
Regulation of Ketone Body Metabolism and the Role of PPARα



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
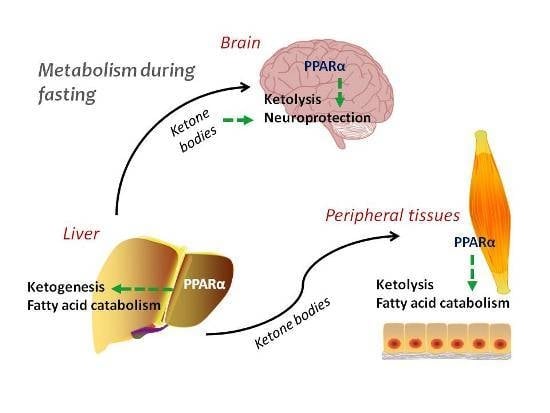
1. Introduction
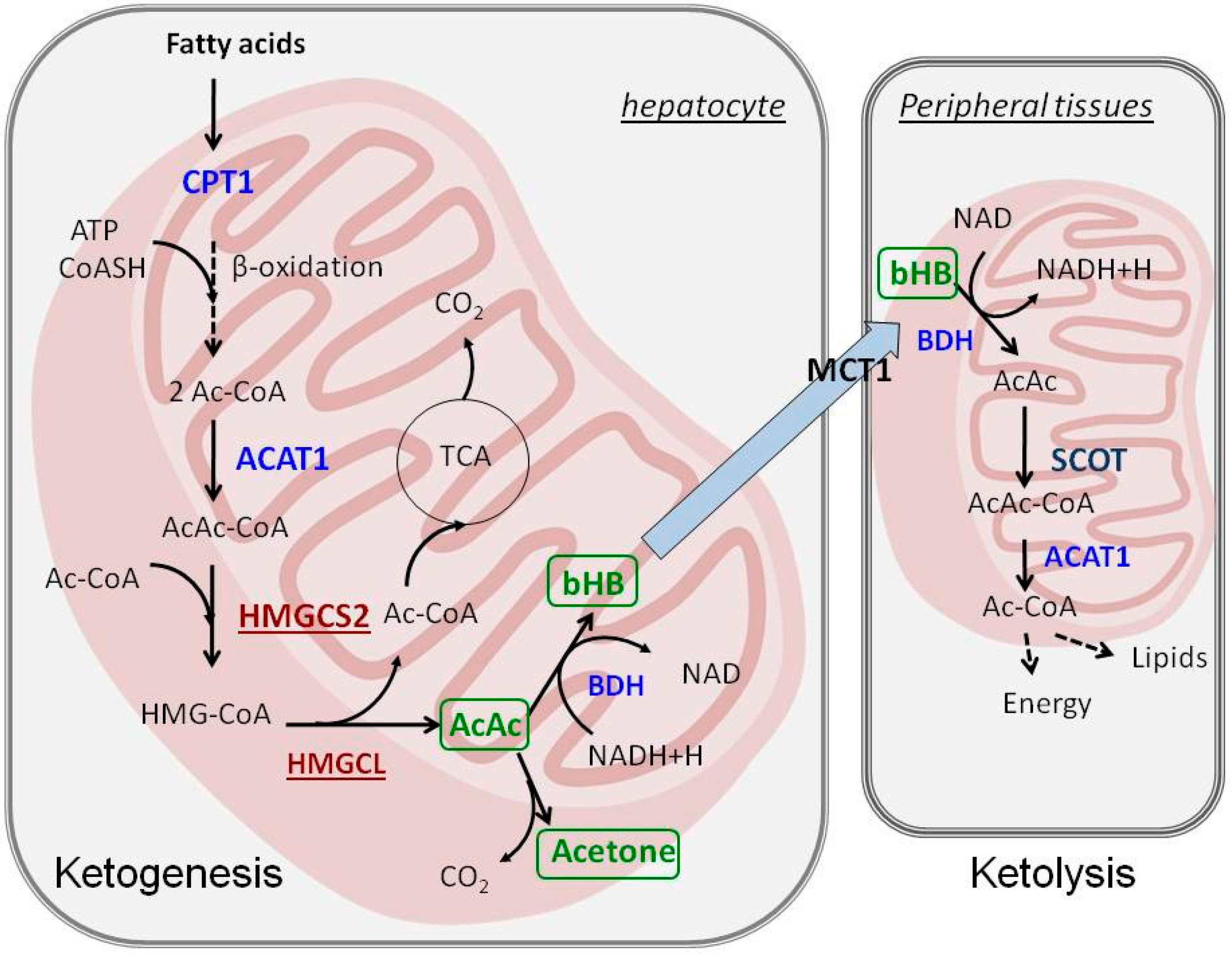
2. Ketogenesis and Ketolysis
3. Regulation of Ketogenesis—The Role of PPARα
3.1. Endocrine Regulation
3.2. Transcriptional Regulation
3.3. Posttranslational Modifications
