
Effects of Particulate Matter on Inflammation and Thrombosis: Past Evidence for Future Prevention

 , and
, and
Abstract
:
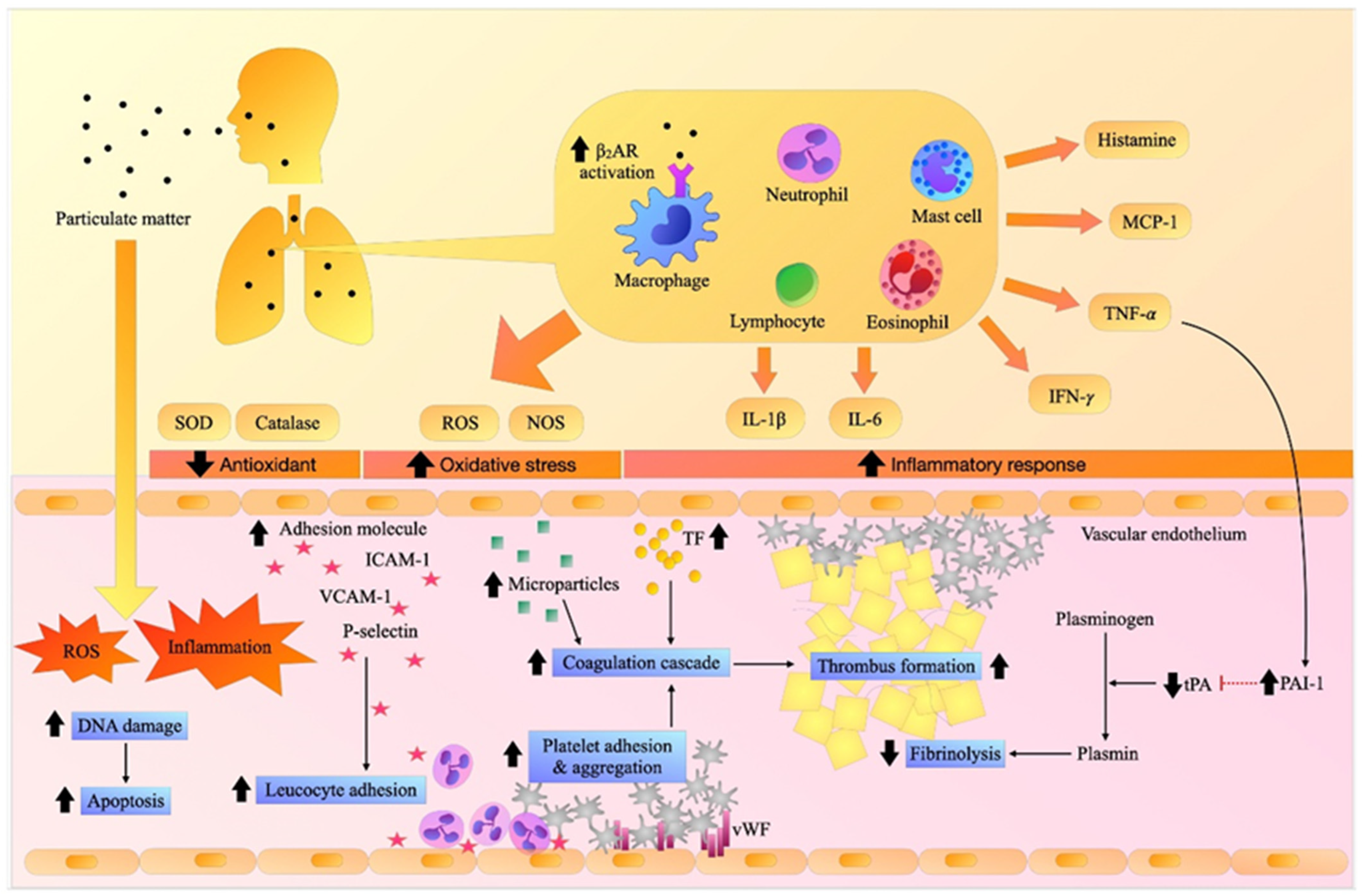
1. Introduction
2. Hemostasis and the Fibrinolytic Pathway
3. The Effects of Particulate Matter on Inflammation, Oxidative Stress, and the Coagulation System: Reports from In Vitro Studies
4. The Effects of Particulate Matter on Inflammation, Oxidative Stress, and the Coagulation System: Reports from In Vivo Studies
5. The effects of Diesel Exhaust Particles (DEP) on Inflammation, Oxidative Stress, and Coagulation Systems: Reports from In Vivo Studies
6. The Effects of Particulate Matter on Inflammation and the Coagulation System: Reports from Clinical Studies

{kind=link}
{kind=link}
| Models | Exposure/Method | Results | Interpretation | References | ||
|---|---|---|---|---|---|---|
| Inflammation | Coagulation & Adhesion Molecules | Blood Parameters | ||||
| Healthy young adults (N = 16) Mean age 25 y | Inhalation of # Petrodiesel exhaust (PDE) # Mixture of biodiesel 30% and 70% of petrodiesel from rapeseed methyl ester (RME30) # Biodiesel 100% from rapeseed methyl ester (RME100) For 1 h Evaluation at 2, 4, 8, 24 h after exposure | ↔ tPA ↔ thrombus formation | ↔ Hb, WBC, platelet | Inhalation exposure of biodiesel formulation (RME30, RME100) did not alter coagulation and blood cell parameters in comparison to PDE. | [77] | |
| # Healthy adults (N = 15) Mean age: 28 y # Metabolic syndrome patients (N = 17) mean age 40 y | Inhalation of # DEP # Filtered fresh air (control) for 2 h Evaluate at pre-exposure, 7 and 22 h after exposure | ↔ MMP-9 ↔ IL-1β ↔ IL-6 ↔ IL-10 | ↔ E-selectin ↔ ICAM-1, VCAM-1 | # Both healthy and metabolic syndrome subjects: ↑ Hct ↔ Hb, RBC ↑ platelet ↔ WBC | Short-term DEP exposure resulted in hemoconcentration & thrombocytosis but did not affect inflammatory response and endothelial cell activation in both healthy and metabolic syndrome subjects. | [71] |
| Healthy adults (N = 73) Mean age 23.3 y | Ambient air pollution in an urban area, Beijing, China Evaluate the PM level at 1, 2, 3, 5, and 7 d MA | # PM2.5: ↑ sRAGE ↑ MIP-1α, β ↔ IL-1β, CRP # BC: ↑ sRAGE ↑ MIP-1α, β ↔ IL-1β, CRP | # PM2.5: ↑ P-selectin ↑ sCD40L ↔ PT ↑ FDP # BC: ↑ P-selectin ↔ sCD40L ↓ PT ↑ FDP | Exposure to higher ambient air pollution was associated with increased inflammatory biomarkers and heightened thrombogenicity. | [69] | |
| Healthy young adults (N = 125) Mean age 24.2 y | Ambient air pollution in Beijing, China # Pre-Olympic period # During Olympics (Strict pollution control) # Post-Olympics period Evaluate the PM level at 1, 12, 24, 48, 96 h | # PM2.5, EC: ↑ P-selectin, sCD40L ↑ fibrinogen, VWF # OC: ↑ P-selectin, ↔ sCD40L ↑ fibrinogen, VWF # Pre- vs. During Olympic period: ↓ P-selectin, sCD40L ↔ fibrinogen ↓ VWF # During vs. post-Olympic period: ↑ P-selectin, ↔ sCD40L ↔ fibrinogen ↔ VWF | # PM, OC, EC: ↔ WBC # Pre- vs. During Olympic period: ↔ WBC # During vs. post-Olympic period: ↔ WBC | The restricted air pollution control markedly reduced PM, which was associated with decreased platelet activation and prothrombotic state. The alteration of PM level did not affect WBC count. | [76] | |
| Elderly individuals with either CVD, or COPD and healthy individuals (N = 47) Mean age 78 y | Ambient air pollution fine PM outside of each individuals’ homes Seattle, WA, USA Evaluation of the PM level at the zero d and 1 d MA | ↔ CRP ↑ MCP-1 | ↔ fibrinogen ↔ D-dimer | The effects of low ambient levels of PM on inflammation or thrombosis were not significant in elderly individuals. | [70] | |
| Healthy elderly (N = 704) Mean age 73.2 y | Ambient Air Pollution in Boston, USA Evaluate the PM level at 4, 24 h, 3, 7, 14, 21, and 28 d MA | # PM2.5: ↔ CRP # BC: ↔ CRP | # PM2.5: ↑ ICAM-1, VCAM-1 ↔ fibrinogen # BC: ↑ ICAM-1, VCAM-1 ↑ fibrinogen | Short-term (1–3 d MA), and intermediate-term (7–28 d MA) exposure to traffic-related air pollution were associated with alteration of adhesion molecules, reflecting acute inflammatory and endothelial responses. | [73] | |
| Adult patients undergoing cardiac catheterization due to stable IHD or ACS (N = 135) Mean age 61.4 y | Ambient air pollution Rochester, NY, USA Evaluate the PM level at 1, 12, 24, 48, 72, and 96 h MA | # PM2.5: ↑ CRP # Delta-C, AMP: ↔ CRP # BC: ↑ CRP # UFP: ↔ CRP | # PM2.5, Delta-C, BC: ↑ PF4 ↑ fibrinogen ↔ VWF, D-dimer # AMP ↑ PF4 ↑ fibrinogen ↔ P-selectin ↔ VWF, D-dimer # UFP: ↓ PF4 ↑ fibrinogen ↔ P-selectin ↔ VWF, D-dimer | The high PM was generally associated with an increase in biomarkers of systemic inflammation and coagulation. | [72] | |
| Patients with CAD or at least two CVD comorbid diseases (HT, DM, hyperlipidemia) (N = 61) Mean age 62.3 y | Ambient air pollution In an urban area, Taipei City, Taiwan Evaluate the PM level at 1 to 3 d MA | # PM2.5, OC, EC: ↔ hsCRP | # PM2.5: ↔ fibrinogen ↔ D-dimer # OC, EC: ↔ fibrinogen ↑ D-dimer | Short-term exposure (1–3 d) to urban pollution triggered systemic inflammatory and thrombotic response in high-risk CVD patients. | [75] | |
| DM type II (N = 30) Mean age 56.5 y | Acute exposure to ambient PM in Rochester, NY, USA Evaluate the PM level at 1, 12, 24, 48, 96 h | # PM2.5, AMP: ↔ TBXB2 ↔ ADP-, and collagen-induced platelet aggregation # UFP: ↔ TBXB2 ↔ ADP-induced platelet aggregation ↓ collagen-induced platelet aggregation at 48–96 h MA # BC: ↓ TBXB2 at 48–96 h MA ↔ ADP-, and collagen-induced platelet aggregation | High UFP levels were associated with reduced platelet response, whereas PM2.5, AMP, and BC resulted in a trend of increased platelet aggregation. | [78] | ||
7. In Vitro Interventional Reports on the Effects of Particulate Matter on Inflammation, Oxidative Stress, and Coagulation System
8. In Vivo and Clinical Interventional Reports on the Effects of Particulate Matter on Inflammation, Oxidative Stress, and the Coagulation System
9. Limitation of the Current Studies and Direction for Future
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lelieveld, J.; Evans, J.S.; Fnais, M.; Giannadaki, D.; Pozzer, A. The contribution of outdoor air pollution sources to premature mortality on a global scale. Nature 2015, 525, 367–371. [Google Scholar] [CrossRef]
- Brook, R.D.; Newby, D.E.; Rajagopalan, S. The Global Threat of Outdoor Ambient Air Pollution to Cardiovascular Health: Time for Intervention. JAMA Cardiol. 2017, 2, 353–354. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.; Miller, M.R. Ambient air pollution and thrombosis. Part. Fibre Toxicol. 2018, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Jilani, M.H.; Simon-Friedt, B.; Yahya, T.; Khan, A.Y.; Hassan, S.Z.; Kash, B.; Blankstein, R.; Blaha, M.J.; Virani, S.S.; Rajagopalan, S.; et al. Associations between particulate matter air pollution, presence and progression of subclinical coronary and carotid atherosclerosis: A systematic review. Atherosclerosis 2020, 306, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Sun, Q. Fine particulate matter air pollution and atherosclerosis: Mechanistic insights. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2016, 1860, 2863–2868. [Google Scholar] [CrossRef]
- Arias-Pérez, R.D.; Taborda, N.A.; Gómez, D.M.; Narvaez, J.F.; Porras, J.; Hernandez, J.C. Inflammatory effects of particulate matter air pollution. Environ. Sci. Pollut. Res. Int. 2020, 27, 42390–42404. [Google Scholar] [CrossRef]
