
Correlations among Antibiotic Resistance Genes, Mobile Genetic Elements and Microbial Communities in Municipal Sewage Treatment Plants Revealed by High-Throughput Sequencing
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. MSTPs and Samples Collection
2.2. PCR of Tetracycline Resistance Genes
2.3. Cloning and Phylogenetic Analysis of Tet Genes
2.4. q-PCRs of Tet Genes
2.5. High-Throughput Sequencing
2.6. Statistics
3. Results
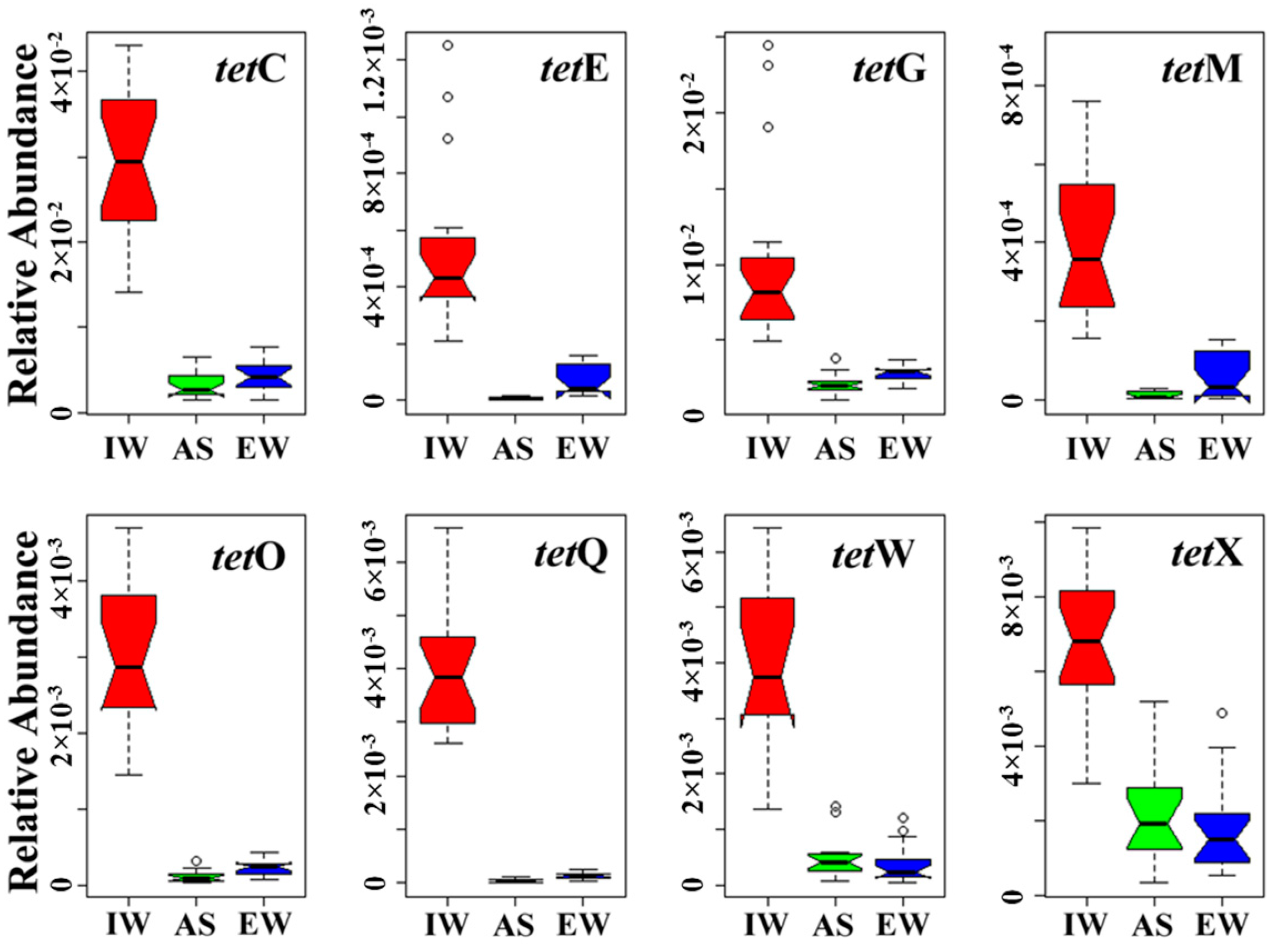
3.1. Occurrence and Abundance of Tet Genes in JXZ-MSTP
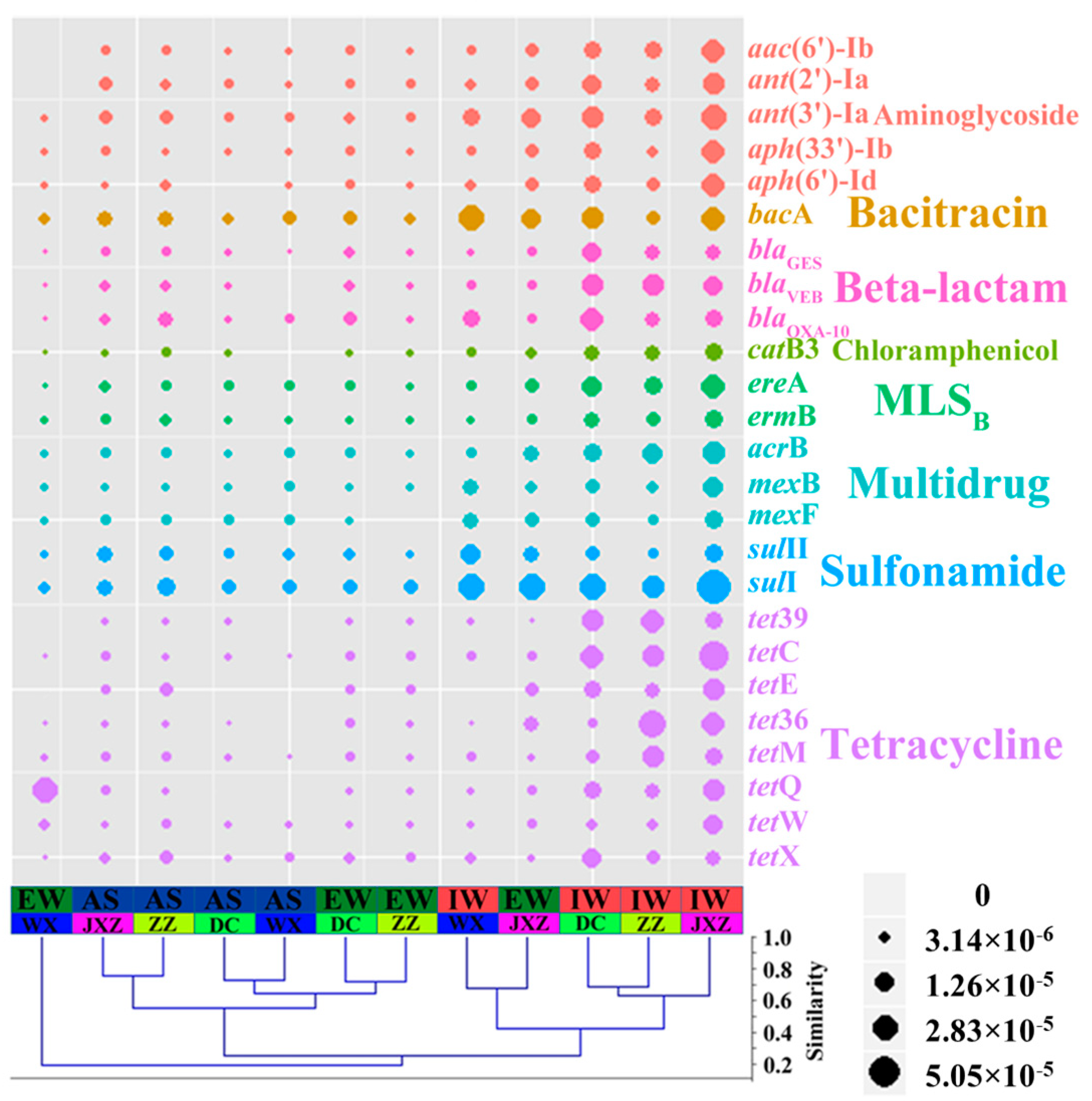
3.2. Abundance and Diversity of ARGs in Four MSTPs
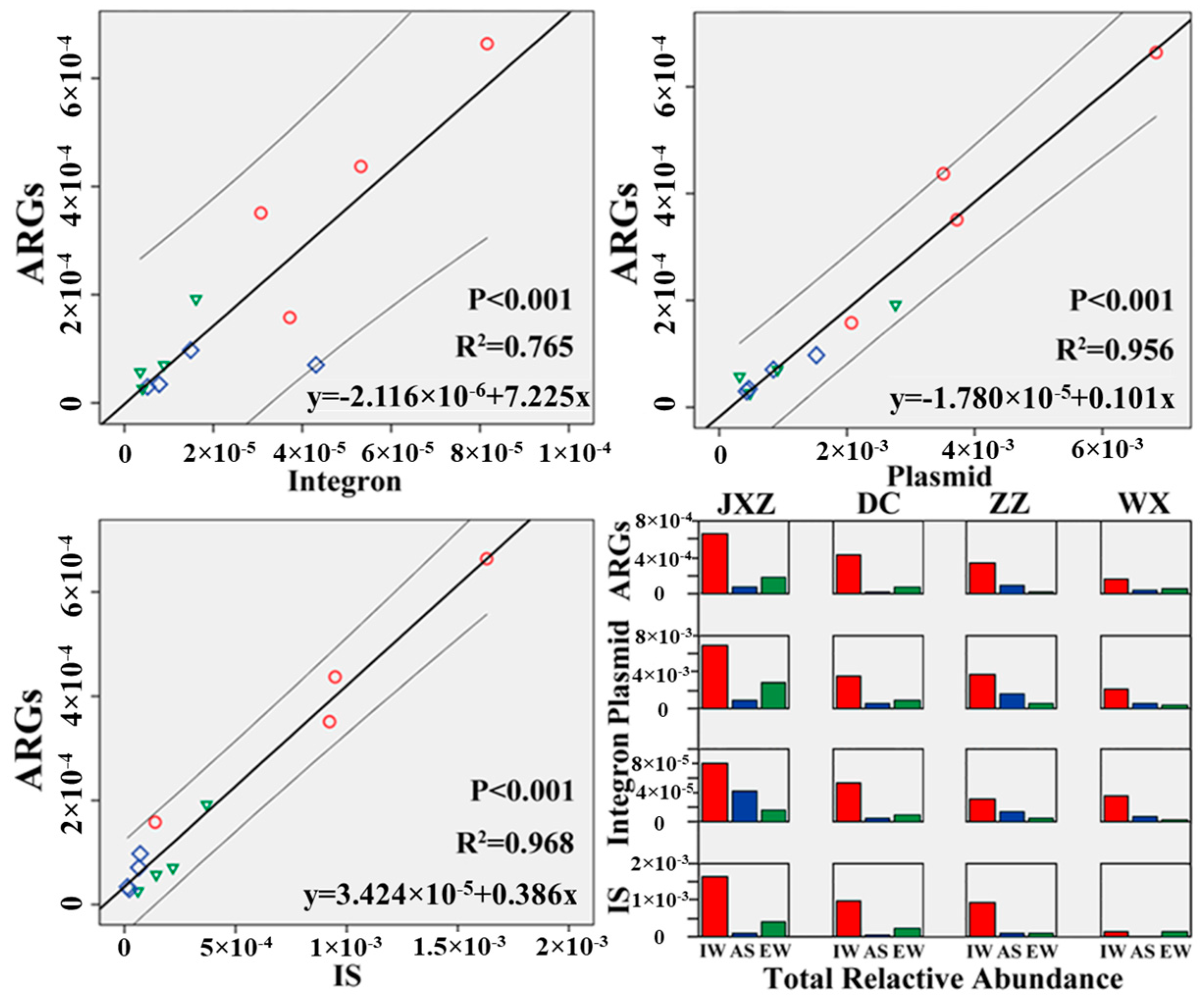
3.3. Correlation between ARGs and MGEs in the MSTPs
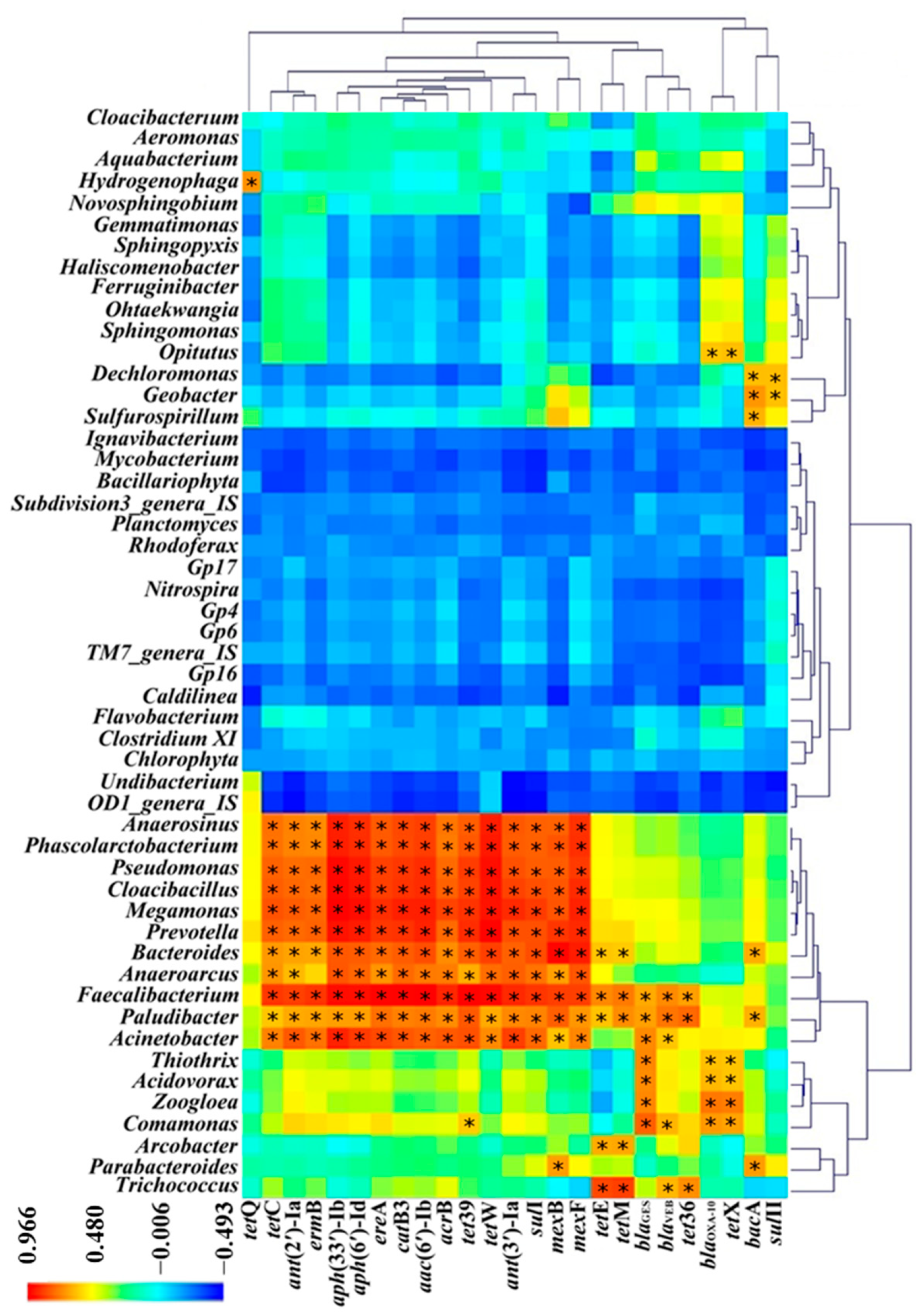
3.4. Correlation between ARGs and Bacterial Communities in MSTPs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Larsson, D.G.J.; Flach, C.-F. Antibiotic Resistance in the Environment. Nat. Rev. Microbiol. 2022, 20, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, Q.E.; Zhou, X.; Wang, F.-H.; Muurinen, J.; Virta, M.P.; Brandt, K.K.; Zhu, Y.-G. Antibiotic Resistome in the Livestock and Aquaculture Industries: Status and Solutions. Crit. Rev. Environ. Sci. Technol. 2021, 51, 2159–2196. [Google Scholar] [CrossRef]
- Rawson, T.M.; Wilson, R.C.; O’Hare, D.; Herrero, P.; Kambugu, A.; Lamorde, M.; Ellington, M.; Georgiou, P.; Cass, A.; Hope, W.W.; et al. Optimizing Antimicrobial Use: Challenges, Advances and Opportunities. Nat. Rev. Microbiol. 2021, 19, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, L.; Manaia, C.; Merlin, C.; Schwartz, T.; Dagot, C.; Ploy, M.C.; Michael, I.; Fatta-Kassinos, D. Urban Wastewater Treatment Plants as Hotspots for Antibiotic Resistant Bacteria and Genes Spread into the Environment: A Review. Sci. Total Environ. 