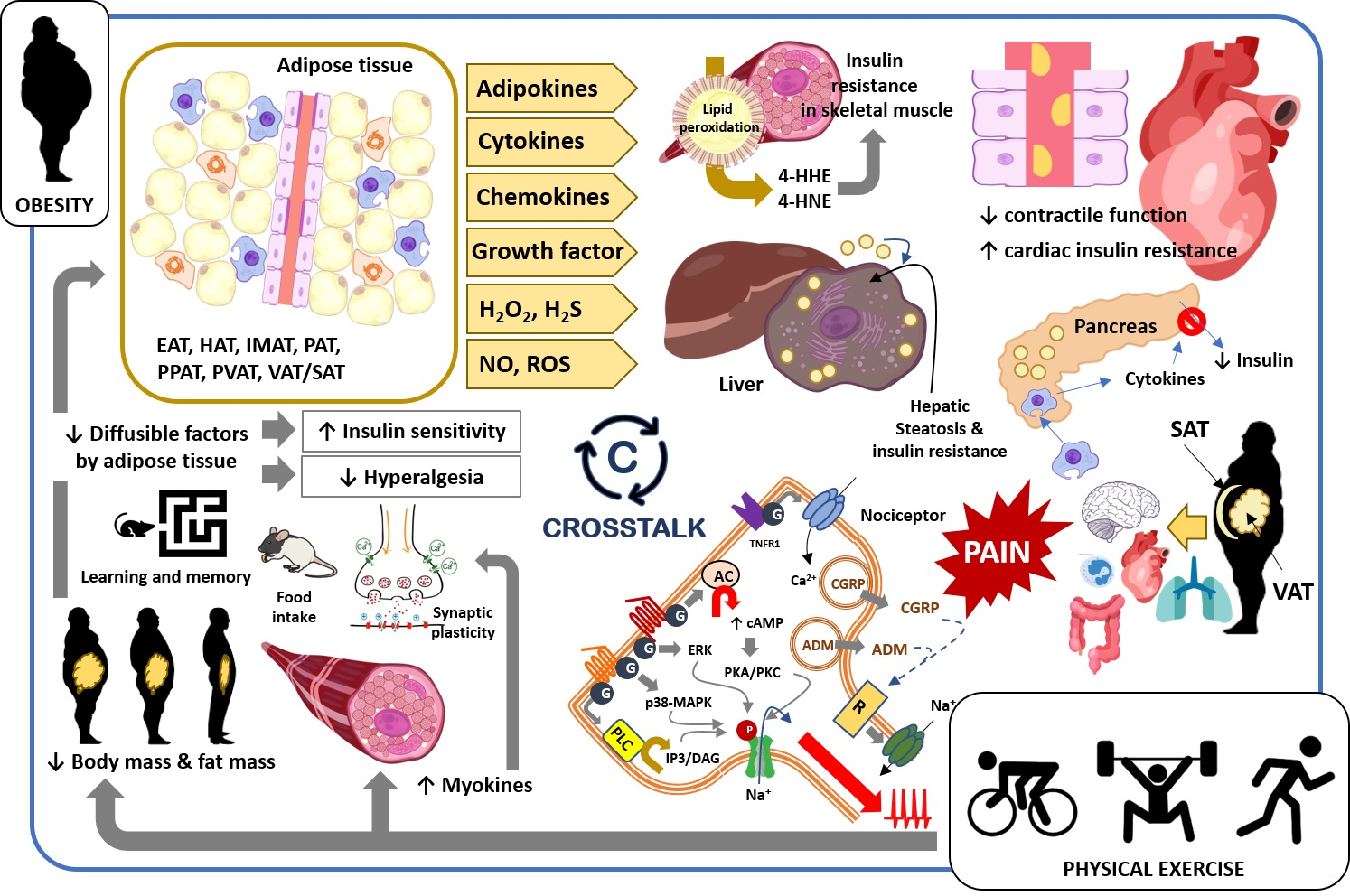{kind=link}
{kind=link}
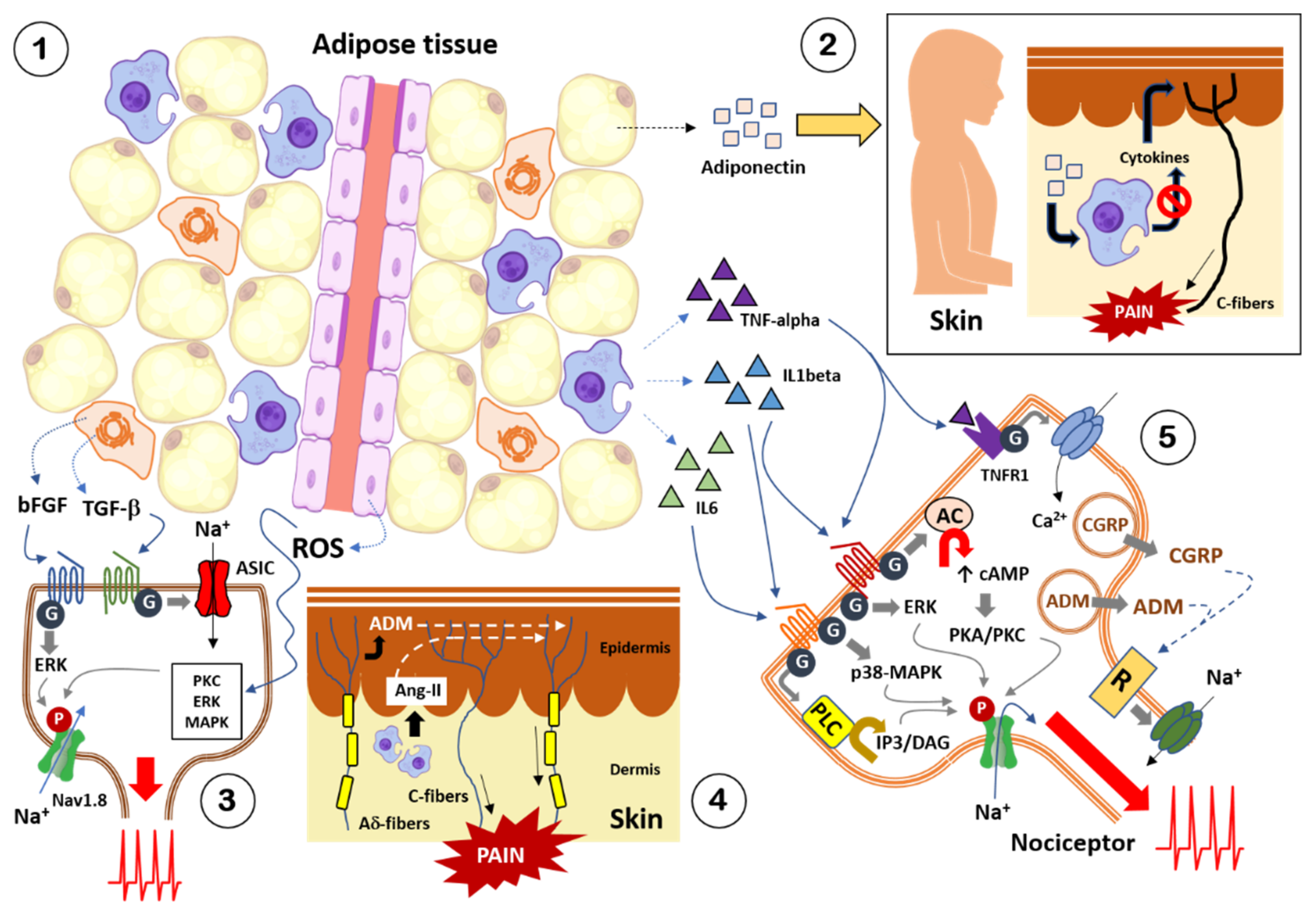{kind=link}
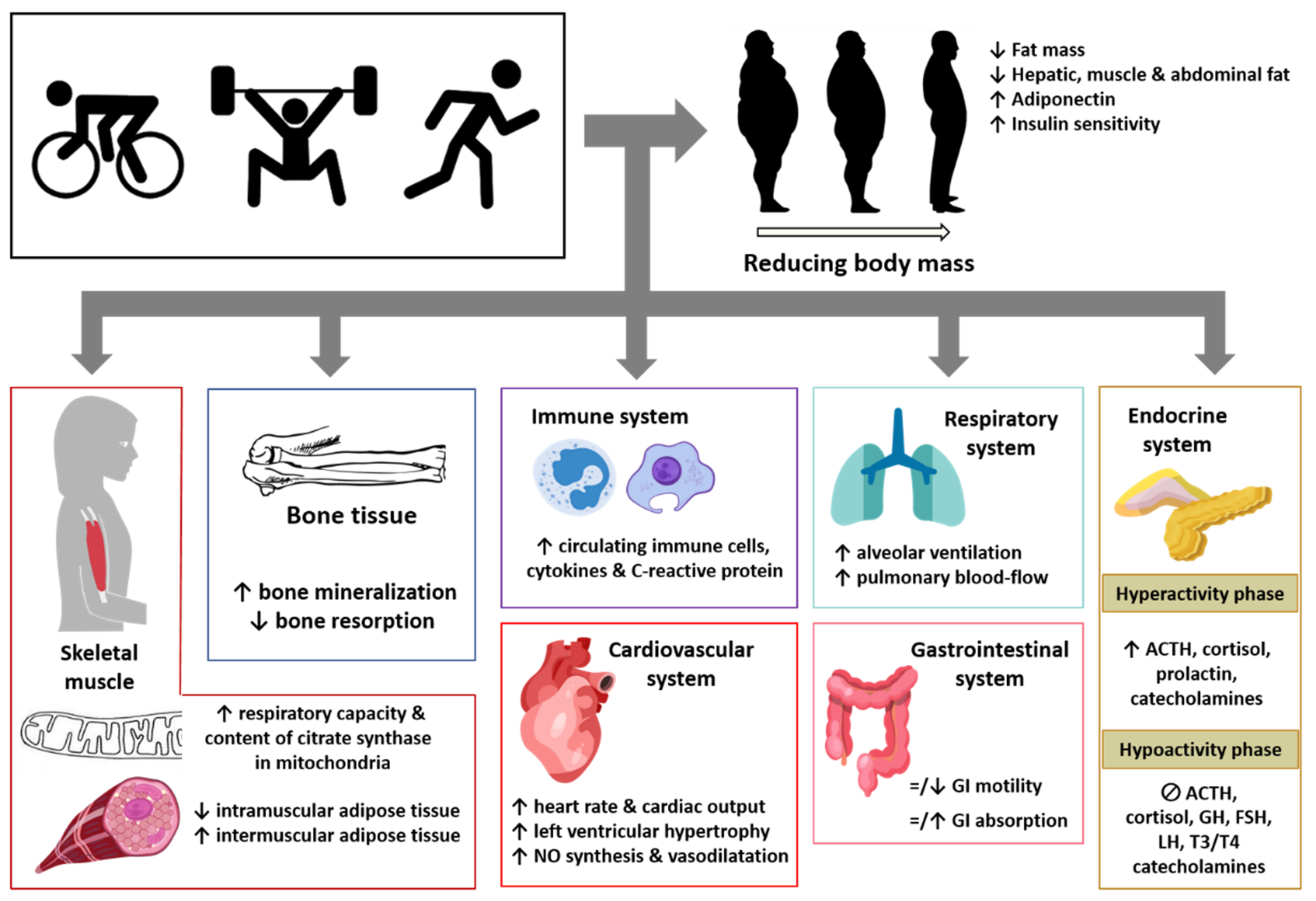{kind=link}
{kind=link}
| Factor/Molecule | EAT | HAT | IMAT | PAT | PPAT | PVAT | VAT/SAT |
|---|---|---|---|---|---|---|---|
| Adiponectin | X | X | |||||
| Adrenomedullin | X | X | |||||
| Angiotensin-II | X | ||||||
| Apelin | X | ||||||
| bFGF | X | ||||||
| CCL1 | X | ||||||
| CCL11 | X | ||||||
| CCL18 | X | ||||||
| CCL21 | X | ||||||
| CCL3 | X | X | |||||
| CCL4 | X | ||||||
| CCL5 | X | X | |||||
| CCL8 | X | ||||||
| Chemerin | X | ||||||
| CTRP-9 | X | ||||||
| CXCL1 | X | ||||||
| CXCL10 | X | ||||||
| CXCL2 | X | X | |||||
| CXCL3 | X | ||||||
| CXCL5/LIX | X | ||||||
| CXCL9 | X | ||||||
| FFA | X | ||||||
| FGF-21 | X | ||||||