- Li, X.Y.; Yu, X.B.; Liang, W.W.; Yu, N.; Wang, L.; Ye, X.J.; Chen, K.; Bian, P.D. Meta-analysis of association between particulate matter and stroke attack. CNS Neurosci. Ther. 2012, 18, 501–508. [Google Scholar] [CrossRef]
- Huang, K.; Liang, F.; Yang, X.; Liu, F.; Li, J.; Xiao, Q.; Chen, J.; Liu, X.; Cao, J.; Shen, C.; et al. Long term exposure to ambient fine particulate matter and incidence of stroke: Prospective cohort study from the China-PAR project. BMJ 2019, 367, l6720. [Google Scholar] [CrossRef] [Green Version]
- Pope, C.A.; Muhlestein, J.B.; Anderson, J.L.; Cannon, J.B.; Hales, N.M.; Meredith, K.G.; Le, V.; Horne, B.D. Short-Term Exposure to Fine Particulate Matter Air Pollution Is Preferentially Associated with the Risk of ST-Segment Elevation Acute Coronary Events. J. Am. Heart Assoc. 2015, 4, e002506. [Google Scholar] [CrossRef] [Green Version]
- Kuźma, Ł.; Pogorzelski, S.; Struniawski, K.; Dobrzycki, S.; Bachórzewska-Gajewska, H. Effect of air pollution on the number of hospital admissions for acute coronary syndrome in elderly patients. Pol. Arch. Intern. Med. 2020, 130, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Zhang, Y.; Yang, K.Q.; Yang, Y.K.; Zhou, X.L. Potential Harmful Effects of PM2.5 on Occurrence and Progression of Acute Coronary Syndrome: Epidemiology, Mechanisms, and Prevention Measures. Int. J. Environ. Res. Public Health 2016, 13, 748. [Google Scholar] [CrossRef]
- Tang, L.; Wang, Q.Y.; Cheng, Z.P.; Hu, B.; Liu, J.D.; Hu, Y. Air pollution and venous thrombosis: A meta-analysis. Sci. Rep. 2016, 6, 32794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baccarelli, A.; Martinelli, I.; Zanobetti, A.; Grillo, P.; Hou, L.F.; Bertazzi, P.A.; Mannucci, P.M.; Schwartz, J. Exposure to particulate air pollution and risk of deep vein thrombosis. Arch. Intern. Med. 2008, 168, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Renzi, M.; Stafoggia, M.; Michelozzi, P.; Davoli, M.; Forastiere, F.; Solimini, A.G. Short-term exposure to PM(2.5) and risk of venous thromboembolism: A case-crossover study. Thromb. Res. 2020, 190, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Secrest, M.H.; Schauer, J.J.; Carter, E.M.; Baumgartner, J. Particulate matter chemical component concentrations and sources in settings of household solid fuel use. Indoor Air 2017, 27, 1052–1066. [Google Scholar] [CrossRef] [PubMed]
- Daellenbach, K.R.; Uzu, G.; Jiang, J.; Cassagnes, L.E.; Leni, Z.; Vlachou, A.; Stefenelli, G.; Canonaco, F.; Weber, S.; Segers, A.; et al. Sources of particulate-matter air pollution and its oxidative potential in Europe. Nature 2020, 587, 414–419. [Google Scholar] [CrossRef]
- Pope, C.A., 3rd. Epidemiology of fine particulate air pollution and human health: Biologic mechanisms and who’s at risk? Environ. Health Perspect. 2000, 108 (Suppl. S4), 713–723. [Google Scholar] [CrossRef]
- Brook, R.D.; Franklin, B.; Cascio, W.; Hong, Y.; Howard, G.; Lipsett, M.; Luepker, R.; Mittleman, M.; Samet, J.; Smith, S.C., Jr.; et al. Air pollution and cardiovascular disease: A statement for healthcare professionals from the Expert Panel on Population and Prevention Science of the American Heart Association. Circulation 2004, 109, 2655–2671. [Google Scholar] [CrossRef]
- Wichmann, H.E. Diesel exhaust particles. Inhal. Toxicol. 2007, 19 (Suppl. S1), 241–244. [Google Scholar] [CrossRef]
- Crobeddu, B.; Aragao-Santiago, L.; Bui, L.C.; Boland, S.; Baeza Squiban, A. Oxidative potential of particulate matter 2.5 as predictive indicator of cellular stress. Environ. Pollut. 2017, 230, 125–133. [Google Scholar] [CrossRef]
- Cattani-Cavalieri, I.; Valenca, S.S.; Lanzetti, M.; Carvalho, G.M.C.; Zin, W.A.; Monte-Alto-Costa, A.; Porto, L.C.; Romana-Souza, B. Acute Exposure to Diesel-Biodiesel Particulate Matter Promotes Murine Lung Oxidative Stress by Nrf2/HO-1 and Inflammation Through the NF-kB/TNF-α Pathways. Inflammation 2019, 42, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Guo, Z.; Zhang, R.; Xu, J.; Dong, W.; Zhuang, G.; Deng, C. Airborne Fine Particulate Matter Induces Oxidative Stress and Inflammation in Human Nasal Epithelial Cells. Tohoku J. Exp. Med. 2016, 239, 117–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riva, D.R.; Magalhães, C.B.; Lopes, A.A.; Lanças, T.; Mauad, T.; Malm, O.; Valença, S.S.; Saldiva, P.H.; Faffe, D.S.; Zin, W.A. Low dose of fine particulate matter (PM2.5) can induce acute oxidative stress, inflammation and pulmonary impairment in healthy mice. Inhal. Toxicol. 2011, 23, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.W.; Lee, T.L.; Chen, Y.C.; Liang, C.J.; Wang, S.H.; Lue, J.H.; Tsai, J.S.; Lee, S.W.; Chen, S.H.; Yang, Y.F.; et al. PM(2.5)-induced oxidative stress increases intercellular adhesion molecule-1 expression in lung epithelial cells through the IL-6/AKT/STAT3/NF-κB-dependent pathway. Part. Fibre Toxicol. 2018, 15, 4. [Google Scholar] [CrossRef]
- Ding, L.R.; Wang, K.; Fahmy, B.; Shen, H.H.; Cormier, S. Airborne fine particulate matter induced pulmonary inflammation as well as oxidative stress in neonate rats. Chin. Med. J. 2010, 123, 2895–2900. [Google Scholar]
- Zhu, X.; Chen, C.; Zhang, B.; Ge, Y.; Wang, W.; Cai, J.; Kan, H. Acute effects of personal exposure to fine particulate matter on salivary and urinary biomarkers of inflammation and oxidative stress in healthy adults. Chemosphere 2021, 272, 129906. [Google Scholar] [CrossRef]
- Gangwar, R.S.; Bevan, G.H.; Palanivel, R.; Das, L.; Rajagopalan, S. Oxidative stress pathways of air pollution mediated toxicity: Recent insights. Redox Biol. 2020, 34, 101545. [Google Scholar] [CrossRef]
- Møller, P.; Danielsen, P.H.; Karottki, D.G.; Jantzen, K.; Roursgaard, M.; Klingberg, H.; Jensen, D.M.; Christophersen, D.V.; Hemmingsen, J.G.; Cao, Y.; et al. Oxidative stress and inflammation generated DNA damage by exposure to air pollution particles. Mutat. Res. Rev. Mutat. Res. 2014, 762, 133–166. [Google Scholar] [CrossRef]
- Danielsen, P.H.; Møller, P.; Jensen, K.A.; Sharma, A.K.; Wallin, H.; Bossi, R.; Autrup, H.; Mølhave, L.; Ravanat, J.L.; Briedé, J.J.; et al. Oxidative stress, DNA damage, and inflammation induced by ambient air and wood smoke particulate matter in human A549 and THP-1 cell lines. Chem. Res. Toxicol. 2011, 24, 168–184. [Google Scholar] [CrossRef]
- Riggs, D.W.; Zafar, N.; Krishnasamy, S.; Yeager, R.; Rai, S.N.; Bhatnagar, A.; O’Toole, T.E. Exposure to airborne fine particulate matter is associated with impaired endothelial function and biomarkers of oxidative stress and inflammation. Environ. Res. 2020, 180, 108890. [Google Scholar] [CrossRef]
- Shkirkova, K.; Lamorie-Foote, K.; Connor, M.; Patel, A.; Barisano, G.; Baertsch, H.; Liu, Q.; Morgan, T.E.; Sioutas, C.; Mack, W.J. Effects of ambient particulate matter on vascular tissue: A review. J. Toxicol. Environ. Health B Crit. Rev. 2020, 23, 319–350. [Google Scholar] [CrossRef] [PubMed]