2013, 447, 345–360. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Ju, F.; Zhang, T. Tracking Human Sewage Microbiome in a Municipal Wastewater Treatment Plant. Appl. Microbiol. Biotechnol. 2014, 98, 3317–3326. [Google Scholar] [CrossRef]
- Abdi, S.N.; Ghotaslou, R.; Ganbarov, K.; Mobed, A.; Tanomand, A.; Yousefi, M.; Asgharzadeh, M.; Kafil, H.S. Acinetobacter Baumannii Efflux Pumps and Antibiotic Resistance. Infect. Drug Resist. 2020, 13, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Salyers, A.; Gupta, A.; Wang, Y. Human Intestinal Bacteria as Reservoirs for Antibiotic Resistance Genes. Trends Microbiol. 2004, 12, 412–416. [Google Scholar] [CrossRef]
- Allen, T.D.; Lawson, P.A.; Collins, M.D.; Falsen, E.; Tanner, R.S. Cloacibacterium Normanense Gen. Nov., Sp. Nov., a Novel Bacterium in the Family Flavobacteriaceae Isolated from Municipal Wastewater. Int. J. Syst. Evol. Microbiol. 2006, 56, 1311–1316. [Google Scholar] [CrossRef] [Green Version]
- Frost, L.S.; Leplae, R.; Summers, A.O.; Toussaint, A. Mobile Genetic Elements: The Agents of Open Source Evolution. Nat. Rev. Microbiol. 2005, 3, 722–732. [Google Scholar] [CrossRef]
- Wright, M.S.; Baker-Austin, C.; Lindell, A.H.; Stepanauskas, R.; Stokes, H.W.; McArthur, J.V. Influence of Industrial Contamination on Mobile Genetic Elements: Class 1 Integron Abundance and Gene Cassette Structure in Aquatic Bacterial Communities. ISME J. 2008, 2, 417–428. [Google Scholar] [CrossRef]
- Cai, L.; Zhang, T. Detecting Human Bacterial Pathogens in Wastewater Treatment Plants by a High-Throughput Shotgun Sequencing Technique. Environ. Sci. Technol. 2013, 47, 5433–5441. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, E.A.; Seyfried, E.E.; McMahon, K.D. Tetracycline Resistance Genes in Activated Sludge Wastewater Treatment Plants. Water Res. 2007, 41, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhang, M.; Zhang, X.; Fang, H.H. Tetracycline Resistance Genes and Tetracycline Resistant Lactose-Fermenting Enterobacteriaceae in Activated Sludge of Sewage Treatment Plants. Environ. Sci. Technol. 2009, 43, 3455–3460. [Google Scholar] [CrossRef] [PubMed]
- Galvin, S.; Boyle, F.; Hickey, P.; Vellinga, A.; Morris, D.; Cormican, M. Enumeration and Characterization of Antimicrobial-Resistant Escherichia Coli Bacteria in Effluent from Municipal, Hospital, and Secondary Treatment Facility Sources. Appl. Environ. Microbiol. 2010, 76, 4772–4779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczepanowski, R.; Linke, B.; Krahn, I.; Gartemann, K.-H.; Gützkow, T.; Eichler, W.; Pühler, A.; Schlüter, A. Detection of 140 Clinically Relevant Antibiotic Resistance Genes in the Plasmid Metagenome of Wastewater Treatment Plant Bacteria Showing Reduced Susceptibility to Selected Antibiotics. Microbiology 2009, 155, 2306–2319. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-X.; Zhang, T. Occurrence, Abundance, and Diversity of Tetracycline Resistance Genes in 15 Sewage Treatment Plants across China and Other Global Locations. Environ. Sci. Technol. 2011, 45, 2598–2604. [Google Scholar] [CrossRef]
- Walsh, F.; Cooke, N.M.; Smith, S.G.; Moran, G.P.; Cooke, F.J.; Ivens, A.; Wain, J.; Rogers, T.R. Comparison of Two DNA Microarrays for Detection of Plasmid-Mediated Antimicrobial Resistance and Virulence Factor Genes in Clinical Isolates of Enterobacteriaceae and Non-Enterobacteriaceae. Int. J. Antimicrob. Agents 2010, 35, 593–598. [Google Scholar] [CrossRef]
