
Core-Proteomics-Based Annotation of Antigenic Targets and Reverse-Vaccinology-Assisted Design of Ensemble Immunogen against the Emerging Nosocomial Infection-Causing Bacterium Elizabethkingia meningoseptica


 , , ,
, , ,
Abstract
:1. Introduction
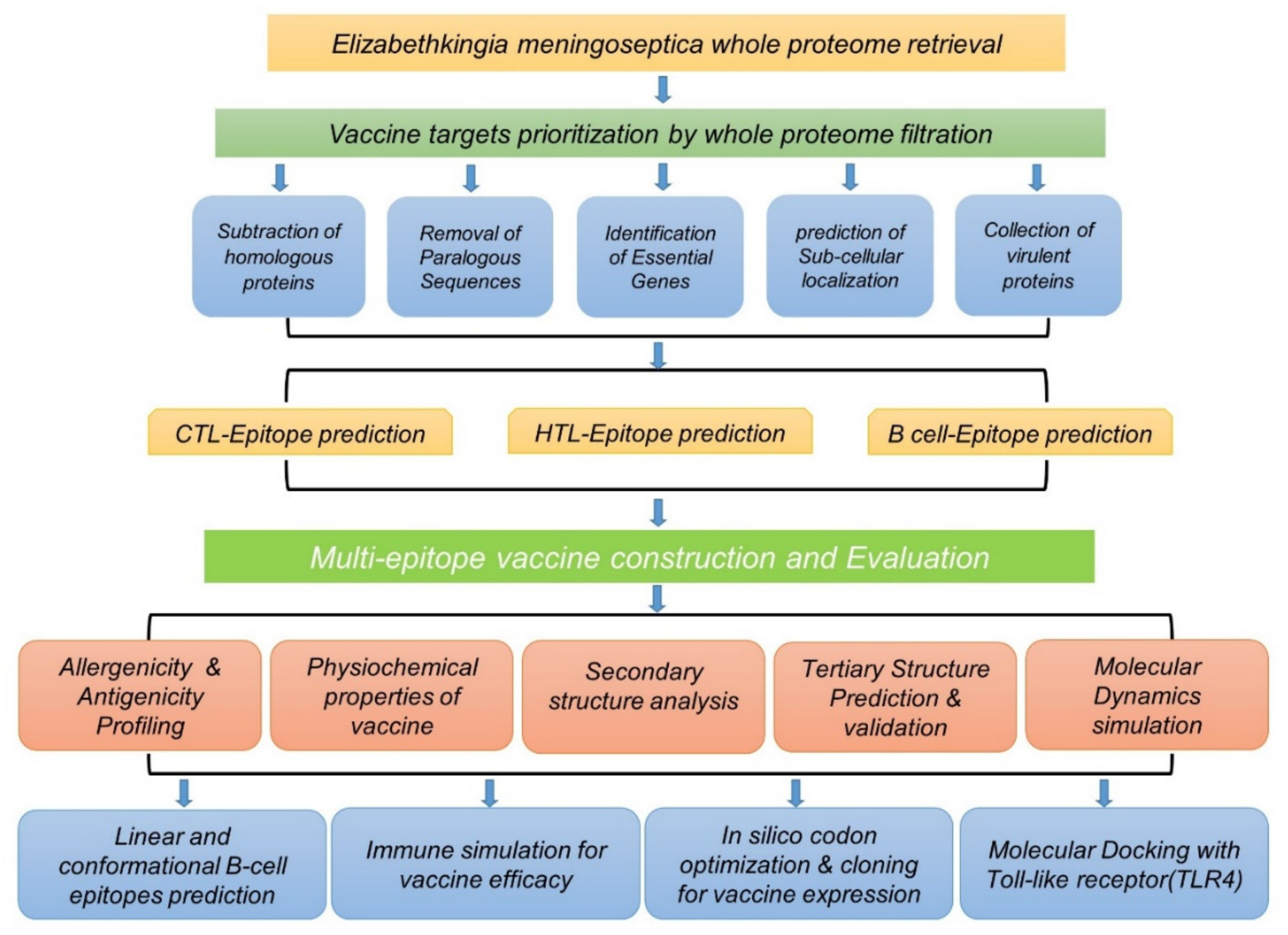
2. Materials and Methods
2.1. Retrieval of Complete Proteome
2.2. Proteome Analysis of Elizabethkingia meningoseptica through Subtractive Proteomics
2.3. Analysis of Subcellular Localization
2.4. Prioritizing Candidates for Potential Insilco Vaccine Construct
2.5. Collection of Virulent Proteins
2.6. Predictions of Epitopes
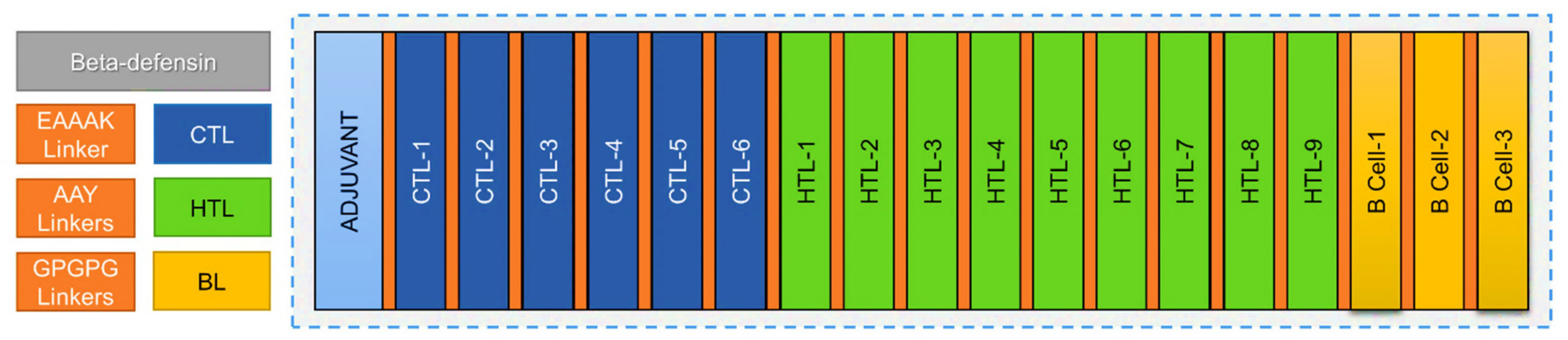
2.7. Multiepitope Vaccine Construction
2.8. Evaluation of Vaccine Construct
2.8.1. Prediction of Allergenicity and Antigenicity
2.8.2. Physiochemical Property Calculations
2.8.3. Tertiary Structure Prediction, Visualization, and Refinement
2.8.4. Validation of the Tertiary Structure
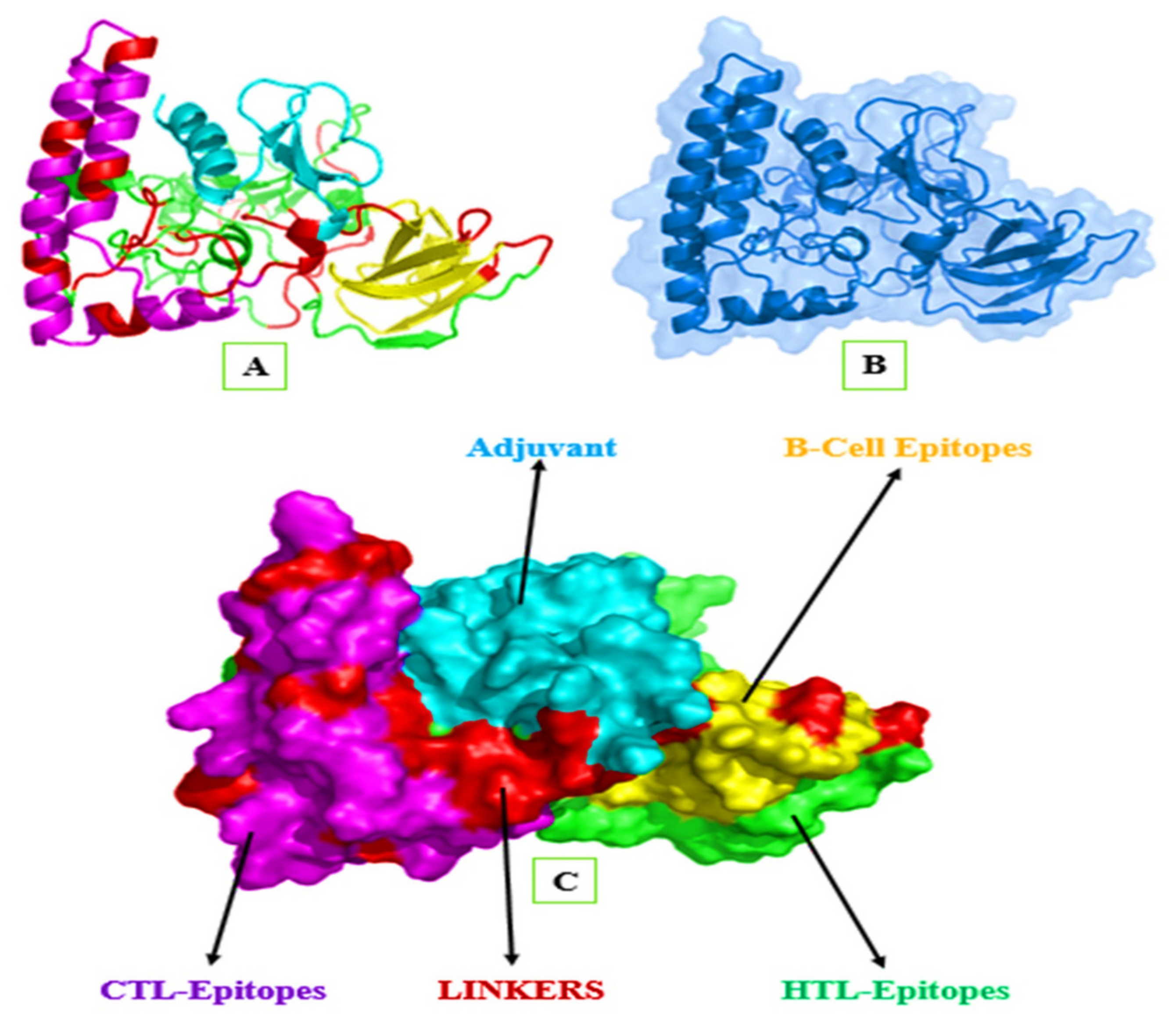
2.8.5. Mapping of the B-Cell Epitopes in the Designed Vaccine’s Tertiary Structure
2.8.6. Molecular Dynamics (MD) Simulations
2.8.7. Immune Simulation for Vaccine Efficacy
2.8.8. Molecular Docking of Our Ligand with TLR4
2.8.9. In Silico Codon Optimization and Cloning of the Vaccine Construct
3. Results and Discussion
3.1. Whole-Proteome Sequence Retrieval
3.2. Removal of Homologous Protein Sequences
3.3. Removal of Paralogous Sequences
3.4. Essential Gene Identification
3.5. Analysis of Sub-Cellular Localization
3.6. Virulent Protein Collection
3.7. Prioritization of Proteins for Final Vaccine Designing and Their Physiochemical Parameters
3.8. Antigenic Epitope Prediction and Prioritization
3.9. Multi-Epitope Immunogen Construction
3.10. Immunogen Property Evaluation
3.10.1. Allergenicity and Antigenicity Status Evaluation
3.10.2. Identification of Physiochemical Properties
3.10.3. Secondary and Tertiary Structure Predictions and Analysis
3.10.4. Modeling of the 3D Structure of MEIC
3.10.5. Refinement of Tertiary Structure
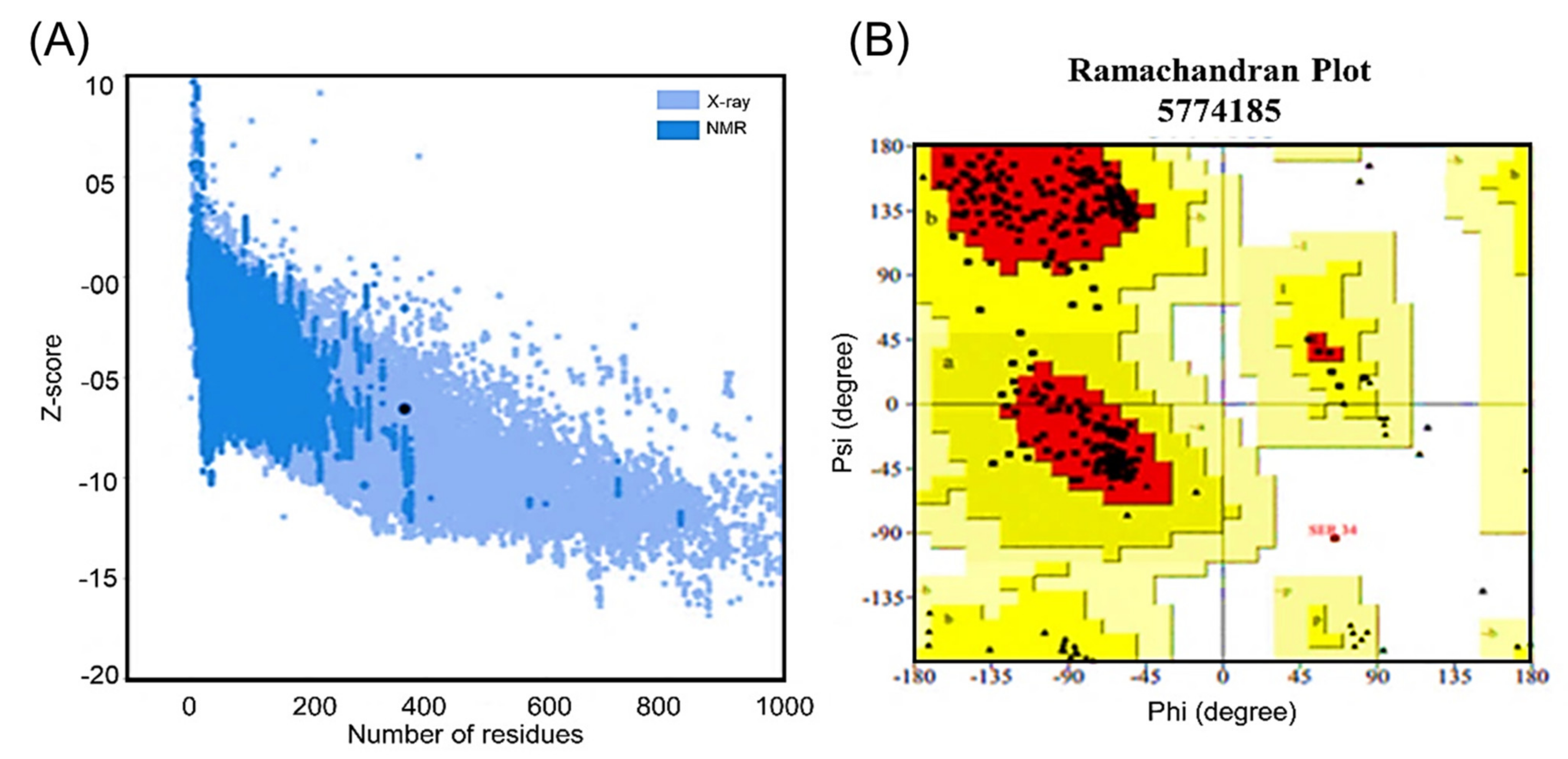
3.10.6. Validation of the Tertiary Structure
3.10.7. Tertiary Immunogen Structure Validation through Molecular Dynamics (MD) Simulation
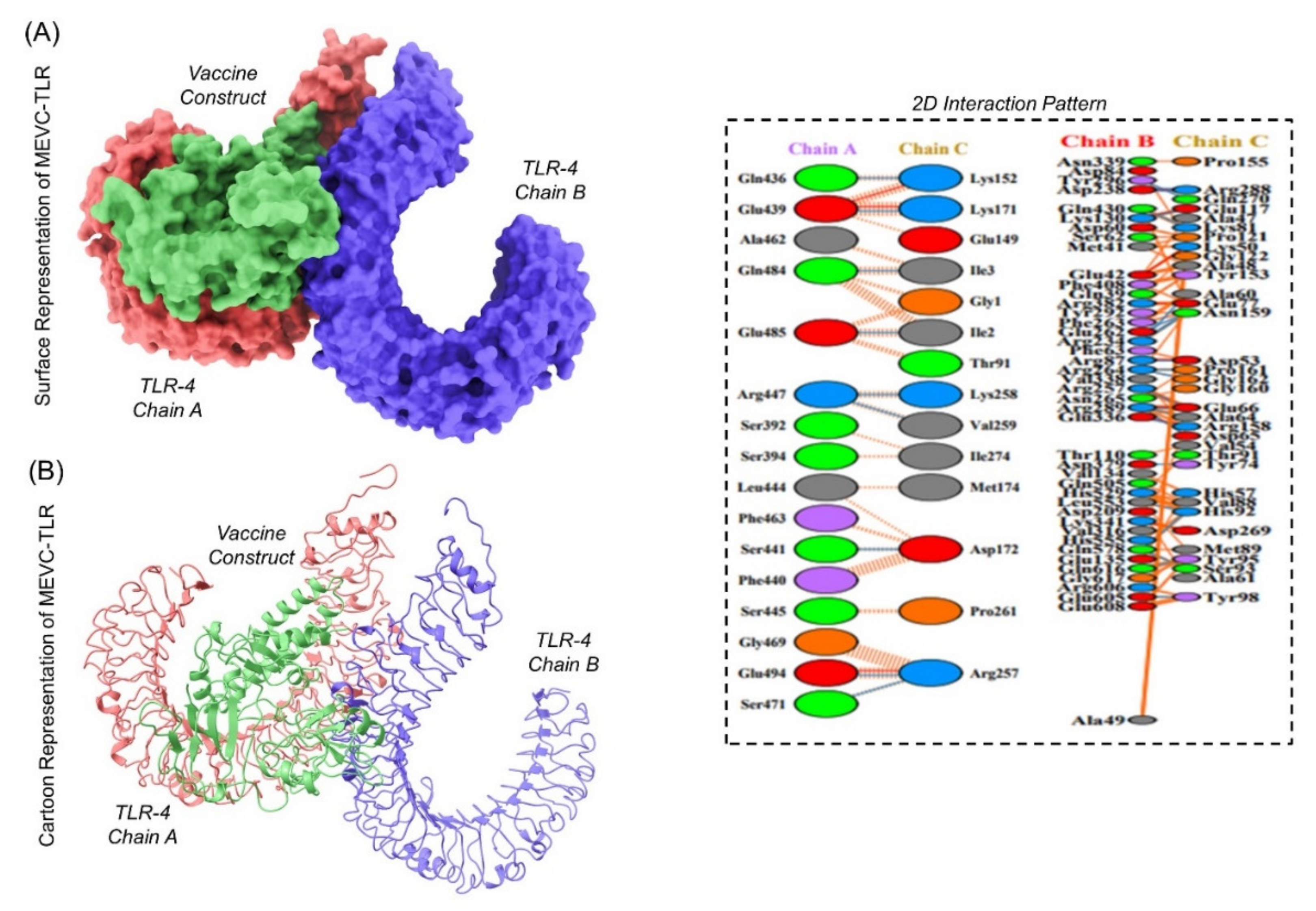
3.10.8. Analysis of Immunogenic Construct Interaction with TLR’s
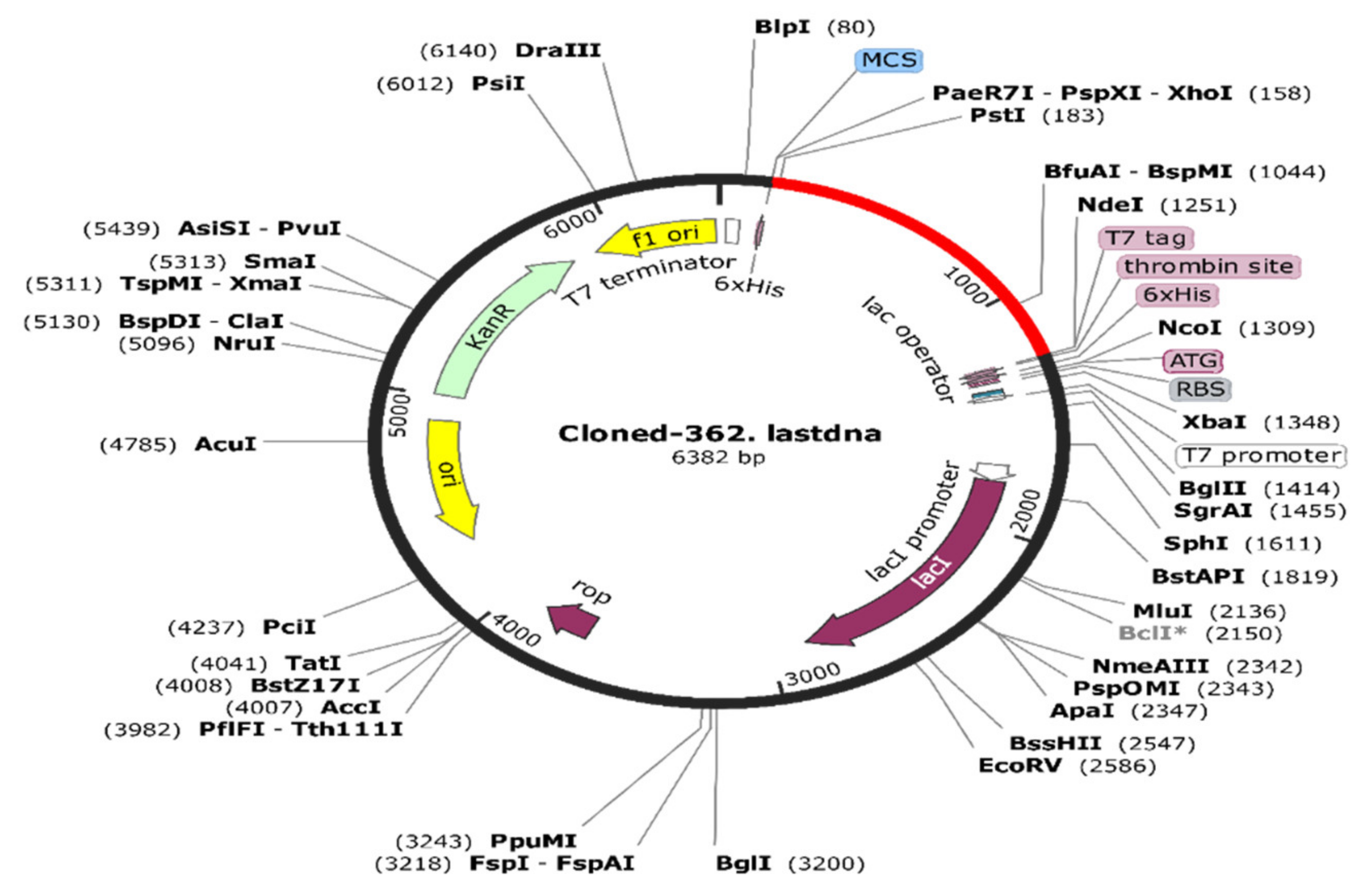
3.10.9. Codon Optimization and In Silico Cloning of the Final Immunogen
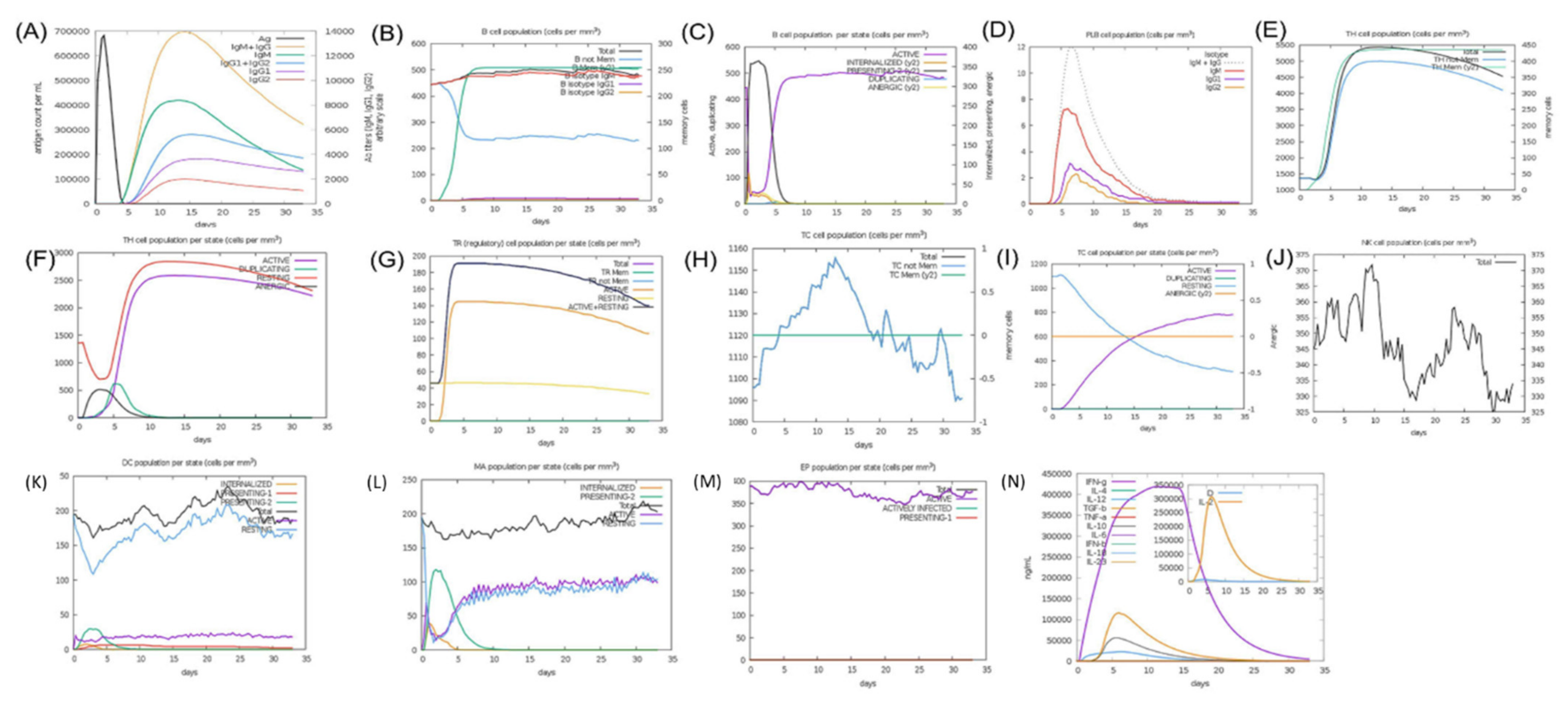