- Forchhammer, L.; Loft, S.; Roursgaard, M.; Cao, Y.; Riddervold, I.S.; Sigsgaard, T.; Møller, P. Expression of adhesion molecules, monocyte interactions and oxidative stress in human endothelial cells exposed to wood smoke and diesel exhaust particulate matter. Toxicol. Lett. 2012, 209, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Calverley, D.C. Platelet Function in Hemostasis and Thrombosis. In Wintrobe’s Clinical Hematology, 14th ed.; Greer, J.P., Rodgers, G.M., Glader, B., Arber, D.A., Jr., Means, R.T., List, A.F., Appelbaum, F.R., Dispenzieri, A., Fehniger, T.A., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2019; pp. 196–418. [Google Scholar]
- Owens, A.P., 3rd; Mackman, N. Microparticles in hemostasis and thrombosis. Circ. Res. 2011, 108, 1284–1297. [Google Scholar] [CrossRef]
- Brummel-Ziedins, K.E.; Orfeo, T.; Everse, S.J.; Mann, K.G. Blood Coagulation and Fibrinolysis. In Wintrobe’s Clinical Hematology, 14th ed.; Greer, J.P., Rodgers, G.M., Glader, B., Arber, D.A., Jr., Means, R.T., List, A.F., Appelbaum, F.R., Dispenzieri, A., Fehniger, T.A., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2019; pp. 419–519. [Google Scholar]
- Peixoto, M.S.; de Oliveira Galvão, M.F.; de Medeiros, S.R.B. Cell death pathways of particulate matter toxicity. Chemosphere 2017, 188, 32–48. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, T.; Tang, M. Ambient particulate matter triggers dysfunction of subcellular structures and endothelial cell apoptosis through disruption of redox equilibrium and calcium homeostasis. J. Hazard. Mater. 2020, 394, 122439. [Google Scholar] [CrossRef] [PubMed]
- Montiel-Davalos, A.; Gonzalez-Villava, A.; Rodriguez-Lara, V.; Montano, L.F.; Fortoul, T.I.; Lopez-Marure, R. Vanadium pentoxide induces activation and death of endothelial cells. J. Appl. Toxicol. 2012, 32, 26–33. [Google Scholar] [CrossRef]
- Gawda, A.; Majka, G.; Nowak, B.; Srottek, M.; Walczewska, M.; Marcinkiewicz, J. Air particulate matter SRM 1648a primes macrophages to hyperinflammatory response after LPS stimulation. Inflamm. Res. 2018, 67, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Chen, H.; Yang, T.; Rui, W.; Liu, F.; Zhang, F.; Zhao, Y.; Ding, W. Direct effects of airborne PM2.5 exposure on macrophage polarizations. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2016, 1860, 2835–2843. [Google Scholar] [CrossRef]
- Yamasaki, K.; Whalen, B.; Van Eeden, S. Impact of particulate matter (PM) exposure on lung macrophage phenotype and phagocytic activity. Eur. Respir. J. 2020, 56, 1968. [Google Scholar] [CrossRef]
- Hiraiwa, K.; van Eeden, S.F. Contribution of lung macrophages to the inflammatory responses induced by exposure to air pollutants. Mediat. Inflamm. 2013, 2013, 619523. [Google Scholar] [CrossRef] [Green Version]
- Sawyer, K.; Mundandhara, S.; Ghio, A.J.; Madden, M.C. The effects of ambient particulate matter on human alveolar macrophage oxidative and inflammatory responses. J. Toxicol. Environ. Health A 2010, 73, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Chiarella, S.E.; Soberanes, S.; Urich, D.; Morales-Nebreda, L.; Nigdelioglu, R.; Green, D.; Young, J.B.; Gonzalez, A.; Rosario, C.; Misharin, A.V.; et al. Beta(2)-Adrenergic agonists augment air pollution-induced IL-6 release and thrombosis. J. Clin. Investig. 2014, 124, 2935–2946. [Google Scholar] [CrossRef] [PubMed]
- Neri, T.; Pergoli, L.; Petrini, S.; Gravendonk, L.; Balia, C.; Scalise, V.; Amoruso, A.; Pedrinelli, R.; Paggiaro, P.; Bollati, V.; et al. Particulate matter induces prothrombotic microparticle shedding by human mononuclear and endothelial cells. Toxicol. Vitr. 2016, 32, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Nemmar, A.; Hoet, P.H.; Dinsdale, D.; Vermylen, J.; Hoylaerts, M.F.; Nemery, B. Diesel exhaust particles in lung acutely enhance experimental peripheral thrombosis. Circulation 2003, 107, 1202–1208. [Google Scholar] [CrossRef] [Green Version]
- Nemmar, A.; Al Dhaheri, R.; Alamiri, J.; Al Hefeiti, S.; Al Saedi, H.; Beegam, S.; Yuvaraju, P.; Yasin, J.; Ali, B.H. Diesel Exhaust Particles Induce Impairment of Vascular and Cardiac Homeostasis in Mice: Ameliorative Effect of Emodin. Cell. Physiol. Biochem. 2015, 36, 1517–1526. [Google Scholar] [CrossRef]
- Nemmar, A.; Al-Salam, S.; Zia, S.; Marzouqi, F.; Al-Dhaheri, A.; Subramaniyan, D.; Dhanasekaran, S.; Yasin, J.; Ali, B.H.; Kazzam, E.E. Contrasting actions of diesel exhaust particles on the pulmonary and cardiovascular systems and the effects of thymoquinone. Br. J. Pharmacol. 2011, 164, 1871–1882. [Google Scholar] [CrossRef] [Green Version]
- Nemmar, A.; Subramaniyan, D.; Yasin, J.; Ali, B.H. Impact of experimental type 1 diabetes mellitus on systemic and coagulation vulnerability in mice acutely exposed to diesel exhaust particles. Part. Fibre Toxicol. 2013, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- Tabor, C.M.; Shaw, C.A.; Robertson, S.; Miller, M.R.; Duffin, R.; Donaldson, K.; Newby, D.E.; Hadoke, P.W. Platelet activation independent of pulmonary inflammation contributes to diesel exhaust particulate-induced promotion of arterial thrombosis. Part.Fibre Toxicol. 2016, 13, 6. [Google Scholar] [CrossRef] [Green Version]
- Nemmar, A.; Hoylaerts, M.F.; Hoet, P.H.M.; Vermylen, J.; Nemery, B. Size effect of intratracheally instilled particles on pulmonary inflammation and vascular thrombosis. Toxicol. Appl. Pharmacol. 2003, 186, 38–45. [Google Scholar] [CrossRef]
- Mutlu, G.M.; Green, D.; Bellmeyer, A.; Baker, C.M.; Burgess, Z.; Rajamannan, N.; Christman, J.W.; Foiles, N.; Kamp, D.W.; Ghio, A.J.; et al. Ambient particulate matter accelerates coagulation via an IL-6-dependent pathway. J. Clin. Investig. 2007, 117, 2952–2961. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Liu, M.C.; Liang, M.; Fu, J. Sirt1 protects against thrombomodulin down-regulation and lung coagulation after particulate matter exposure. Blood 2012, 119, 2422–2429. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Zhao, T.; Hu, H.; Shi, Y.; Xu, Q.; Miller, M.R.; Duan, J.; Sun, Z. Repeat dose exposure of PM2.5 triggers the disseminated intravascular coagulation (DIC) in SD rats. Sci. Total Environ. 2019, 663, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Snow, S.J.; De Vizcaya-Ruiz, A.; Osornio-Vargas, A.; Thomas, R.F.; Schladweiler, M.C.; McGee, J.; Kodavanti, U.P. The effect of composition, size, and solubility on acute pulmonary injury in rats following exposure to Mexico city ambient particulate matter samples. J. Toxicol. Environ. Health A 2014, 77, 1164–1182. [Google Scholar] [CrossRef]
- Budinger, G.R.; McKell, J.L.; Urich, D.; Foiles, N.; Weiss, I.; Chiarella, S.E.; Gonzalez, A.; Soberanes, S.; Ghio, A.J.; Nigdelioglu, R.; et al. Particulate matter-induced lung inflammation increases systemic levels of PAI-1 and activates coagulation through distinct mechanisms. PLoS ONE 2011, 6, e18525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemmar, A.; Al-Salam, S.; Al Ansari, Z.; Alkharas, Z.A.; Al Ahbabi, R.M.; Beegam, S.; Yuvaraju, P.; Yasin, J.; Ali, B.H. Impact of Pulmonary Exposure to Cerium Oxide Nanoparticles on Experimental Acute Kidney Injury. Cell. Physiol. Biochem. 2019, 52, 439–454. [Google Scholar] [CrossRef] [Green Version]
- Frederix, K.; Kooter, I.M.; van Oerle, R.; Fens, D.; Hamulyak, K.; Gerlofs-Nijland, M.E.; Cate, H.T.; Spronk, H.M. A new method to determine tissue specific tissue factor thrombomodulin activities: Endotoxin and particulate air pollution induced disbalance. Thromb. J. 2008, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Emmerechts, J.; De Vooght, V.; Haenen, S.; Loyen, S.; Van kerckhoven, S.; Hemmeryckx, B.; Vanoirbeek, J.A.; Hoet, P.H.; Nemery, B.; Hoylaerts, M.F. Thrombogenic changes in young and old mice upon subchronic exposure to air pollution in an urban roadside tunnel. Thromb. Haemost. 2012, 108, 756–768. [Google Scholar] [CrossRef]