- Looft, T.; Johnson, T.A.; Allen, H.K.; Bayles, D.O.; Alt, D.P.; Stedtfeld, R.D.; Sul, W.J.; Stedtfeld, T.M.; Chai, B.; Cole, J.R.; et al. In-Feed Antibiotic Effects on the Swine Intestinal Microbiome. Proc. Natl. Acad. Sci. USA 2012, 109, 1691–1696. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhang, X.-X.; Huang, K.; Miao, Y.; Shi, P.; Liu, B.; Long, C.; Li, A. Metagenomic Profiling of Antibiotic Resistance Genes and Mobile Genetic Elements in a Tannery Wastewater Treatment Plant. PLoS ONE 2013, 8, e76079. [Google Scholar] [CrossRef]
- Yang, Y.; Li, B.; Ju, F.; Zhang, T. Exploring Variation of Antibiotic Resistance Genes in Activated Sludge over a Four-Year Period through a Metagenomic Approach. Environ. Sci. Technol. 2013, 47, 10197–10205. [Google Scholar] [CrossRef]
- Dethlefsen, L.; Relman, D.A. Incomplete Recovery and Individualized Responses of the Human Distal Gut Microbiota to Repeated Antibiotic Perturbation. Proc. Natl. Acad. Sci. USA 2011, 108, 4554–4561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemec, A. Diversity of Aminoglycoside-Resistance Genes and Their Association with Class 1 Integrons among Strains of Pan-European Acinetobacter Baumannii Clones. J. Med. Microbiol. 2004, 53, 1233–1240. [Google Scholar] [CrossRef] [PubMed]
- Segal, H.; Garny, S.; Elisha, B.G. Is ISABA-1Customized for Acinetobacter? FEMS Microbiol. Lett. 2005, 243, 425–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Claesson, M.J.; O’Sullivan, O.; Wang, Q.; Nikkilä, J.; Marchesi, J.R.; Smidt, H.; de Vos, W.M.; Ross, R.P.; O’Toole, P.W. Comparative Analysis of Pyrosequencing and a Phylogenetic Microarray for Exploring Microbial Community Structures in the Human Distal Intestine. PLoS ONE 2009, 4, e6669. [Google Scholar] [CrossRef] [Green Version]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Mark Welch, D.