3.10.10. Immune Simulation for Immunogenic Efficacy Prediction
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sztajnbok, J.; Troster, E. Community-acquired Chryseobacterium meningosepticum pneumonia and sepsis in a previously healthy child. J. Infect. 1998, 37, 310–312. [Google Scholar] [CrossRef]
- Hsu, M.-S.; Liao, C.-H.; Huang, Y.-T.; Liu, C.-Y.; Yang, C.-J.; Kao, K.-L.; Hsueh, P.-R. Clinical features, antimicrobial susceptibilities, and outcomes of Elizabethkingia meningoseptica (Chryseobacterium meningosepticum) bacteremia at a medical center in Taiwan, 1999–2006. Eur. J. Clin. Microbiol. Infect. Dis. 2011, 30, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Jean, S.; Lee, W.; Chen, F.; Ou, T.Y.; Hsueh, P. Elizabethkingia meningoseptica: An important emerging pathogen causing healthcare-associated infections. J. Hosp. Infect. 2014, 86, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Breurec, S.; Criscuolo, A.; Diancourt, L.; Rendueles, O.; Vandenbogaert, M.; Passet, V.; Caro, V.; Rocha, E.P.; Touchon, M.; Brisse, S. Genomic epidemiology and global diversity of the emerging bacterial pathogen Elizabethkingia anophelis. Sci. Rep. 2016, 6, 30379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, S.K.; Chow, W.-N.; Foo, C.-H.; Curreem, S.O.; Lo, G.C.-S.; Teng, J.L.; Chen, J.H.; Ng, R.H.; Wu, A.K.; Cheung, I.Y. Elizabethkingia anophelis bacteremia is associated with clinically significant infections and high mortality. Sci. Rep. 2016, 6, 26045. [Google Scholar] [CrossRef] [PubMed]
- Tuon, F.F.; Campos, L.; de Almeida, G.D.; Gryschek, R.C. Chryseobacterium meningosepticum as a cause of cellulitis and sepsis in an immunocompetent patient. J. Med. Microbiol. 2007, 56, 1116–1117. [Google Scholar] [CrossRef]
- Lin, Y.-T.; Chiu, C.-H.; Chan, Y.-J.; Lin, M.-L.; Yu, K.-W.; Wang, F.-D.; Liu, C.-Y. Clinical and microbiological analysis of Elizabethkingia meningoseptica bacteremia in adult patients in Taiwan. Scand. J. Infect. Dis. 2009, 41, 628–634. [Google Scholar] [CrossRef]
- Chiu, C.-H.; Waddingdon, M.; Greenberg, D.; Schreckenberger, P.C.; Carnahan, A. Atypical Chryseobacterium meningosepticum and meningitis and sepsis in newborns and the immunocompromised, Taiwan. Emerg. Infect. Dis. 2000, 6, 481. [Google Scholar] [CrossRef]
