
Metagenomic Analysis of Microbial Contamination in the U.S. Portion of the Tijuana River Watershed


Abstract
:1. Introduction
2. Materials and Methods
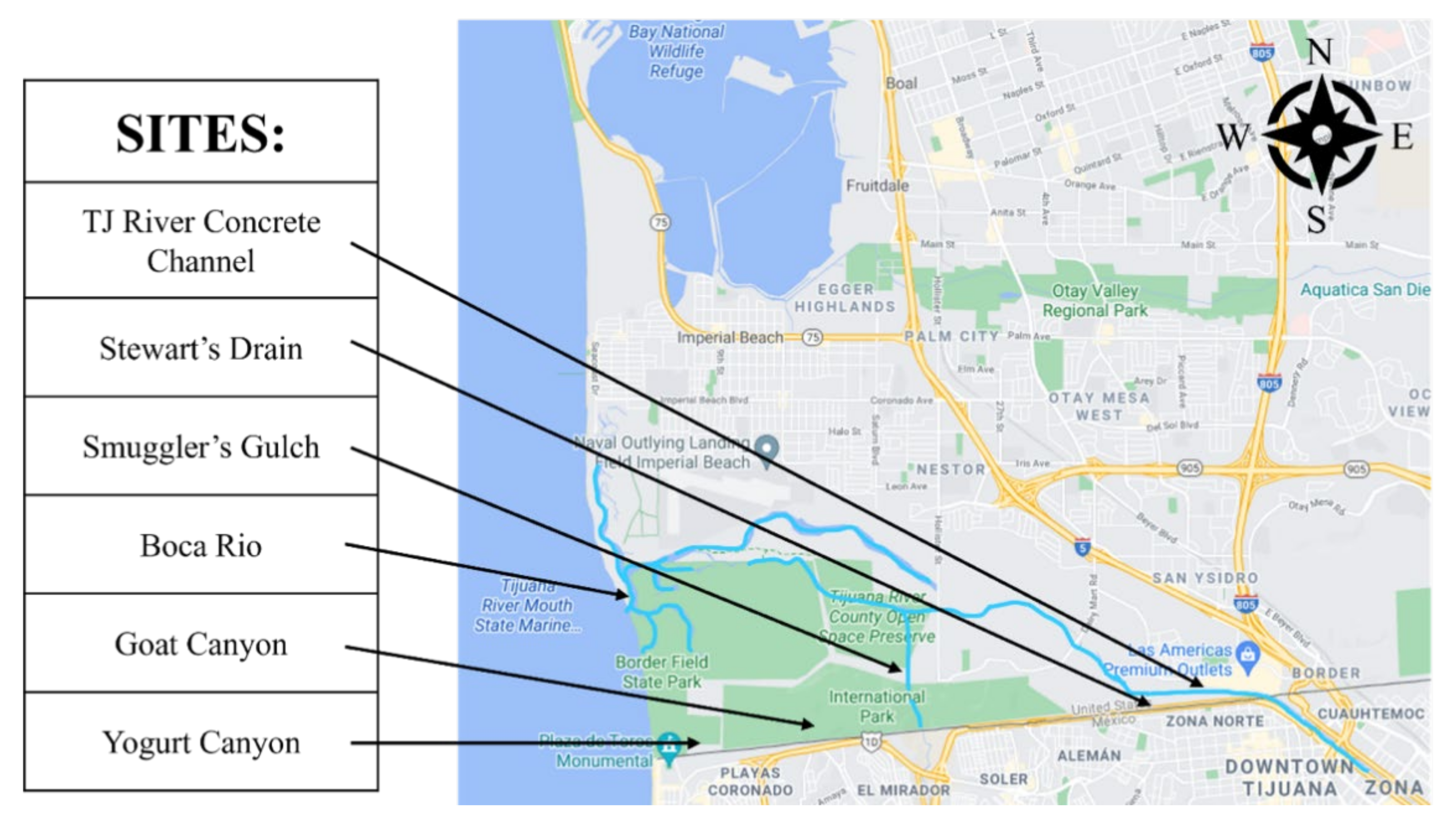
2.1. Sample Collection, E. coli and Coliform Measurements
2.2. DNA Extraction, Purification, and Sequencing
2.3. Processing and Taxonomic Classification
2.4. OTU Table Creation and Modification
2.5. Disease-Causing Microbe and Virus Identification
2.6. Breadth of Coverage
2.7. Diversity Analyses
2.8. Fecal Contamination Analysis
2.9. Antibiotic Resistance Genes (ARGs)
2.10. Scripts and Data
3. Results
3.1. DNA Concentration and Sequence Quality
3.2. Metagenomic Results with Disease-Causing Microbes and Viruses
3.3. Breadth of Coverage
3.4. Diversity Analysis
3.5. Alpha Diversity
3.6. Fecal Contamination Analysis
3.7. Antibiotic Resistance Genes (ARGs)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization; United Nations Children’s Fund (UNICEF). Progress on Household Drinking Water, Sanitation and Hygiene 2000–2020: Five Years into the SDGs; World Health Organization: Geneva, Switzerland, 2021.
- International Database (IDB). United States Census Bureau, and World Population Clock; International Database (IDB): Washington, DC, USA, 2017. [Google Scholar]
- WHO/UNICEF Joint Monitoring Programme (JMP). Water Supply and Sanitation (2000–2020). Available online: https://washdata.org/data (accessed on 1 May 2022).
- Troeger, C.; Blacker, B.F.; Khalil, I.A.; Rao, P.C.; Cao, S.; Zimsen, S.R.M.; Albertson, S.B.; Stanaway, J.D.; Deshpande, A.; Abebe, Z.; et al. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of diarrhoea in 195 countries: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 2018, 18, 1211–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, R.C.; Kelly, M.; Tam, J.M.; Akter, A.; Hossain, M.; Islam, K.; Biswas, R.; Kamruzzaman, M.; Chowdhury, F.; Khan, A.I.; et al. Humans Surviving Cholera Develop Antibodies against Vibrio cholerae O-Specific Polysaccharide That Inhibit Pathogen Motility. mBio 2020, 11, e02847-20. [Google Scholar] [CrossRef] [PubMed]