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial Diversity in the Deep Sea and the Underexplored “Rare Biosphere. ” Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Tang, J.; Zhang, X.-X.; Xu, K.; Ren, H. A Comprehensive Insight into Tetracycline Resistant Bacteria and Antibiotic Resistance Genes in Activated Sludge Using Next-Generation Sequencing. Int. J. Mol. Sci. 2014, 15, 10083–10100. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Li, B. Occurrence, Transformation, and Fate of Antibiotics in Municipal Wastewater Treatment Plants. Crit. Rev. Environ. Sci. Technol. 2011, 41, 951–998. [Google Scholar] [CrossRef]
- Zhang, X.-X.; Zhang, T.; Zhang, M.; Fang, H.H.P.; Cheng, S.-P. Characterization and Quantification of Class 1 Integrons and Associated Gene Cassettes in Sewage Treatment Plants. Appl. Microbiol. Biotechnol. 2009, 82, 1169–1177. [Google Scholar] [CrossRef]
- Li, B.; Zhang, T. Biodegradation and Adsorption of Antibiotics in the Activated Sludge Process. Environ. Sci. Technol. 2010, 44, 3468–3473. [Google Scholar] [CrossRef]
- Shanks, O.C.; Newton, R.J.; Kelty, C.A.; Huse, S.M.; Sogin, M.L.; McLellan, S.L. Comparison of the Microbial Community Structures of Untreated Wastewaters from Different Geographic Locales. Appl. Environ. Microbiol. 2013, 79, 2906–2913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reischer, G.H.; Ebdon, J.E.; Bauer, J.M.; Schuster, N.; Ahmed, W.; Åström, J.; Blanch, A.R.; Blöschl, G.; Byamukama, D.; Coakley, T.; et al. Performance Characteristics of QPCR Assays Targeting Human- and Ruminant-Associated Bacteroidetes for Microbial Source Tracking across Sixteen Countries on Six Continents. Environ. Sci. Technol. 2013, 47, 8548–8556. [Google Scholar] [CrossRef] [PubMed]