- Lin, P.-Y.; Chu, C.; Su, L.-H.; Huang, C.-T.; Chang, W.-Y.; Chiu, C.-H. Clinical and microbiological analysis of bloodstream infections caused by Chryseobacterium meningosepticum in nonneonatal patients. J. Clin. Microbiol. 2004, 42, 3353–3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceyhan, M.; Yıldırım, I.; Tekelı, A.; Yurdakok, M.; Us, E.; Altun, B.; Kutluk, T.; Cengiz, A.B.; Gurbuz, V.; Barın, C. A Chryseobacterium meningosepticum outbreak observed in 3 clusters involving both neonatal and non-neonatal pediatric patients. Am. J. Infect. Control 2008, 36, 453–457. [Google Scholar] [CrossRef]
- Lambiase, A.; Del Pezzo, M.; Raia, V.; Sepe, A.; Ferri, P.; Rossano, F. Chryseobacterium respiratory tract infections in patients with cystic fibrosis. J. Infect. 2007, 55, 518–523. [Google Scholar] [CrossRef] [PubMed]
- HY, C.C.; Chiu, N.-C.; Li, W.-C.; Huang, F.-Y. Characteristics of neonatal bacterial meningitis in a teaching hospital in Taiwan from 1984–1997. J. Microbiol. Immunol. Infect. Wei Mian Yu Gan Ran Za Zhi 2000, 33, 100–104. [Google Scholar]
- Joshi, P.; Shah, B.; Joshi, V.; Kumar, A.; Singhal, T. Treatment of Elizabethkingia meningoseptica Neonatal Meningitis with Combination Systemic and Intraventricular Therapy. Indian J. Pediatrics 2019, 86, 379–381. [Google Scholar] [CrossRef]
- Steinberg, J.P. Other gram-negative and gram-variable bacilli. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases; Elsevier: Amsterdam, The Netherlands, 2009; p. 3023. [Google Scholar]
- Issack, M.I.; Neetoo, Y. An outbreak of Elizabethkingia meningoseptica neonatal meningitis in Mauritius. J. Infect. Dev. Ctries. 2011, 5, 834–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, G.; Yu, X.; Bao, C.; Wang, L.; Li, M.; Gan, J.; Qu, D.; Ma, J.; Chen, L. Identification and characterization of a novel prokaryotic peptide: N-glycosidase from Elizabethkingia meningoseptica. J. Biol. Chem. 2015, 290, 7452–7462. [Google Scholar] [CrossRef] [Green Version]
- Barnawi, A.I.; Kordy, F.N.; Almuwallad, O.K.; Kassarah, K.A. Early neonatal sepsis and meningitis caused by Elizabethkingia meningoseptica in Saudi Arabia. Saudi Med. J. 2020, 41, 753–756. [Google Scholar] [CrossRef]
- Umair, A.; Nasir, N. Clinical Features and Outcomes of Patients with Elizabethkingia meningoseptica Infection: An Emerging Pathogen. 2020. Available online: https://www.accjournal.org/journal/view.php?doi=10.4266/acc.2020.01158 (accessed on 7 August 2021).
- Ahmed, K.; Qudsia, S.A.; Rehman, A.; Abidi, S.H. Fatal Elizabethkingia meningoseptica Cholangitis Following Biliary Stent Placement. 2018. Available online: https://ecommons.aku.edu/pakistan_fhs_mc_bbs/335/ (accessed on 7 August 2021).