- Nemmar, A.; Hoylaerts, M.F.; Hoet, P.H.; Nemery, B. Possible mechanisms of the cardiovascular effects of inhaled particles: Systemic translocation and prothrombotic effects. Toxicol. Lett. 2004, 149, 243–253. [Google Scholar] [CrossRef]
- Huang, C.H.; Lin, L.Y.; Tsai, M.S.; Hsu, C.Y.; Chen, H.W.; Wang, T.D.; Chang, W.T.; Cheng, T.J.; Chen, W.J. Acute cardiac dysfunction after short-term diesel exhaust particles exposure. Toxicol. Lett. 2010, 192, 349–355. [Google Scholar] [CrossRef]
- Yan, Y.H.; Huang, C.H.; Chen, W.J.; Wu, M.F.; Cheng, T.J. Effects of diesel exhaust particles on left ventricular function in isoproterenol-induced myocardial injury and healthy rats. Inhal. Toxicol. 2008, 20, 199–203. [Google Scholar] [CrossRef] [Green Version]
- Nightingale, J.A.; Maggs, R.; Cullinan, P.; Donnelly, L.E.; Rogers, D.F.; Kinnersley, R.; Chung, K.F.; Barnes, P.J.; Ashmore, M.; Newman-Taylor, A. Airway inflammation after controlled exposure to diesel exhaust particulates. Am. J. Respir. Crit. Care Med. 2000, 162, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Pandya, R.J.; Solomon, G.; Kinner, A.; Balmes, J.R. Diesel exhaust and asthma: Hypotheses and molecular mechanisms of action. Environ. Health Perspect. 2002, 110 (Suppl. S1), 103–112. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.G.; Lee, P.H.; Lee, S.H.; Kim, Y.E.; Shin, M.Y.; Kang, Y.; Bae, S.H.; Kim, M.J.; Rhim, T.; Park, C.S.; et al. Long-Term Effects of Diesel Exhaust Particles on Airway Inflammation and Remodeling in a Mouse Model. Allergy Asthma Immunol. Res. 2016, 8, 246–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemmar, A.; Subramaniyan, D.; Ali, B.H. Protective effect of curcumin on pulmonary and cardiovascular effects induced by repeated exposure to diesel exhaust particles in mice. PLoS ONE 2012, 7, e39554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemmar, A.; Nemery, B.; Hoet, P.H.; Vermylen, J.; Hoylaerts, M.F. Pulmonary inflammation and thrombogenicity caused by diesel particles in hamsters: Role of histamine. Am. J. Respir. Crit. Care Med. 2003, 168, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- Nemmar, A.; Hoet, P.H.; Vermylen, J.; Nemery, B.; Hoylaerts, M.F. Pharmacological stabilization of mast cells abrogates late thrombotic events induced by diesel exhaust particles in hamsters. Circulation 2004, 110, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, T.; Liu, S.; Brook, R.D.; Feng, B.; Zhao, Q.; Song, X.; Yi, T.; Chen, J.; Zhang, Y.; et al. Extreme Levels of Air Pollution Associated with Changes in Biomarkers of Atherosclerotic Plaque Vulnerability and Thrombogenicity in Healthy Adults. Circ. Res. 2019, 124, e30–e43. [Google Scholar] [CrossRef]
- Sullivan, J.H.; Hubbard, R.; Liu, S.L.; Shepherd, K.; Trenga, C.A.; Koenig, J.Q.; Chandler, W.L.; Kaufman, J.D. A community study of the effect of particulate matter on blood measures of inflammation and thrombosis in an elderly population. Environ. Health 2007, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, R.M.; Sullivan, J.H.; Carlsten, C.; Wilkerson, H.W.; Beyer, R.P.; Bammler, T.; Farin, F.; Peretz, A.; Kaufman, J.D. A randomized cross-over study of inhalation of diesel exhaust, hematological indices, and endothelial markers in humans. Part. Fibre Toxicol. 2013, 10, 7. [Google Scholar] [CrossRef] [Green Version]
- Croft, D.P.; Cameron, S.J.; Morrell, C.N.; Lowenstein, C.J.; Ling, F.; Zareba, W.; Hopke, P.K.; Utell, M.J.; Thurston, S.W.; Thevenet-Morrison, K.; et al. Associations between ambient wood smoke and other particulate pollutants and biomarkers of systemic inflammation, coagulation and thrombosis in cardiac patients. Environ. Res. 2017, 154, 352–361. [Google Scholar] [CrossRef]
- Bind, M.A.; Baccarelli, A.; Zanobetti, A.; Tarantini, L.; Suh, H.; Vokonas, P.; Schwartz, J. Air pollution and markers of coagulation, inflammation, and endothelial function: Associations and epigene-environment interactions in an elderly cohort. Epidemiology 2012, 23, 332–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyczalkowska-Tomasik, A.; Czarkowska-Paczek, B.; Zielenkiewicz, M.; Paczek, L. Inflammatory Markers Change with Age, but do not Fall Beyond Reported Normal Ranges. Arch. Immunol. Ther. Exp. 2016, 64, 249–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.Y.; Chan, C.C.; Su, T.C. Particulate and gaseous pollutants on inflammation, thrombosis, and autonomic imbalance in subjects at risk for cardiovascular disease. Environ. Pollut. 2017, 223, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Rich, D.Q.; Kipen, H.M.; Huang, W.; Wang, G.; Wang, Y.; Zhu, P.; Ohman-Strickland, P.; Hu, M.; Philipp, C.; Diehl, S.R.; et al. Association between changes in air pollution levels during the Beijing Olympics and biomarkers of inflammation and thrombosis in healthy young adults. JAMA 2012, 307, 2068–2078. [Google Scholar] [CrossRef]
- Unosson, J.; Kabéle, M.; Boman, C.; Nyström, R.; Sadiktsis, I.; Westerholm, R.; Mudway, I.S.; Purdie, E.; Raftis, J.; Miller, M.R.; et al. Acute cardiovascular effects of controlled exposure to dilute Petrodiesel and biodiesel exhaust in healthy volunteers: A crossover study. Part. Fibre Toxicol. 2021, 18, 22. [Google Scholar] [CrossRef]
- Becerra, A.Z.; Georas, S.; Brenna, J.T.; Hopke, P.K.; Kane, C.; Chalupa, D.; Frampton, M.W.; Block, R.; Rich, D.Q. Increases in ambient particulate matter air pollution, acute changes in platelet function, and effect modification by aspirin and omega-3 fatty acids: A panel study. J. Toxicol. Environ. Health A 2016, 79, 287–298. [Google Scholar] [CrossRef] [Green Version]
- Rydz, N.; Grabell, J.; Lillicrap, D.; James, P.D. Changes in von Willebrand factor level and von Willebrand activity with age in type 1 von Willebrand disease. Haemophilia 2015, 21, 636–641. [Google Scholar] [CrossRef] [Green Version]
- Borghi, M.; Guglielmini, G.; Mezzasoma, A.M.; Falcinelli, E.; Bury, L.; Malvestiti, M.; Gresele, P. Increase of von Willebrand factor with aging in type 1 von Willebrand disease: Fact or fiction? Haematologica 2017, 102, e431–e433. [Google Scholar] [CrossRef] [Green Version]
- Abou-Ismail, M.Y.; Ogunbayo, G.O.; Secic, M.; Kouides, P.A. Outgrowing the laboratory diagnosis of type 1 von Willebrand disease: A two decade study. Am. J. Hematol. 2018, 93, 232–237. [Google Scholar] [CrossRef] [Green Version]
- Schultz, D.R.; Arnold, P.I. Properties of four acute phase proteins: C-reactive protein, serum amyloid A protein, alpha 1-acid glycoprotein, and fibrinogen. Semin. Arthritis Rheum. 1990, 20, 129–147. [Google Scholar] [CrossRef]
- Gabay, C.; Kushner, I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef]
- Hu, W.; Wang, Y.; Wang, T.; Ji, Q.; Jia, Q.; Meng, T.; Ma, S.; Zhang, Z.; Li, Y.; Chen, R.; et al. Ambient particulate matter compositions and increased oxidative stress: Exposure-response analysis among high-level exposed population. Environ. Int. 2021, 147, 106341. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Lu, J.; Ning, J.; Su, X.; Tong, Y.; Chen, J.; Ding, Y. Exposure to fine particulate matter induces self-recovery and susceptibility of oxidative stress and inflammation in rat lungs. Environ. Sci. Pollut. Res. Int. 2020, 27, 40262–40276. [Google Scholar] [CrossRef]