- Sanapareddy, N.; Hamp, T.J.; Gonzalez, L.C.; Hilger, H.A.; Fodor, A.A.; Clinton, S.M. Molecular Diversity of a North Carolina Wastewater Treatment Plant as Revealed by Pyrosequencing. Appl. Environ. Microbiol. 2009, 75, 1688–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, V.; Poirel, L.; Marie, C.; Arpin, C.; Nordmann, P.; Quentin, C. Molecular Characterization of a Novel Class 1 Integron Containing BlaGES-1 and a Fused Product of Aac(3)-Ib/Aac(6″)-Ib″ Gene Cassettes in Pseudomonas Aeruginosa . Antimicrob. Agents Chemother. 2002, 46, 638–645. [Google Scholar] [CrossRef] [Green Version]
- Dintner, S.; Staroń, A.; Berchtold, E.; Petri, T.; Mascher, T.; Gebhard, S. Coevolution of ABC Transporters and Two-Component Regulatory Systems as Resistance Modules against Antimicrobial Peptides in Firmicutes Bacteria. J. Bacteriol. 2011, 193, 3851–3862. [Google Scholar] [CrossRef] [Green Version]
- Vila, J.; Martí, S.; Sánchez-Céspedes, J. Porins, Efflux Pumps and Multidrug Resistance in Acinetobacter Baumannii. J. Antimicrob. Chemother. 2007, 59, 1210–1215. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Zhang, X.-X.; Cheng, S.; Zhang, Z.; Shi, P.; Liu, B.; Wu, B.; Zhang, Y. Occurrence, Abundance and Elimination of Class 1 Integrons in One Municipal Sewage Treatment Plant. Ecotoxicology 2011, 20, 968–973. [Google Scholar] [CrossRef]
- Tennstedt, T.; Szczepanowski, R.; Braun, S.; Pühler, A.; Schlüter, A. Occurrence of Integron-Associated Resistance Gene Cassettes Located on Antibiotic Resistance Plasmids Isolated from a Wastewater Treatment Plant. FEMS Microbiol. Ecol. 2003, 45, 239–252. [Google Scholar] [CrossRef]
- Zhang, X.-X.; Zhang, T.; Fang, H.H.P. Antibiotic Resistance Genes in Water Environment. Appl. Microbiol. Biotechnol. 2009, 82, 397–414. [Google Scholar] [CrossRef]