- Chawla, K.; Gopinathan, A.; Varma, M.; Mukhopadhyay, C. Elizabethkingia meningoseptica outbreak in intensive care unit. J. Glob. Infect. Dis. 2015, 7, 43. [Google Scholar] [CrossRef]
- Alfouzan, W.; Dhar, R.; Al-Hashemi, H.; Al-Sweih, N.; Albert, M.J. Clinical and microbiological characteristics of Chryseobacterium spp. isolated from neonates in Kuwait. JMM Case Rep. 2014, 1, e001008. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-H.; Lin, C.-H.; Lin, J.-S. Bacteremia caused by Elizabethkingia meningoseptica in a mechanically ventilated patient successfully treated with imipenem-cilastatin and ciprofloxacin. Rev. Inst. Med. Trop. São Paulo 2017, 59, e26. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Soehnlen, M.; Blom, J.; Terrapon, N.; Henrissat, B.; Walker, E.D. Comparative genomic analyses reveal diverse virulence factors and antimicrobial resistance mechanisms in clinical Elizabethkingia meningoseptica strains. PLoS ONE 2019, 14, e0222648. [Google Scholar] [CrossRef] [Green Version]
- Meza, B.; Ascencio, F.; Sierra-Beltrán, A.P.; Torres, J.; Angulo, C. A novel design of a multi-antigenic, multistage and multi-epitope vaccine against Helicobacter pylori: An in silico approach. Infect. Genet. Evol. 2017, 49, 309–317. [Google Scholar] [CrossRef]
- Lin, J.-N.; Lai, C.-H.; Yang, C.-H.; Huang, Y.-H. Elizabethkingia infections in humans: From genomics to clinics. Microorganisms 2019, 7, 295. [Google Scholar] [CrossRef] [Green Version]
- Yum, J.H.; Lee, E.Y.; Hur, S.-H.; Jeong, S.H.; Lee, H.; Yong, D.; Chong, Y.; Lee, E.-W.; Nordmann, P.; Lee, K. Genetic diversity of chromosomal metallo-β-lactamase genes in clinical isolates of Elizabethkingia meningoseptica from Korea. J. Microbiol. 2010, 48, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-X.; Zhang, R.; Zhou, H.W. Heterogeneity of metallo-β-lactamases in clinical isolates of Chryseobacterium meningosepticum from Hangzhou, China. J. Antimicrob. Chemother. 2006, 57, 750–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vessillier, S.; Docquier, J.-D.; Rival, S.; Frere, J.-M.; Galleni, M.; Amicosante, G.; Rossolini, G.M.; Franceschini, N. Overproduction and biochemical characterization of the Chryseobacterium meningosepticum BlaB metallo-β-lactamase. Antimicrob. Agents Chemother. 2002, 46, 1921–1927. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.-H.; Xu, Y.-H.; Sun, X.-H.; Huang, Y.; Li, J.-B. Genetic diversity analyses of antimicrobial resistance genes in clinical Chryseobacterium meningosepticum isolated from Hefei, China. Int. J. Antimicrob. Agents 2012, 40, 186–188. [Google Scholar] [CrossRef] [PubMed]