- Lawal, A.O. Air particulate matter induced oxidative stress and inflammation in cardiovascular disease and atherosclerosis: The role of Nrf2 and AhR-mediated pathways. Toxicol. Lett. 2017, 270, 88–95. [Google Scholar] [CrossRef]
- White, M.V. The role of histamine in allergic diseases. J. Allergy Clin. Immunol. 1990, 86, 599–605. [Google Scholar] [CrossRef]
- Asako, H.; Kurose, I.; Wolf, R.; DeFrees, S.; Zheng, Z.L.; Phillips, M.L.; Paulson, J.C.; Granger, D.N. Role of H1 receptors and P-selectin in histamine-induced leukocyte rolling and adhesion in postcapillary venules. J. Clin. Investig. 1994, 93, 1508–1515. [Google Scholar] [CrossRef] [Green Version]
- Eppihimer, M.J.; Wolitzky, B.; Anderson, D.C.; Labow, M.A.; Granger, D.N. Heterogeneity of expression of E- and P-selectins in vivo. Circ. Res. 1996, 79, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.P.; Brandes, L.J.; Becker, A.B.; Simons, K.J.; LaBella, F.S.; Gerrard, J.M. Histamine is an intracellular messenger mediating platelet aggregation. Science 1989, 243, 1596–1599. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.; Blomberg, A.; Rudell, B.; Kelly, F.; Sandström, T.; Holgate, S.T.; Frew, A. Acute inflammatory responses in the airways and peripheral blood after short-term exposure to diesel exhaust in healthy human volunteers. Am. J. Respir. Crit. Care Med. 1999, 159, 702–709. [Google Scholar] [CrossRef] [Green Version]
- Devouassoux, G.; Saxon, A.; Metcalfe, D.D.; Prussin, C.; Colomb, M.G.; Brambilla, C.; Diaz-Sanchez, D. Chemical constituents of diesel exhaust particles induce IL-4 production and histamine release by human basophils. J. Allergy Clin. Immunol. 2002, 109, 847–853. [Google Scholar] [CrossRef]
| Models | Exposure/Method | Results | Interpretation | References | ||
|---|---|---|---|---|---|---|
| Inflammation and Oxidative Stress | Coagulation and Adhesion Molecules | Morphology and Cell Proliferation | ||||
| HUVECs | Vanadium oxide (V2O5) 3.12, 6.25, 12.5, 25 µg/cm2 for 1, 2, 3, 24, 48, 72 h | ↑ ROS at 25 µg/cm2 ↑ NO at 25 µg/cm2 (time-dependent) | ↑ VCAM-1 ↑ ICAM-1 ↑↑ PECAM-1 | Morphology changed to fibroblast-like cells ↓ cell proliferation at 25 µg/cm2 ↑ annexin V, PI | Exposure to V2O5 induced oxidative stress, enhanced the expression of adhesion molecules, and affected cell survival by diminishing cell proliferation, shape changes, and apoptosis. | [38] |
| MH-S, Human alveolar macrophages | PM (SRM 1649a) 10 µg/cm2 for 24 h | # MH-S: ↑ IL-6 ↑ cAMP # Human alveolar macrophage: ↑ IL-6 | β2AR encoding from the Adrb2 gene had an important role in PM-induced IL-6 release and activation of β2AR enhanced inflammatory response in both cell lines. | [44] | ||
| PM (SRM 1649a) 10 µg/cm2 for 24 h Pretreated with β2AR agonist; albuterol 10−7 M | ↑↑ IL-6 | |||||
| Alveolar macrophages from Adrb2−/− mice | PM (SRM 1649a) 10 µg/cm2 for 24 h Pretreated with β2AR agonist; albuterol 10−7 M | ↓ IL-6 | ||||
| HUVECs, PBMC | PM (SRM 1648a) 62.5, 125, 250 and 500 µg/mL for 1, 4, 24, 48 h | ↑ MP (dose-, and time-dependent) ↑ intracellular Ca concentration | ↑ TF activity | PM-induced MP release, which, mediated by calcium mobilization, resulted in the prothrombotic state in both cell lines. | [45] | |
| HUVECs | DEP 10–150 µg/mL for 16 h ±thrombin stimulation 1 U/mL | # Without thrombin: ↓ tPA ↓ PAI-1 # With thrombin stimulation: ↔ tPA ↑ PAI-1 | DEP enhanced arterial thrombus formation through decreased fibrinolytic function but did not affect cell survival. | [50] | ||
| Venous blood of hamsters | DEP (SRM 1650) 0.1, 0.5, 1, 5 µg/mL for 5 min | ↓ PFA100 closure time, dose-dependent | DEP promoted thrombosis via platelet activation in a dose-dependent manner. | [46] | ||
| Venous blood of TO mice | DEP 1 µg/mL for 3 min | ↑ platelet aggregation ↓ PT ↓ PTT | DEP promoted thrombosis by enhancing platelet aggregation and coagulation. | [47] | ||
| Venous blood of TO mice | DEP (SRM 2975) 0.1, 0.25, 0.5, 1 µg/mL for 3 min | ↑ platelet aggregation at 0.5 and 1 µg/mL, dose-dependent | DEP promoted thrombosis by enhancing platelet aggregation. | [48] | ||
| Venous blood of TO mice Non-DM and DM mice | DEP (SRM 2975) 0.25, 0.5, 1 µg/mL for 3 min | # Non-DM mice: ↑ platelet aggregation at 1 µg/mL # DM mice: ↑↑ platelet aggregation, dose-dependent | DEP promoted thrombosis by enhancing platelet aggregation, which was more obvious in DM mice. | [49] | ||
| Venous blood of hamsters (Pfd Gold) | Polystyrene particles: # 60 nm UFP - unmodified - carboxylated - amined at 1 or 3 µg/mL # 400 nm: Amine-polystyrene particles at 3 or 9 µg/mL for 5 min | # Unmodified and carboxylated UFP: ↔ PFA100 closure time # Amine-UFP (60 nm): ↓ PFA100 closure time (3 µg/mL) # Amine-particles (400 nm): ↓ PFA100 closure time (9 µg/mL) | Exposure to positively charged UFP (60 & 400 nm) augmented platelet function, leading to thrombosis. | [51] | ||
| Models | Exposure/Method | Results | Interpretation | References | ||
|---|---|---|---|---|---|---|
| Inflammation and Oxidative Stress | Coagulation and Adhesion Molecules | Blood Parameters | ||||
| Male Wistar rats 10–12 wk-old Cisplatin-induced AKI rats | Intratracheal instillation of Cerium oxide nanoparticles (CeO2 NPs) 1 mg/kg | Normal rats: # Kidney ↑ TNF-α, IL-6, GSH ↑ DNA damage # Lung tissue ↔ TNF-α, IL-6 ↓ catalase activity AKI rats: # Kidney ↑ TNF-α, IL-6, GSH ↑ DNA damage # Lung tissue ↑ TNF-α, IL-6 ↓ catalase activity | Pulmonary exposure to CeO2 NPs induced inflammation and oxidative stress, and damaged DNA in the kidney. These effects were enhanced in kidney injury models. | [57] | ||
| Male mice C57Bl6/j 8–12 wk-old IL-6+/+ IL-6−/− | Inhalation exposure to concentrated ambient particles (CAPs) from downtown Chicago for 8 h/d for 3 d Evaluate at 24 h after exposure | # IL-6+/+ vs. non-PM): Lung tissue ↑ IL-6/18s mRNA ↑ SP-B/18s mRNA BALF ↑ IL-6 ↑ TNF-α ↑ MCP-1 # IL-6−/−: Lung tissue ↓ IL-6/18s mRNA ↓ SP-B/18s mRNA BALF ↓ IL-6 ↔ TNF-α ↔ MCP-1 | # IL-6+/+ vs. non- PM): Lung tissue ↑ TF/18s mRNA Plasma ↑ TAT complexes White adipose tissue ↑ PAI-1/18s mRNA # IL-6−/−: Lung tissue ↓ TF/18s mRNA Plasma ↓ TAT complexes White adipose tissue ↔ PAI-1/18s mRNA | Exposure to all types of PM could activate inflammatory response, coagulation system and inhibit fibrinolysis, resulting in a prothrombotic state. PM-induced coagulation through IL-6 production and blocking IL-6 signaling could alleviate the thrombotic process. | [56] | |
| Intratracheal instillation of urban PM (SRM1649a) 10, 100, 200 µg/animal Evaluate at 24 h after exposure | # IL-6+/+ vs. non- PM): BALF ↑ protein ↑ macrophage, PMN ↑ IL-6 (dose-dependent) ↑ TNF-α # IL-6−/−: BALF ↔ protein ↔ macrophage, PMN ↓ IL-6 ↔ TNF-α | # IL-6+/+ vs. non- PM): ↑ TF, ↑TF mRNA in lung tissue ↑ BALF D-dimer ↑ TAT complexes ↓ Bleeding time ↓ PT, ↓ PTT ↑ PAI-1/18s mRNA in the lung, adipose tissue ↑ PAI-1 in BALF # IL-6−/−: ↓ TF level, ↓TF mRNA in lung tissue ↓ BALF D-dimer ↓ TAT complexes ↔ PAI-1/18s mRNA in the lung, adipose tissue ↔ PAI-1 in BALF | ||||