- Obaro, S.K.; Iroh Tam, P.-Y.; Mintz, E.D. The unrecognized burden of typhoid fever. Expert Rev. Vaccines 2017, 16, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Kelderman, K.; Phillips, A.; Pelton, T.; Schaeffer, E.; MacGillis-Falcon, P.; Bernhardt, C. The Clean Water Act at 50: Promises Half Kept at the Half-Century Mark; Environmental Integrity Project: Washington, DC, USA, 2022. [Google Scholar]
- Mueller, J.T.; Gasteyer, S. The widespread and unjust drinking water and clean water crisis in the United States. Nat. Commun. 2021, 12, 3544. [Google Scholar] [CrossRef] [PubMed]
- Tijuana River National Estuarine Research Reserve. Tijuana Estuary—Tijuana River Watershed. Available online: https://trnerr.org/about/tijuana-river-watershed/ (accessed on 1 May 2022).
- San Diego Regional Water Quality Control Board. Sewage Pollution within the Tijuana River Watershed. Available online: www.waterboards.ca.gov/sandiego/water_issues/programs/tijuana_river_valley_strategy/sewage_issue.html (accessed on 1 May 2022).
- United States Customs and Border Protection. Water Quality Analysis: Sampling Period January–June 2018; United States Customs and Border Protection: San Diego, CA, USA, 2019; p. 10. [Google Scholar]
- County of San Diego Health & Human Services Agency; Diego, L.W.S. 3-4-50: Chronic Disease Deaths in San Diego County—South Region, 2000–2018; County of San Diego: San Diego, CA, USA, 2020; p. 5. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Tange, O. GNU Parallel 2018; Zenodo: Genève, Switzerland, 2018. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [Green Version]
- Sisk-Hackworth, L. Kaiju_Table_to_OTU_table. 2020. Available online: https://github.com/residentjordan/SourceTracker2-diagnostics/tree/master/Kaiju_Table_to_OTU_table (accessed on 1 September 2020).