- González, L.J.; Vila, A.J. Carbapenem resistance in Elizabethkingia meningoseptica is mediated by metallo-β-lactamase BlaB. Antimicrob. Agents Chemother. 2012, 56, 1686–1692. [Google Scholar] [CrossRef] [Green Version]
- Güngör, S.; Özen, M.; Akinci, A.; Durmaz, R. A Chryseobacterium meningosepticum outbreak in a neonatal ward. Infect. Control Hosp. Epidemiol. 2003, 24, 613–617. [Google Scholar] [CrossRef]
- Hoque, S.; Graham, J.; Kaufmann, M.; Tabaqchali, S. Chryseobacterium (Flavobacterium) meningosepticum outbreak associated with colonization of water taps in a neonatal intensive care unit. J. Hosp. Infect. 2001, 47, 188–192. [Google Scholar] [CrossRef]
- Hsueh, P.-R.; Teng, L.-J.; Yang, P.-C.; Ho, S.-W.; Luh, K.-T. Susceptibilities of Chryseobacterium indologenes and Chryseobacterium meningosepticum to cefepime and cefpirome. J. Clin. Microbiol. 1997, 35, 3323–3324. [Google Scholar] [CrossRef] [Green Version]
- Vakili, B.; Nezafat, N.; Hatam, G.R.; Zare, B.; Erfani, N.; Ghasemi, Y. Proteome-scale identification of Leishmania infantum for novel vaccine candidates: A hierarchical subtractive approach. Comput. Biol. Chem. 2018, 72, 16–25. [Google Scholar] [CrossRef]
- Doria-Rose, N.A.; Joyce, M.G. Strategies to guide the antibody affinity maturation process. Curr. Opin. Virol. 2015, 11, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Kumar Pandey, R.; Ojha, R.; Mishra, A.; Kumar Prajapati, V. Designing B-and T-cell multi-epitope based subunit vaccine using immunoinformatics approach to control Zika virus infection. J. Cell. Biochem. 2018, 119, 7631–7642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Air, G.M. Influenza virus antigenicity and broadly neutralizing epitopes. Curr. Opin. Virol. 2015, 11, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Solanki, V.; Tiwari, M.; Tiwari, V. Prioritization of potential vaccine targets using comparative proteomics and designing of the chimeric multi-epitope vaccine against Pseudomonas aeruginosa. Sci. Rep. 2019, 9, 5240. [Google Scholar] [CrossRef] [PubMed]