| Male mice (C57BL/6) 8–12 wk-old | Inhalation exposure to concentrated ambient particles (CAPs) from downtown Chicago for 8 h/d for 3 d | ↑ NE in the lung, BAT, adrenal gland ↑ IL-6 in BALF | ↑ TAT complexes ↑ thrombus formation ↓ thrombotic occlusion time | Inhalation of PM caused catecholamine release and promoted IL-6-mediated thrombosis. | [44] | |
| Adrb1+/+Adrb2+/+ Adrb1−/−Adrb2+/+ Adrb1+/+Adrb2−/− Adrb1−/−Adrb2−/− | Intratracheal instillation of urban PM (SRM1649a) 200 µg/animal Evaluate at 24 h after exposure | BALF # Adrb1+/+Adrb2+/+ (vs. non-PM): ↑ IL-6 ↔ TNF-α, MCP-1 # Adrb1−/− Adrb2+/+ (vs. non-PM): ↑ IL-6 ↔ TNF-α, MCP-1 # Adrb1+/+Adrb2−/−: ↓ IL-6 ↔ TNF-α, MCP-1 # Adrb1−/−Adrb2−/−: ↓ IL-6 ↔ TNF-α, MCP-1 | Plasma # Adrb1+/+Adrb2+/+ (vs. non-PM): ↑ TAT complexes ↓ thrombotic occlusion time # Adrb1−/− Adrb2+/+ (vs. non-PM): ↑ TAT complexes # Adrb1+/+Adrb2−/−: ↓ TAT complexes ↑ thrombotic occlusion time # Adrb1−/−Adrb2−/−: ↓ TAT complexes | β2AR encoded by the Adrb2 gene in alveolar macrophages was necessary for PM-induced upregulation of IL-6, and enhanced susceptibility to thrombotic events. | ||
| Adrb1+/+Adrb2+/+ Adrb1+/+Adrb2−/− | Inhalation exposure to concentrated ambient particles (CAPs) from downtown Chicago for 8 h/d for 3 d | # Adrb1+/+Adrb2−/−: ↓ IL-6/18s mRNA | # Adrb1+/+Adrb2−/−: ↓ TAT complexes ↓ TF | |||
| Lyms-Cre Adrb2flox/flox mice (macrophage-specific deletion of β2AR) vs. Adrb2flox/flox | Inhalation exposure to concentrated ambient particles (CAPs) from downtown Chicago for 8 h/d for 3 d Pretreated with formoterol (long-acting β2AR agonist) 1 × 10−5 M via inhalation twice every 12 h | BALF # Adrb2flox/flox without formoterol: ↑ IL-6 in BALF # Adrb2flox/flox with formoterol: ↑↑ IL-6 # Lyms-Cre Adrb2flox/flox: ↓ IL-6 # Lyms-Cre Adrb2flox/flox with formoterol: ↓ IL-6 (vs. Adrb2flox/flox) ↔ IL-6 (vs. without formoterol) | Plasma # Adrb2flox/flox without formoterol: ↑ TAT complexes ↑ factor II, TF mRNA ↓ thrombotic occlusion time # Adrb2flox/flox with formoterol: ↑ factor II, TF mRNA ↓ thrombotic occlusion time # Lyms-Cre Adrb2flox/flox: ↓ factor II, TF mRNA ↓ TAT complexes ↑ thrombotic occlusion time # Lyms-Cre Adrb2flox/flox with formoterol vs. Adrb2flox/flox: ↓ factor II, TF mRNA ↑ thrombotic occlusion time vs. without formoterol: ↔ factor II, TF mRNA ↔ thrombotic occlusion time | |||
| Male mice C57Bl6/j Old mice (20 mo-old) vs. Young mice (10 wk-old) | Inhalation of ambient PM2.5 and PM10 at the roadside tunnel for 25–26 d (A) tunnel-filtered (B) tunnel-exposed in urban roadside tunnel (C) control in clean facility | # Young mice (vs. non-PM): ↔ WBC in BALF # Old mice (vs. young mice) in clean air: ↑ WBC in BALF # Old mice (vs. young mice) with PM: ↔ WBC in BALF | # Young mice (vs. non-PM): ↔ lung vWF ↔ plasma vWF ↓ lung TM ↑ P-selectin ↔ PF4 # Old mice (vs. young mice) in clean air: ↑ lung VWF ↑ plasma VWF ↔ lung TM ↑ P-selectin ↔ PF4 # Old mice (vs. young mice) with PM: ↑ lung vWF ↔ plasma VWF ↔ lung TM ↔ P-selectin ↔ PF4 | # Young mice (vs. non-PM): ↑ RBC, Hb ↑ platelets ↔ WBC # Old mice (vs. young mice) in clean air: ↑ RBC, Hb ↑ platelets ↑ WBC # Old mice (vs. young mice) with PM: ↔ RBC, Hb ↔ platelets ↔ WBC | Continuous inhalation of particulate matter air pollution triggered inflammatory response, and activated platelets, and endothelial cells. The older mice had higher inflammatory biomarkers at baseline, therefore the PM-mediated effects were not demonstrated in the old mice. | [59] |
| Male mice C57Bl6/j with spontaneous hypertension 11–12 wk-old | Intratracheal instillation particulate matter # Road tunnel dust (RTD): 0.3, 1, 3, and 10 mg/kg # Urban dust (EHC-93) from Environmental Health Center in Ottawa, Canada 10 mg/kg Evaluation of lung tissue at 4, and 48 h after PM exposure | # RTD (at 10 mg/kg): - at 4 h: ↑ TF ↑ thrombus formation - at 48 h: ↑ TF ↑↑ thrombus formation # EHC-93: - at 4 h: ↔ TF ↑ thrombus formation - at 48 h: ↑↑ TF ↑↑ thrombus formation | PM induced procoagulant activity in the lungs, via increased TF expression and aggravated thrombus formation. | [58] | ||
| Hamsters (Pfd Gold) 100–110 g | Intratracheal instillation of polystyrene particles: # 60 nm UFP -unmodified 500 μg/animal -carboxylated 500 μg/animal -amined 5, 50, 500 μg/animal # 400 nm: Amine-modified polystyrene particles 500 μg/animal Evaluation of BALF at 1 h after UFP exposure | # Unmodified and carboxylated UFP: ↔ PMN influx # Amine-UFP (60 nm): ↑ PMN influx (50 and 500 µg/animal) ↑ protein, histamine (500 μg/animal) # Amine-particles (400 nm): ↑ PMN influx ↑ BALF protein ↔ BALF histamine | # Unmodified and carboxylated UFP: ↔ thrombus formation # Amine-UFP (60 nm): ↑ thrombus formation (at 50 and 500 µg/animal) # Amine-particles (400 nm): ↔ thrombus formation | Exposure to both positively charged UFP (60 & 400 nm) resulted in inflammation in the respiratory tract, but only the UFP (60 nm) rapidly activated the clotting system within an hour, leading to thrombosis. | [51] | |
| Hamster 100–110 g | Intratracheal instillation of polystyrene particles: # 60 nm UFP - unmodified 500 μg/animal - carboxylated 500 μg/animal - amined 5, 50, 500 μg/animal # 400 nm amined- polystyrene particles 500 μg/animal Evaluation of BALF at 1 h after UFP exposure | # Unmodified and carboxylated UFP: ↔ PMN influx # Amine-particles (60 nm and 400 nm): ↑ PMN influx (50 μg) ↑↑ PMN influx (500 μg) | # Unmodified and carboxylated UFP: ↔ thrombus formation # Amine-particles (60 nm): ↑↑ thrombus formation (50 μg) ↑ thrombus formation (500 μg) # Amine-particles (400 nm): ↔ thrombus formation | UFP induced pulmonary inflammation and promoted thrombosis, but the degree of lung inflammation did not show a correlation with the extent of thrombosis. | [60] | |
| Intratracheal instillation of DEP (SRM 1650) 5, 50, 500 μg/animal Evaluate at 1 h after UFP exposure | BALF ↑ PMN influx ↑ protein ↑ histamine (at 50 and 500 μg/animal) | ↑ thrombus formation (50 μg) ↑↑ thrombus formation (500 μg) ↓ PFA100 closure time | DEP exposure activated platelet and thrombin generation, leading to thrombosis. | |||
| Female mice (C57BL/6) 8–10 wk-old sex-age-matched Sirt1 +/+ Sirt1 −/− Sirt1 overexpression in WT mice (vs. WT mice) | Intranasal instillation of PM2.5 (SRM 8785) 100 µg/animal for 24 h | # Sirt1 +/+: ↑ lung NF-ĸB ↑ BALF albumin, PMN ↑ BALF TNF-α & IL-6 # Sirt1 −/−: ↑↑ lung NF-κB ↑↑ BALF albumin, PMN ↑↑ BALF TNF-α & IL-6 | # Sirt1 +/+: ↑ lung fibrin formation ↓ TFPI ↑ TF ↑ lung PAI-1 ↔ plasma PAI-1 ↓ lung TM # Sirt1 −/−: ↑ ↑ lung fibrin formation ↓ ↓ TFPI ↑ TF ↑ ↑ lung PAI-1 ↔ plasma PAI-1 ↓↓ lung TM # Sirt1 overexpression: ↓ lung fibrin formation ↑ lung TM | PM2.5 exposure promoted pulmonary vascular injury and enhanced inflammation, coagulation, and inhibited fibrinolysis, which was regulated by Sirt1 and NF-κB pathways. | [53] | |
| Male SD rats 8–12 wk-old | Intratracheal instillation of PM2.5 once every 3 d for 30 d Doses: - Low dose: 1.8 mg/kg - Middle dose: 5.4 mg/kg - High dose: 16.2 mg/kg PM2.5 was collected from central Beijing, China | ↑ Alveolar wall thickening ↑ IL-6, IL-1β, CRP ↔ MCP-1 | ↓ Aortic valve peak blood flow ↑ thrombus formation ↑ TF ↑ TAT complexes ↑ Factor Xa ↑↑ D-dimer ↓ TM ↔ TFPI ↑ tPA ↓ vWF ↑ PT, PTT, TT ↔ fibrinogen ↑↑ ICAM-1, VCAM-1 | ↓ platelets | PM2.5 induced vascular endothelial injury, systemic inflammatory response, altered coagulation factors, anticoagulant pathway, and fibrinolytic system, resulting in the prothrombotic state, and DIC. | [54] |