- County of San Diego Health and Human Services Agency Epidemiology and Immunization Services Branch. Reportable Diseases and Conditions by Year 2015–2019; County of San Diego Health and Human Services Agency: San Diego, CA, USA, 2020. [Google Scholar]
- Scholz, M. SAMtools: Get Breadth of Coverage. Available online: https://www.metagenomics.wiki/tools/samtools/breadth-of-coverage (accessed on 1 June 2021).
- Langmead, B.; Wilks, C.; Antonescu, V.; Charles, R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 2018, 35, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Palarea-Albaladejo, J.; Martín-Fernández, J.A. zCompositions—R package for multivariate imputation of left-censored data under a compositional approach. Chemom. Intell. Lab. Syst. 2015, 143, 85–96. [Google Scholar] [CrossRef]
- Gloor, G.B.; Macklaim, J.M.; Pawlowsky-Glahn, V.; Egozcue, J.J. Microbiome Datasets Are Compositional: And This Is Not Optional. Front. Microbiol. 2017, 8, 2224. [Google Scholar] [CrossRef] [Green Version]
- McGhee, J.J.; Rawson, N.; Bailey, B.A.; Fernandez-Guerra, A.; Sisk-Hackworth, L.; Kelley, S.T. Meta-SourceTracker: Application of Bayesian source tracking to shotgun metagenomics. PeerJ 2020, 8, e8783. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package, R Package Version 2.5-7. 2020. Available online: https://CRAN.R-project.org/package=vegan (accessed on 1 June 2020).
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Nshimyimana, J.P.; Ekklesia, E.; Shanahan, P.; Chua, L.H.C.; Thompson, J.R. Distribution and abundance of human-specific Bacteroides and relation to traditional indicators in an urban tropical catchment. J. Appl. Microbiol. 2014, 116, 1369–1383. [Google Scholar] [CrossRef] [Green Version]
- Morozova, V.; Fofanov, M.; Tikunova, N.; Babkin, I.; Morozov, V.V.; Tikunov, A. First crAss-Like Phage Genome Encoding the Diversity-Generating Retroelement (DGR). Viruses 2020, 12, 573. [Google Scholar] [CrossRef]
- Brown, B.P.; Jaspan, H.B.; Varsani, A. CrAssphage sp. C0521BD4, Direct Submission: October 3, 2020. The Biodesign Center of Fundamental and Applied Microbiomics, Arizona State University, 2020. Available online: https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=2780377 (accessed on 1 June 2021).
- Shkoporov, A.N.; Stockdale, S.R.; Guerin, E.; Ross, R.P.; Hill, C. uncultured phage cr7_1, Direct Submission: July 16, 2020. APC Microbiome Ireland, University College Cork, BioSciences Institute, 2020. Available online: https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=2772086 (accessed on 1 June 2021).
- Shkoporov, A.N.; Stockdale, S.R.; Guerin, E.; Ross, R.P.; Hill, C. uncultured phage cr85_1, Direct Submission: July 16, 2020. APC Microbiome Ireland, University College Cork, BioSciences Institute, 2020. Available online: https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=2772074 (accessed on 1 June 2021).
- Shkoporov, A.N.; Stockdale, S.R.; Guerin, E.; Ross, R.P.; Hill, C. uncultured phage cr8_1, Direct Submission: July 16, 2020. APC Microbiome Ireland, University College Cork, BioSciences Institute, 2020. Available online: https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/wwwtax.cgi?id=2772068 (accessed on 1 June 2021).