- Magrane, M. UniProt Knowledgebase: A hub of integrated protein data. Database 2011, 2011, bar009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, R.; Siddiqui, Q.N.; Azam, S.S.; Saima, B.; Wadood, A. Identification and characterization of potential druggable targets among hypothetical proteins of extensively drug resistant Mycobacterium tuberculosis (XDR KZN 605) through subtractive genomics approach. Eur. J. Pharm. Sci. 2018, 114, 13–23. [Google Scholar] [CrossRef]
- Mahram, A.; Herbordt, M.C. Fast and Accurate NCBI BLASTP: Acceleration with Multiphase FPGA-Based Prefiltering. In Proceedings of the 24th ACM International Conference on Supercomputing, Tsukuba, Japan, 2–4 June 2010; pp. 73–82. [Google Scholar]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Ou, H.Y.; Zhang, C.T. DEG: A database of essential genes. Nucleic Acids Res. 2004, 32, D271–D272. [Google Scholar] [CrossRef]
- Bakheet, T.M.; Doig, A.J. Properties and identification of antibiotic drug targets. BMC Bioinform. 2010, 11, 195. [Google Scholar] [CrossRef] [Green Version]
- Shuvo, M.S.R.; Shakil, S.K.; Ahmed, F. Potential Drug Target Identification of Legionella pneumophila by Subtractive Genome Analysis: An In Silico Approach. Bangladesh J. Microbiol. 2018, 35, 102–107. [Google Scholar] [CrossRef]
- Schirmer, E.C.; Florens, L.; Guan, T.; Yates, J.R.; Gerace, L. Nuclear membrane proteins with potential disease links found by subtractive proteomics. Science 2003, 301, 1380–1382. [Google Scholar] [CrossRef] [PubMed]
- Gul, H.; Ali, S.S.; Saleem, S.; Khan, S.; Khan, J.; Wadood, A.; Rehman, A.U.; Ullah, Z.; Ali, S.; Khan, H. Subtractive proteomics and immunoinformatics approaches to explore Bartonella bacilliformis proteome (virulence factors) to design B and T cell multi-epitope subunit vaccine. Infect. Genet. Evol. 2020, 85, 104551. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Ali, S.S.; Zaheer, I.; Saleem, S.; Ziaullah; Zaman, N.; Iqbal, A.; Suleman, M.; Wadood, A.; Rehman, A.U. Proteome-wide mapping and reverse vaccinology-based B and T cell multi-epitope subunit vaccine designing for immune response reinforcement against Porphyromonas gingivalis. J. Biomol. Struct. Dyn. 2020, 1–15. [Google Scholar] [CrossRef]
- Chen, L.; Yang, J.; Yu, J.; Yao, Z.; Sun, L.; Shen, Y.; Jin, Q. VFDB: A reference database for bacterial virulence factors. Nucleic Acids Res. 2005, 33, D325–D328. [Google Scholar] [CrossRef] [Green Version]