- Ng, L.-K.; Martin, I.; Alfa, M.; Mulvey, M. Multiplex PCR for the Detection of Tetracycline Resistant Genes. Mol. Cell. Probes 2001, 15, 209–215. [Google Scholar] [CrossRef]
- Lee, C.; Langlois, B.E.; Dawson, K.A. Detection of Tetracycline Resistance Determinants in Pig Isolates from Three Herds with Different Histories of Antimicrobial Agent Exposure. Appl. Environ. Microbiol. 1993, 59, 1467–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Gutiérrez, J.C.; Henry, S.; Hallet, S.; Martin-Laurent, F.; Catroux, G.; Philippot, L. Quantification of a Novel Group of Nitrate-Reducing Bacteria in the Environment by Real-Time PCR. J. Microbiol. Methods 2004, 57, 399–407. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, F.; Wang, B.; Huang, K.; Yin, J.; Ren, X.; Wang, Z.; Zhang, X.-X. Correlations among Antibiotic Resistance Genes, Mobile Genetic Elements and Microbial Communities in Municipal Sewage Treatment Plants Revealed by High-Throughput Sequencing. Int. J. Environ. Res. Public Health 2023, 20, 3593. https://doi.org/10.3390/ijerph20043593
Zhao F, Wang B, Huang K, Yin J, Ren X, Wang Z, Zhang X-X. Correlations among Antibiotic Resistance Genes, Mobile Genetic Elements and Microbial Communities in Municipal Sewage Treatment Plants Revealed by High-Throughput Sequencing. International Journal of Environmental Research and Public Health. 2023; 20(4):3593. https://doi.org/10.3390/ijerph20043593
Chicago/Turabian StyleZhao, Fuzheng, Bo Wang, Kailong Huang, Jinbao Yin, Xuechang Ren, Zhu Wang, and Xu-Xiang Zhang. 2023. "Correlations among Antibiotic Resistance Genes, Mobile Genetic Elements and Microbial Communities in Municipal Sewage Treatment Plants Revealed by High-Throughput Sequencing" International Journal of Environmental Research and Public Health 20, no. 4: 3593. https://doi.org/10.3390/ijerph20043593