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; McVeigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef]
- Fox, A.N. Assessment of Bacterial Contamination in the U.S. Portion of the Tijuana River and Tijuana River Estuary. Master of Public Health Thesis, San Diego State University, San Diego, CA, USA, 2021. [Google Scholar]
- Li, H.; Buffalo, V.; Murray, K.D.; Langhorst, B.; Rik; zachcp; Klötzl, F.; Jain, C. lh3/seqtk: Toolkit for Processing Sequences in FASTA/Q Formats. 2018. Available online: https://github.com/lh3/seqtk (accessed on 1 June 2020).
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2019, 48, D517–D525. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Dinno, A. dunn.test: Dunn’s Test of Multiple Comparisons Using Rank Sums, v.1.3.5. 2017. Available online: https://cran.r-project.org/web/packages/dunn.test/index.html (accessed on 15 February 2022).
- Drancourt, M. 38—Acute Diarrhea. In Infectious Diseases, 4th ed.; Cohen, J., Powderly, W.G., Opal, S.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 335–340.e332. [Google Scholar]
- Barboza, K.; Cubillo, Z.; Castro, E.; Redondo-Solano, M.; Fernández-Jaramillo, H.; Echandi, M.L.A. First isolation report of Arcobacter cryaerophilus from a human diarrhea sample in Costa Rica. Rev. Inst. Med. Trop. Sao Paulo 2017, 59, e72. [Google Scholar] [CrossRef]
- Schlaberg, R.; Chiu, C.Y.; Miller, S.; Procop, G.W.; Weinstock, G. Validation of Metagenomic Next-Generation Sequencing Tests for Universal Pathogen Detection. Arch. Pathol. Lab. Med. 2017, 141, 776–786. [Google Scholar] [CrossRef]
- Kowalchuk, G.A.; Speksnijder, A.G.C.L.; Zhang, K.; Goodman, R.M.; van Veen, J.A. Finding the Needles in the Metagenome Haystack. Microb. Ecol. 2007, 53, 475–485. [Google Scholar] [CrossRef] [Green Version]
- Levy, S.B. Active efflux, a common mechanism for biocide and antibiotic resistance. J. Appl. Microbiol. 2002, 92, 65S–71S. [Google Scholar] [CrossRef] [PubMed]
- Webber, M.A.; Piddock, L.J.V. The importance of efflux pumps in bacterial antibiotic resistance. J. Antimicrob. Chemother. 2003, 51, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Multiple antibiotic resistance and efflux. Curr. Opin. Microbiol. 1998, 1, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Majiduddin, F.K.; Materon, I.C.; Palzkill, T.G. Molecular analysis of beta-lactamase structure and function. Int. J. Med. Microbiol. 2002, 292, 127–137. [Google Scholar] [CrossRef]
- Thompson, J.R.; Nancharaiah, Y.V.; Gu, X.; Lee, W.L.; Rajal, V.B.; Haines, M.B.; Girones, R.; Ng, L.C.; Alm, E.J.; Wuertz, S. Making waves: Wastewater surveillance of SARS-CoV-2 for population-based health management. Water Res. 2020, 184, 116181. [Google Scholar] [CrossRef]
- Rocha, A.Y.; Verbyla, M.E.; Sant, K.E.; Mladenov, N. Detection, Quantification, and Simplified Wastewater Surveillance Model of SARS-CoV-2 RNA in the Tijuana River. ACS ES T Water 2022, 2, 2134–2143. [Google Scholar] [CrossRef]
- Roy, M.A.; Arnaud, J.M.; Jasmin, P.M.; Hamner, S.; Hasan, N.A.; Colwell, R.R.; Ford, T.E. A Metagenomic Approach to Evaluating Surface Water Quality in Haiti. Int. J. Environ. Res. Public Health 2018, 15, 2211. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Average Number of Hits * |