- Pandey, R.K.; Bhatt, T.K.; Prajapati, V.K. Novel immunoinformatics approaches to design multi-epitope subunit vaccine for malaria by investigating Anopheles salivary protein. Sci. Rep. 2018, 8, 1125. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Khan, A.; Rehman, A.U.; Ahmad, I.; Ullah, S.; Khan, A.A.; Ali, S.S.; Afridi, S.G.; Wei, D.-Q. Immunoinformatics and structural vaccinology driven prediction of multi-epitope vaccine against Mayaro virus and validation through in-silico expression. Infect. Genet. Evol. 2019, 73, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Rapin, N.; Lund, O.; Bernaschi, M.; Castiglione, F. Computational immunology meets bioinformatics: The use of prediction tools for molecular binding in the simulation of the immune system. PLoS ONE 2010, 5, e9862. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession/Cellular Location | Sequence Length | Bit-Sore | Percent Identity | Molecular Weight | Antigenicity Value | Allergenicity |
|---|---|---|---|---|---|---|
| A0A1T3FLU2 | 207aa | 158 bits | 40% | 23.32 kDa | 0.5802 | Non-allergenic |
| A0A1T3INK9 | 243aa | 127 bits | 36% | 27.48 kDa | 0.5383 | Non-allergenic |
| A0A1V3U124 | 505aa | 237 bits | 37% | 54.29 kDa | 0.5170 | Non-allergenic |
| Cellular Location/Accession | Type of Epitope | Epitope Sequence | Epitope Length | (MHC) Binding Affinity | Combined Score |
|---|---|---|---|---|---|
| periplasmic (A0A1T3FLU) | CTL | GVDVWEHAY | 9 | 0.5794 | 2.7407 |
| CTL | LADEINAAF | 9 | 0.2387 | 1.2355 | |
| outer membrane (A0A1T3INK9) | CTL | TIEKLSKIY | 9 | 0.2651 | 1.4033 |
| CTL | IVMVTHSSY | 9 | 0.1531 | 0.9540 | |
| outer membrane (A0A1V3U124) | CTL | ATTVGTLHY | 9 | 0.6437 | 3.0377 |
| CTL | SSTGYYEGY | 9 | 0.4328 | 2.1323 |
| Cellular Location/Accession | HTL S. No | Type of Allele | Epitope Sequence | Start-End | Percentile Ranks | Allergenicity Values |
|---|---|---|---|---|---|---|
| periplasmic (A0A1T3FLU) | 1st | HLA-DRB3*02:02 | MDTQLTSRGFPILGV | 151–165 | 22 | −1.068 |
| 2nd | HLA-DRB3*02:02 | EILPEISKYSPAVRN | 56–70 | 33 | −0.567 | |
| 3rd | HLA-DRB1*03:01 | LEPHFDKDTMSIHHQ | 16–30 | 55 | −0.521 | |
| outer membrane (A0A1T3INK9) | 1st | HLA-DRB3*02:02 | LPLIYNGVSSSERRK | 106–120 | 0.75 | −0.720 |
| 2nd | HLA-DRB1*07:01 | LLAELHREGSTIVMV | 183–197 | 14 | −0.898 | |
| 3rd | HLA-DRB3*01:01 | LSKIYRTEDVQTTAL | 7–21 | 9.7 | −0.658 | |
| outer membrane (A0A1V3U124) | 1st | HLA-DRB5*01:01 | EGYGFAVPSNLARKV | 272–286 | 6.9 | −0.886 |
| 2nd | HLA-DRB3*01:01 | TSFADQSLAIGSKRP | 362–376 | 28 | −0.591 | |
| 3rd | HLA-DRB4*01:01 | AKGRSIDLLSQQSKT | 215–229 | 4.1 | −0.542 |