| Male Wistar Kyoto (WKY) rats 12–15 wk-old | Intratracheal instillation of PM2.5 and PM10 from The Northern and Southern Mexico - Total fraction - Insoluble fraction - Soluble fraction (control) of each PM2.5 and PM10 3.3 mg/kg Evaluation at 24 or 72 h after PM exposure | # Total fraction and insoluble fraction of PM2.5 & PM10: ↑ BALF cell count ↓ alveolar macrophages Lung tissue ↑ total protein, ↑ albumin, ↓ ascorbic acid ↑ MIP-2, TNF-α mRNA ↑ BALF MIP-2, TNF-α ↑ HO-1 ↑ LOX-1R, ↑ NOS | # Total fraction and insoluble fraction of PM2.5 & PM10: ↑ lung TF mRNA ↓ tPA mRNA ↑ PAI-1 mRNA | # Total fraction and insoluble fraction of PM2.5 & PM10: ↔ RBC, Hb, Hct, platelet, and WBC | Exposure to PM aggravated pulmonary inflammation and oxidative stress, as well as disruption in the procoagulant and fibrinolytic pathways of the lung. | [55] |
| Male mice (C57BL/6) 8–12 wk-old IL-6+/+ IL-6−/− IL-6+/+ depleted alveolar macrophages | Intratracheal instillation of PM10 from ambient air in Düsseldorf, Germany 10 μg/animal for 24 h # Pretreated with Intratracheally instillation of liposomal clodronate 120 mg/animal for 48 h before PM exposure (Setting of WT mice depleted of alveolar macrophages) | BALF # IL-6+/+ vs. non-PM10: ↑ macrophage, PMN ↑ IL-6, TNF-α, IFN-γ ↔ MCP-1, IL-10, IL-12 # IL-6−/− vs. non-PM10: ↑ macrophage, PMN ↔ IL-6 ↑ TNF-α ↔ MCP-1, IL-10, IL-12, IFN-γ # IL-6−/− vs. IL-6+/+: ↓ IL-6 ↔ TNF-α, MCP-1, IL-10, IL-12, IFN-γ # IL-6+/+ depleted alveolar macrophages: ↓ macrophage ↔ PMN ↓ IL-6 ↔ TNF-α, MCP-1, IL-10, IL-12, IFN-γ | Plasma # IL-6+/+ vs. non-PM10: ↑ Factor II, VIII, X ↑ Fibrinogen ↓ Bleeding time ↓ PT, ↓ PTT ↓ thrombotic occlusion time ↑ TAT complexes # IL-6−/− vs. non-PM10: ↔ Factor VIII ↔ Bleeding time ↔ PT, ↔ PTT ↔ thrombotic occlusion time ↔TAT complexes # IL-6+/+ depleted alveolar macrophages: ↓ Factor VIII ↑ Bleeding time ↑ PT, ↑ PTT ↓ TAT complexes ↑ thrombotic occlusion time | # IL-6+/+ vs. non-PM10: ↑ Platelet # IL-6−/− vs. non-PM10: ↔ Platelet # IL-6+/+ depleted alveolar macrophages: ↓ Platelet | PM10 exposure-induced pulmonary inflammation, and IL-6 release. IL-6 was the key mediator, which enhanced coagulation factor function, resulted in shortening of clotting time, and led to thrombosis. Blocking either the macrophage function or IL-6 signal could alleviate PM-induced prothrombotic state. | [52] |
| Models | Exposure/Method | Results | Interpretation | References | ||
|---|---|---|---|---|---|---|
| Inflammation and Oxidative Stress | Coagulation and Adhesion Molecules | Blood Parameters | ||||
| Male Wistar rats 175–275 g | Intratracheal instillation of # DEP (SRM 2975) 0.5 mg/animal # Black carbon (BC) 0.5 mg/animal # DQ12 quartz microparticles 0.125 mg/animal Evaluation at 2, 6, and 24 h after exposure | BALF # DEP: ↑ PMN influx ↑ IL-6 ↔ TNF-α, CRP # BC: ↑↑ PMN influx ↑ IL-6 ↔ TNF-α ↑ CRP # DQ12 quarts: ↑↑ PMN influx ↔ IL-6 ↔ TNF-α ↑ CRP | Plasma # DEP ↓ thrombotic occlusion time ↑ PAI-1 ↓ tPA ↓ tPA:PAI-1 ratio ↑ platelet-monocyte aggregation # BC, DQ12 quarts: ↔ thrombotic occlusion time ↑ PAI-1 ↓ tPA ↓ tPA:PAI-1 ratio ↔ platelet-monocyte aggregation | Pulmonary exposure of DEP caused lung inflammation and accelerated arterial thrombus formation through increasing platelet activation, and impaired fibrinolytic function, while IV injection of DEP promoted thrombosis, without evidence of pulmonary inflammatory response. | [50] | |
| Intravenous injection (IV) of DEP or BC 0.5 mg/kg Evaluation at 2, 6, and 24 h after exposure | # DEP: ↔ inflammatory cells in BALF ↔ BALF TNF-α, CRP ↔ plasma TNF-α, CRP, IL-6 # BC: ↔ inflammatory cells in BALF ↔ BALF TNF-α, CRP ↔ plasma TNF-α, IL-6 ↑ plasma CRP | # DEP: ↓ thrombotic occlusion time ↑ PAI-1 ↓ tPA ↓ tPA:PAI-1 ratio ↑ platelet-monocyte aggregation # BC: ↓ thrombotic occlusion time ↑ PAI-1 ↓ tPA ↓ tPA:PAI-1 ratio ↔ platelet-monocyte aggregation | ||||
| Hamsters (Pfd Gold) 100–110 g | Intratracheal instillation of DEP (SRM 1650) 5, 50, 500 µg/animal Evaluate at 1 h after PM exposure | BALF ↑ PMN influx ↑ protein, histamine, in a dose-dependent manner ↔ LDH | ↑ venous thrombus formation, dose-dependent manner ↑ arterial thrombus formation ↓ PFA100 closure time | ↔ platelet | DEP enhanced lung inflammation, platelet activation, and peripheral vascular thrombosis. | [46] |
| Hamsters 100–110 g | Intratracheal instillation of DEP (SRM 1650) 50 µg/animal Evaluation at 1, 6, 24 h after exposure | ↑ BALF PMN influx, time-dependent manner ↑ BALF histamine ↑ plasma histamine | ↑ thrombus formation ↓ PFA100 closure time | ↔ platelet | Histamine involved in the process of DEP-induced lung inflammation and platelet activation led to a prothrombotic state. | [67] |
| Hamsters (Pfd Gold) 100–110 g | Intratracheal instillation of # DEP (SRM 1650) 50 µg/animal # Polystyrene particles 400 nm 500 µg/animal Evaluation at 24 h after exposure | # DEP: ↑↑ PMN influx # Polystyrene particles: ↑ PMN influx # DEP and polystyrene particles: ↑ histamine in BALF and plasma | # DEP and polystyrene particles: ↑ thrombus formation ↔ VWF | DEP triggered mast cell degranulation by histamine release and enhanced thrombus formation. | [68] | |
| Male TO mice (HsdOla: TO) 10–12 wk-old DM vs. non-DM mice (Intraperitoneal injection of streptozotocin 200 mg/kg to induced Type 1 DM) | Intratracheal instillation of DEP (SRM 2975) 0.4 mg/kg Evaluation of plasma at 24 h after exposure | # Non-DM mice: ↔ CRP ↔ 8-isoprostane # DM mice: ↑ CRP ↑ 8-isoprostane | # Non-DM mice: ↔ thrombotic occlusion time ↑ PAI-1 ↔ VWF # DM mice: ↓ thrombotic occlusion time ↑↑ PAI-1 ↑ VWF | # Non-DM mice: ↔ WBC ↔ platelet # DM mice: ↑ WBC ↓ platelet | Particulate air pollution activated systemic inflammation, oxidative stress, hypoxemia, hepatotoxicity, coagulation, and interfered with fibrinolytic function, resulting in a procoagulant state. These results were more enhanced in DM mice. | [49] |
| Male TO mice (HsdOla: TO) 30–35 g | Intratracheal instillation of DEP (SRM 2975) 15 µg/animal on day 0, 2, 4, 6 Evaluation at 48 h after the last exposure | BALF: ↑ PMN influx ↑ macrophages ↑ TNF-α ↔ IL-6 Plasma: ↑ CRP ↑ TNF-α ↔ IL-6 | ↓ thrombotic occlusion time ↑ D-dimer ↑ PAI-1 ↔ VWF | ↓ platelet | Repeated DEP exposure activated systemic inflammation, thrombotic events, and platelet aggregation. | [66] |
| Male TO mice (HsdOla: TO) 30–35 g | Intratracheal instillation of DEP (SRM 2975) 30 µg/animal Evaluation at 4, and 18 h after exposure | BALF ↑↑ PMN, macrophages ↑↑ IL-6 ↑↑ total protein ↓ superoxide dismutase | ↑ IL-6 ↓ thrombotic occlusion time | ↑ WBC ↓ platelet | DEP exposure-induced pulmonary inflammation, and enhanced platelet aggregation and thrombosis. | [48] |
| Male TO mice | Intratracheal instillation of DEP 1 mg/kg Evaluation at 24 h after exposure | ↑ TNF-α ↑ IL-1β ↓ superoxide dismutase ↑ glutathione reductase | ↓ thrombotic occlusion time | ↑ Hb, Hct, RBC, WBC, platelet | DEP exposure activated inflammation, oxidative stress, and promoted thrombosis. | [47] |
| Models | Exposure | Intervention | Results | Interpretation | References | |
|---|---|---|---|---|---|---|
| Inflammation and Oxidative Stress | Coagulation and Adhesion Molecules | |||||