|---|---|
| Arcobacter cryaerophilus | 140,491.1 |
| Pseudoarcobacter acticola | 98,044.3 |
| Simplicispira metamorpha | 76,102.6 |
| Bacteroides graminisolvens | 59,197.3 |
| Tolumonas auensis | 57,818.1 |
| Arcobacter suis | 50,397.2 |
| Acinetobacter johnsonii | 39,478.1 |
| Aeromonas media | 36,381.4 |
| Arcobacter butzleri | 32,238.7 |
| Rheinheimera sp. LHK132 | 32,046.6 |
| Bacterial Species | Average Hits * | Eukaryotic Species | Average Hits |
|---|---|---|---|
| Salmonella enterica | 1890.4 | Trichomonas vaginalis | 97.8 |
| Vibrio parahaemolyticus | 1602.4 | Plasmodium ovale | 36.2 |
| Streptococcus pneumoniae | 1009.0 | Plasmodium vivax | 29.1 |
| Vibrio alginolyticus | 462.8 | Cyclospora cayetanensis | 25.2 |
| Bordetella pertussis | 459.6 | Plasmodium falciparum | 23.8 |
| Francisella tularensis | 360.3 | Plasmodium yoelii | 21.7 |
| Vibrio vulnificus | 340.5 | Entamoeba histolytica | 18.4 |
| Neisseria meningitidis | 312.0 | Viruses | Average Hits |
| Yersinia enterocolitica | 273.5 | HIV-1 | 14.2 |
| Mycobacterium tuberculosis | 268.0 | Hepatitis C | 6.1 |
| Listeria monocytogenes | 101.0 | Hepatitis B | 3.7 |
| Species | Sample 1 2/12/20 | Sample 8 11/22/19 | Sample 14 12/6/19 | Sample 20 2/24/20 |
|---|---|---|---|---|
| S. enterica 45157 | 2.12% | 9.77% | 6.21% | 3.48% |
| S. enterica DA34827 | 2.62% | 10.09% | 6.77% | 3.31% |
| S. enterica FDAARGOS 94 | 2.50% | 10.14% | 6.71% | 3.72% |
| S. enterica FDAARGOS 878 | 2.47% | 10.00% | 6.59% | 3.63% |
| S. enterica LT2 | 2.48% | 10.01% | 6.63% | 3.66% |
| S. enterica SA20021456 | 1.85% | 8.06% | 5.27% | 2.88% |
| S. enterica SA20100201 | 1.97% | 8.79% | 5.68% | 3.15% |
| Escherichia coli | 11.87% | 78.56% | 55.51% | 26.03% |
| Arcobacter cryaerophilus | 91.27% | 79.60% | 94.81% | 94.20% |
| HIV-1 | 0% | 0% | 0% | 0% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allsing, N.; Kelley, S.T.; Fox, A.N.; Sant, K.E. Metagenomic Analysis of Microbial Contamination in the U.S. Portion of the Tijuana River Watershed. Int. J. Environ. Res. Public Health 2023, 20, 600. https://doi.org/10.3390/ijerph20010600
Allsing N, Kelley ST, Fox AN, Sant KE. Metagenomic Analysis of Microbial Contamination in the U.S. Portion of the Tijuana River Watershed. International Journal of Environmental Research and Public Health. 2023; 20(1):600. https://doi.org/10.3390/ijerph20010600
Chicago/Turabian StyleAllsing, Nicholas, Scott T. Kelley, Alexandra N. Fox, and Karilyn E. Sant. 2023. "Metagenomic Analysis of Microbial Contamination in the U.S. Portion of the Tijuana River Watershed" International Journal of Environmental Research and Public Health 20, no. 1: 600. https://doi.org/10.3390/ijerph20010600
APA StyleAllsing, N., Kelley, S. T., Fox, A. N., & Sant, K. E. (2023). Metagenomic Analysis of Microbial Contamination in the U.S. Portion of the Tijuana River Watershed. International Journal of Environmental Research and Public Health, 20(1), 600. https://doi.org/10.3390/ijerph20010600