| Cellular Location/Accession | Type of Epitope | Epitope Sequence | Position | aa Length | Combined Scores |
|---|---|---|---|---|---|
| periplasmic (A0A1T3FLU) | B-cell | TMSIHHQRHHQAYVDN | 24 | 16 | 0.96 |
| B-cell | ATANQDNPLMDTQLTS | 142 | 16 | 0.92 | |
| outer membrane (A0A1T3INK9) | B-cell | THSSYDASFSSRIVNM | 198 | 16 | 0.88 |
| B-cell | EIMEKINIKHRAKHYP | 124 | 16 | 0.88 | |
| outer membrane (A0A1V3U124) | B-cell | DGYIITNNHVVANANK | 129 | 16 | 0.96 |
| B-cell | DVVKVTYLRNGKESTT | 378 | 16 | 0.91 |
| Models | GDT-HAs | RMSDs | Mol-Probity | Clash Scores | Poor Rotamers | Rama Favored |
|---|---|---|---|---|---|---|
| Initial model | 1.0000 | 0.000 | 1.175 | 3.9 | 0.0 | 98.1 |
| MODEL 1 | 0.9883 | 0.318 | 1.690 | 15.4 | 0.7 | 98.3 |
| MODEL 2 | 0.9779 | 0.336 | 1.639 | 13.5 | 0.7 | 98.3 |
| MODEL 3 | 0.9855 | 0.332 | 1.644 | 13.7 | 0.7 | 98.3 |
| MODEL 4 | 0.9807 | 0.338 | 1.639 | 13.5 | 0.4 | 98.3 |
| MODEL 5 | 0.9786 | 0.357 | 1.709 | 16.1 | 0.4 | 98.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Idrees, M.; Noorani, M.Y.; Altaf, K.U.; Alatawi, E.A.; Aba Alkhayl, F.F.; Allemailem, K.S.; Almatroudi, A.; Ali Khan, M.; Hamayun, M.; Khan, T.; et al. Core-Proteomics-Based Annotation of Antigenic Targets and Reverse-Vaccinology-Assisted Design of Ensemble Immunogen against the Emerging Nosocomial Infection-Causing Bacterium Elizabethkingia meningoseptica. Int. J. Environ. Res. Public Health 2022, 19, 194. https://doi.org/10.3390/ijerph19010194
Idrees M, Noorani MY, Altaf KU, Alatawi EA, Aba Alkhayl FF, Allemailem KS, Almatroudi A, Ali Khan M, Hamayun M, Khan T, et al. Core-Proteomics-Based Annotation of Antigenic Targets and Reverse-Vaccinology-Assisted Design of Ensemble Immunogen against the Emerging Nosocomial Infection-Causing Bacterium Elizabethkingia meningoseptica. International Journal of Environmental Research and Public Health. 2022; 19(1):194. https://doi.org/10.3390/ijerph19010194
Chicago/Turabian StyleIdrees, Muhammad, Muhammad Yasir Noorani, Kalim Ullah Altaf, Eid A. Alatawi, Faris F. Aba Alkhayl, Khaled S. Allemailem, Ahmad Almatroudi, Murad Ali Khan, Muhammad Hamayun, Taimoor Khan, and et al. 2022. "Core-Proteomics-Based Annotation of Antigenic Targets and Reverse-Vaccinology-Assisted Design of Ensemble Immunogen against the Emerging Nosocomial Infection-Causing Bacterium Elizabethkingia meningoseptica" International Journal of Environmental Research and Public Health 19, no. 1: 194. https://doi.org/10.3390/ijerph19010194