| Fractalkine | X | ||||||
| HGF | X | ||||||
| H2O2 | X | ||||||
| H2S | X | ||||||
| IGFB-3 | X | ||||||
| IL1-beta | X | X | X | ||||
| IL10 | X | X | |||||
| IL12 | X | ||||||
| IL13 | X | ||||||
| IL15 | X | ||||||
| IL17 | X | ||||||
| IL18 | X | ||||||
| IL32 | X | ||||||
| IL33 | X | ||||||
| IL6 | X | X | X | ||||
| IL7 | X | ||||||
| IL8 | X | ||||||
| INF-γ | X | ||||||
| Irisin | X | ||||||
| Leptin | X | X | X | ||||
| Lipocalin-2 | X | ||||||
| MCP-1/CCL2 | X | X | X | X | |||
| Methyl palmitate | X | ||||||
| Micro-RNAs | X | X | |||||
| MIP-1α/CCL3 | X | ||||||
| MIP1 | |||||||
| MIP3 | |||||||
| Nitric Oxide | X | ||||||
| Omentin | X | ||||||
| Osteopontin | |||||||
| Osteoprotegerin | |||||||
| PDGF | X | X | |||||
| RANTES | X | ||||||
| Resistin | X | X | X | ||||
| ROS | X | ||||||
| Serpin-E1 | X | ||||||
| TGF-β | X | X | |||||
| Thrombospondin-1 | X | ||||||
| TNF-alpha | X | X | X | ||||
| TWEAK | |||||||
| Vaspin | X | ||||||
| VEGF | X | X | |||||
| Visfatin | X |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verdú, E.; Homs, J.; Boadas-Vaello, P. Physiological Changes and Pathological Pain Associated with Sedentary Lifestyle-Induced Body Systems Fat Accumulation and Their Modulation by Physical Exercise. Int. J. Environ. Res. Public Health 2021, 18, 13333. https://doi.org/10.3390/ijerph182413333
Verdú E, Homs J, Boadas-Vaello P. Physiological Changes and Pathological Pain Associated with Sedentary Lifestyle-Induced Body Systems Fat Accumulation and Their Modulation by Physical Exercise. International Journal of Environmental Research and Public Health. 2021; 18(24):13333. https://doi.org/10.3390/ijerph182413333
Chicago/Turabian StyleVerdú, Enrique, Judit Homs, and Pere Boadas-Vaello. 2021. "Physiological Changes and Pathological Pain Associated with Sedentary Lifestyle-Induced Body Systems Fat Accumulation and Their Modulation by Physical Exercise" International Journal of Environmental Research and Public Health 18, no. 24: 13333. https://doi.org/10.3390/ijerph182413333
APA StyleVerdú, E., Homs, J., & Boadas-Vaello, P. (2021). Physiological Changes and Pathological Pain Associated with Sedentary Lifestyle-Induced Body Systems Fat Accumulation and Their Modulation by Physical Exercise. International Journal of Environmental Research and Public Health, 18(24), 13333. https://doi.org/10.3390/ijerph182413333