| Human alveolar macrophages, MH-S | PM (SRM 1649a) 10 µg/cm2 for 24 h | # β2AR agonists; albuterol 10−7 M # βAR antagonist; propranolol 10 µM # Albuterol + propranolol Added 1 h after PM | ↑ IL-6 ↓ IL-6 ↓ IL-6 | Activation of β2AR enhanced PM-mediated IL-6 release, while βAR blockade inhibited the release of IL-6 in response to PM. | [44] | |
| MH-S | PM (SRM 1649a) 10 µg/cm2 for 1 h | # An adenylyl cyclase activator, Forskolin 50 µM # Forskolin 50 µM and adenylyl cyclase inhibitor (SQ2253) 300 µM # Forskolin + PDE inhibitors; IBMX 1 µM # Forskolin + Aminophylline 10 µM # Antioxidant; Mito-Q # Superoxide dismutase/catalase mimetic Eukarion 134 (EUK-134) # Forskolin + EUK-134 | # Forskolin: ↑↑ IL-6 ↑ cAMP # Forskolin + SQ2253: ↑ IL-6 # Forskolin + IBMX: ↑ cAMP # Forskolin + Aminophylline: ↑ cAMP # Mito-Q: ↓ IL-6 # EUK-134: ↓ IL-6 ↔ cAMP # Forskolin + EUK-134: ↓cAMP | PM exposure enhanced IL-6 release and activated systemic inflammation via adenylyl cyclase and CREB functions. | ||
| CREB shRNA-transfected MH-S cells p65-shRNA-transfected MH-S cells | PM (SRM 1649a) 10 µg/cm2 for 1 h | # Albuterol 10−7 M for 1 h | ↓ IL-6 | |||
| HUVECs, PBMC | PM (SRM1648a) 500 µg/mL for 1 h | # Phospholipase C inhibitor (U73122) 1 µM for 30 min | ↓ MP | PM-induced MP release was mediated through phospholipase C. | [45] | |
| Venous blood of TO mice | DEP (SRM 2975) 1 µg/mL for 3 min | # Thymoquinone 0.1 mg/mL for 3 min | ↔ platelet aggregation | Thymoquinone did not prevent DEP-induced platelet aggregation. | [48] | |
| Venous blood of TO mice | DEP 1 µg/mL for 3 min | # Emodin 1 µg/mL for 3 min | ↓ platelet aggregation ↑ PT ↑ PTT | Emodin prevented the effects of DEP-induced platelet aggregation and thrombosis. | [47] | |
| Models | Exposure | Intervention | Results | Interpretation | References | ||
|---|---|---|---|---|---|---|---|
| Inflammation and Oxidative Stress | Coagulation and Adhesion Molecules | Blood Parameters | |||||
| Male mice (C57BL/6) 8–12 wk-old | Intratracheal instillation of urban PM (SRM1649a) 200 µg/animal Evaluated at 24 h after exposure | Pretreated with # Vesicular monoamine transporter: Reserpine (chemical sympathectomy) # Propranolol 3 mg/kg IP q 8 h for 48 h | ↓ NE in BALF, BAT, adrenal gland, lung ↓ IL-6 in BALF ↓ IL-6 in BALF | ↓ plasma TAT complexes ↓ plasma TAT complexes ↑ thrombotic occlusion time | Blocking of the sympathetic nervous system and β2AR signaling alleviated IL-6 release, lung inflammation, and reduced thrombosis. | [44] | |
| Male mice C57Bl6/j 8–12 wk-old | Inhalation exposure to concentrated ambient particles (CAPs) from downtown Chicago for 8 h/d for 3 d Evaluated at 24 h after exposure | Pretreated with TNF-α inhibitor, Etanercept 10 mg/kg IP. 3 days before, and on the first day of exposure to CAPs | ↓ PAI-1/18s mRNA | Blocking TNF-α could promote normal fibrinolytic function, but not alter the PM-induced clotting formation. | [56] | ||
| Intratracheal instillation of urban PM (SRM1649a) 200 µg/animal Evaluate at 24 h after exposure | Pretreated with TNF-α inhibitor, Etanercept 10 mg/kg IP. 3 days before, and on the first day of exposure to PM | ↔ Bleeding time ↔ PT, ↔ PTT ↔ TAT complexes ↓ PAI-1/18s mRNA ↓ PAI-1 in BALF | |||||
| Hamsters 100–110 g | Intratracheal instillation of DEP (SRM 1650) 50 µg/animal For 1, 3, 6, 24 h after exposure | Pretreated with Antihistamine: Diphenhydramine IP 30 mg/kg for 1 h | BALF ↓ cell count, PMN influx ↓ histamine plasma ↓ histamine | ↓ thrombus formation ↑ PFA100 closure time | Pretreatment with diphenhydramine reduced the effects of DEP-induced pulmonary inflammation and peripheral thrombosis. | [67] | |
| Hamsters (Pfd Gold) 100–110 g | Intratracheal instillation of DEP (SRM 1650) 50 µg/animal Evaluation at 24 h after exposure | # Pretreated dexamethasone IP 5 mg/kg # Pretreated dexamethasone IT 0.1 or 0.5 mg/kg # Pretreated Sodium Cromoglycate IP 40 mg/kg for 1 h | # Dexamethasone IP: ↓ BALF cell count, PMN ↓ BALF and plasma histamine # Dexamethasone IT (0.5 mg/kg): ↓ BALF cell count, PMN ↓ BALF histamine ↔ plasma histamine # Sodium cromoglycate: ↓ BALF cell count, PMN ↓ BALF and plasma histamine | # Dexamethasone IP: ↓ thrombus formation # Dexamethasone IT (0.5 mg/kg): ↓ thrombus formation # Sodium cromoglycate: ↑ PFA100 closure time | Dexamethasone prevented PM-induced lung inflammation, histamine release, and thrombosis. Anti-inflammatory pretreatment also helped prevent PM-induced histamine release and reduced the prothrombotic state. | [68] | |
| Male TO mice (HsdOla: TO) 30–35 g | Intratracheal instillation of DEP (SRM 2975) 15 µg/animal on day 0, 2, 4, 6 Evaluation at 48 h after the last exposure | Pretreated with Curcumin (200 µl) oral gavage for 1 h | BALF: ↓ PMN, macrophages ↓ TNF-α ↔ IL-6 Plasma: ↓ CRP ↓ TNF-α ↔ IL-6 | ↑ thrombotic occlusion time ↓ D-dimer ↓ PAI-1 ↔ VWF | ↑ Platelet | Curcumin pretreatment prevented DEP-induced inflammation and promoted fibrinolytic activity, which diminished the prothrombotic state. | [66] |
| Male TO mice (HsdOla: TO) 30–35 g | Intratracheal instillation of DEP (SRM 2975) 30 µg/animal Evaluation at 4, and 18 h after exposure | Pretreated with anti-inflammatory agent: Thymoquinone IP 6 mg/kg for 1 and 24 h | BALF ↓ PMN, macrophages ↓ IL-6 ↓ total protein Plasma ↓ IL-6 ↑ superoxide dismutase | ↑ thrombotic occlusion time | ↓ WBC ↑ Platelets | Thymoquinone pretreatment significantly prevented DEP-induced inflammatory response, oxidative stress, and thrombosis. | [48] |
| Male TO mice | Intratracheal instillation of DEP 1 mg/kg Evaluation at 24 h after exposure | Emodin (antioxidant/anti-inflammation) IP 4 mg/kg twice, 1 h before, and 7 h after exposure | ↓ TNF-α ↓ IL-1β ↑ superoxide dismutase ↓ glutathione reductase | ↑ thrombotic occlusion time | ↓ Hb, Hct, RBC ↓ WBC | Administration of antioxidants prevented DEP-induced inflammatory response, oxidative stress, and thrombotic complications. | [47] |
| DM type II patients (N = 30) Mean age 56.5 y | Acute exposure to ambient PM in Rochester, NY, USA Evaluated the PM level at 1, 12, 24, 48, 96 h | 8 wk sequential therapy with # ASA 81 mg/d for 7 d # Fish oil 4 g/d for 28 d # Combined for 7 d | # ASA, and/or fish oil: ↔ TBXB2 ↔ ADP-, and collagen-induced platelet aggregation | ASA/fish oil blunted the effect of pollution on platelet function and TBXB2. | [78] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hantrakool, S.; Kumfu, S.; Chattipakorn, S.C.; Chattipakorn, N. Effects of Particulate Matter on Inflammation and Thrombosis: Past Evidence for Future Prevention. Int. J. Environ. Res. Public Health 2022, 19, 8771. https://doi.org/10.3390/ijerph19148771
Hantrakool S, Kumfu S, Chattipakorn SC, Chattipakorn N. Effects of Particulate Matter on Inflammation and Thrombosis: Past Evidence for Future Prevention. International Journal of Environmental Research and Public Health. 2022; 19(14):8771. https://doi.org/10.3390/ijerph19148771
Chicago/Turabian StyleHantrakool, Sasinee, Sirinart Kumfu, Siriporn C. Chattipakorn, and Nipon Chattipakorn. 2022. "Effects of Particulate Matter on Inflammation and Thrombosis: Past Evidence for Future Prevention" International Journal of Environmental Research and Public Health 19, no. 14: 8771. https://doi.org/10.3390/ijerph19148771