3.4. Biochemical Regulation
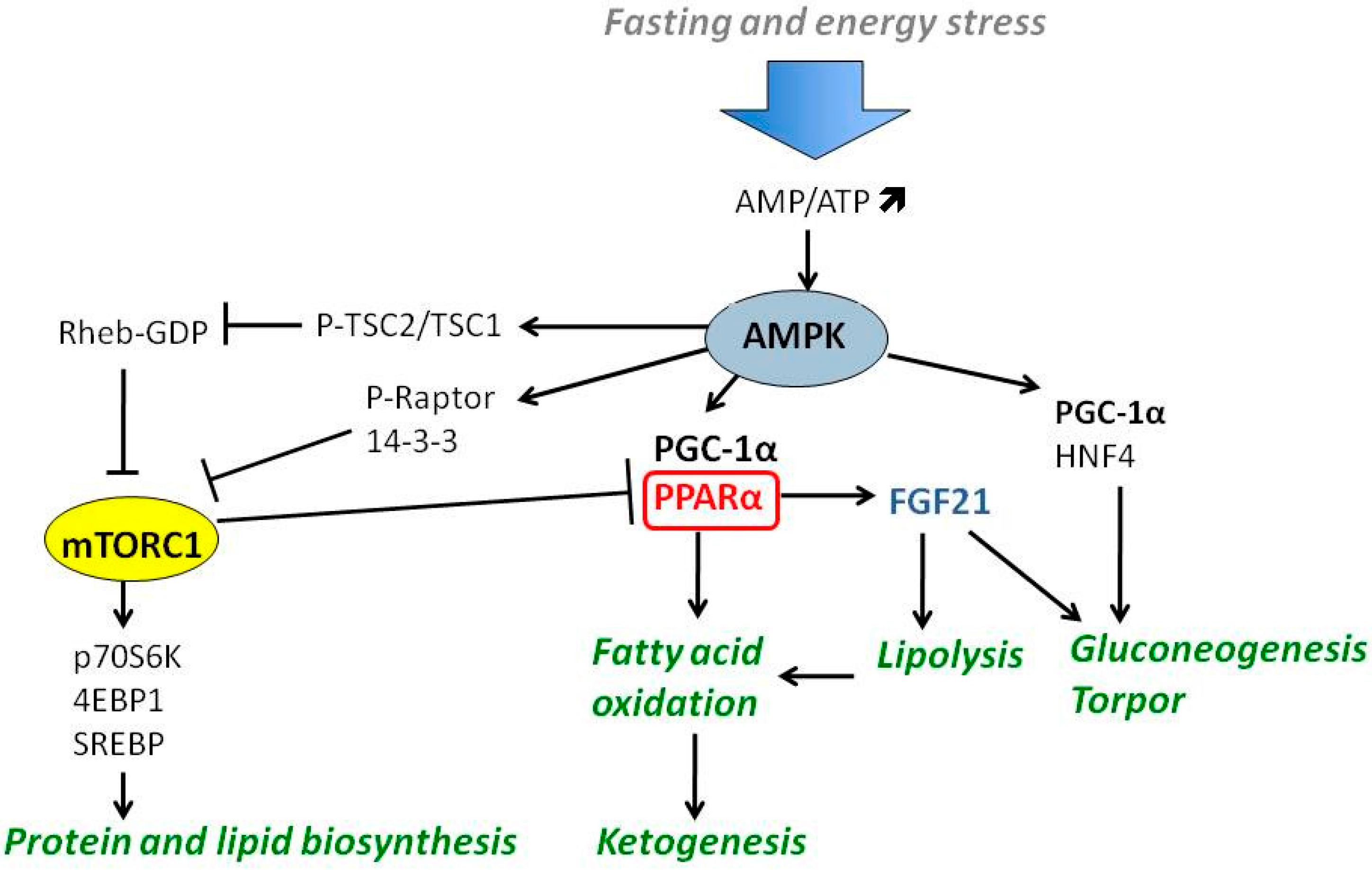
4. Nutrient-Responsive Intracellular Signaling in the Regulation of Ketogenesis
4.1. PGC-1α-PPARα-FGF21 Axis
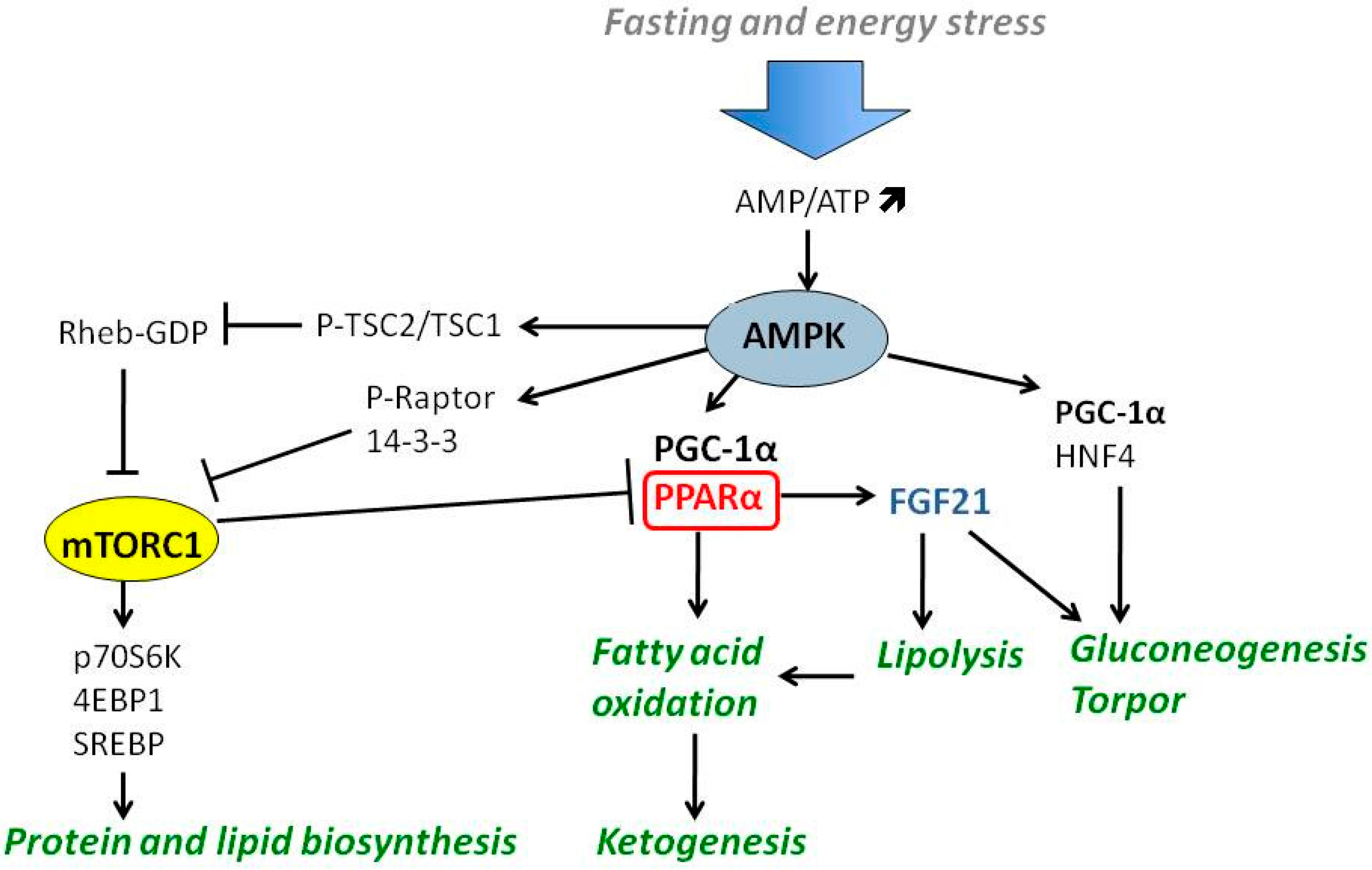
4.2. The Role of AMPK and mTOR
4.3. Ketone Bodies as Signaling Intermediates
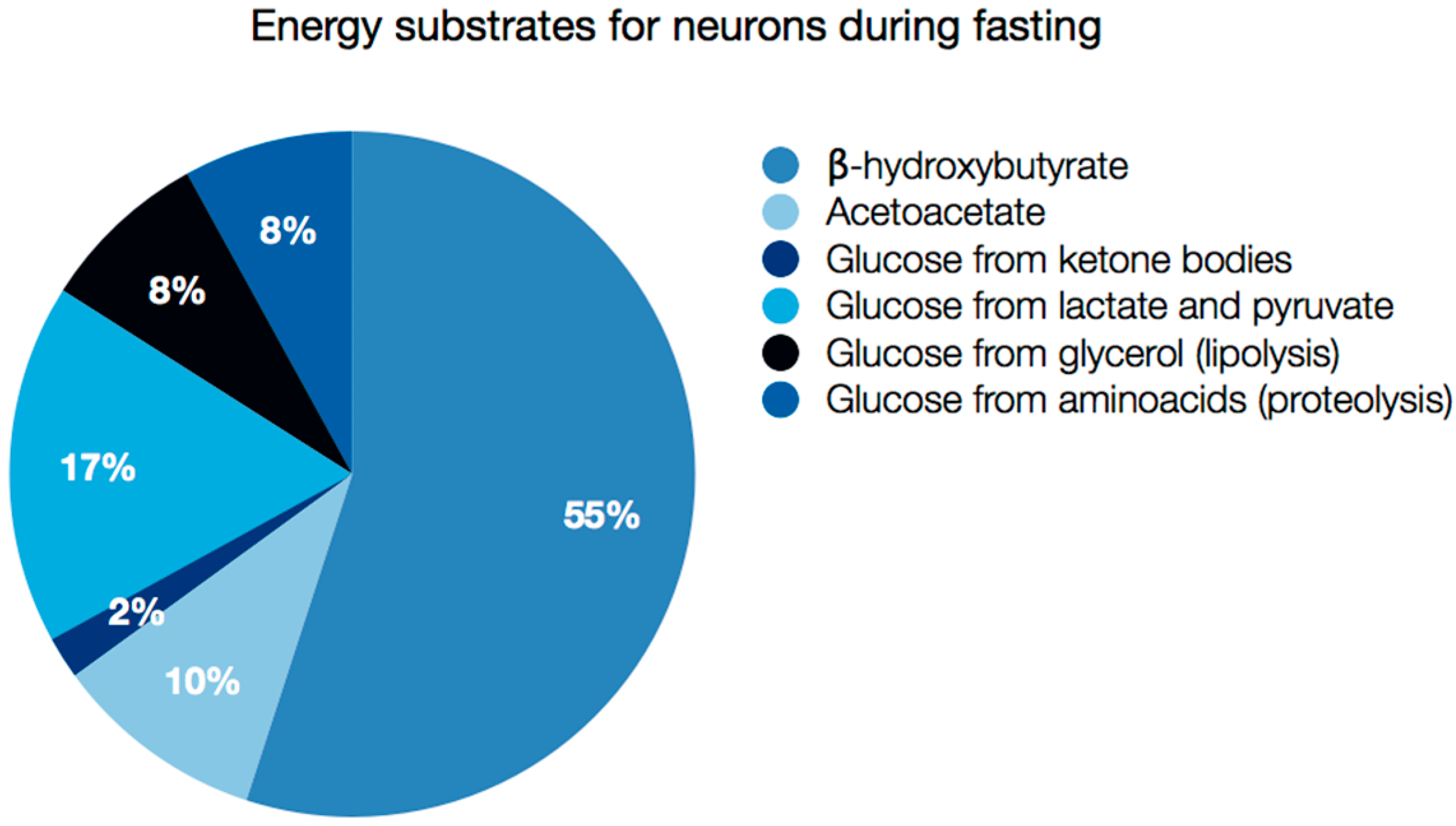
5. Brain as an Example of Ketolytic Organ
6. Neuroprotective and Therapeutic Activity of Ketone Bodies in Central Nervous System Pathologies
6.1. Epilepsy
6.2. Neurodegenerative Diseases
6.3. Traumatic Brain Injuries
6.4. Anti-Inflammatory Actions of PPARα and Ketone Bodies
7. Ketogenesis and Ketolysis in Cancer Cells
8. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ACAT1 | Acetoacetyl-Coa Thiolase 1 |
| ACC | Acetyl-CoA carboxylase |
| AD | Alzheimer’s disease |
| AMPK | AMP-activated kinase |
| BCAA | Branched chain amino acids |
| BCKD | Branched keto acid dehydrogenase |
| BDH | β-Hydroxybutyrate dehydrogenase |
| bHB | β-Hydroxy butyrate |
| CNS | Central nervous system |
| COUP-TF | Chicken ovoalbumin-upstream promoter transcription factor |
| COX-2 | Cyclooxygenase 2 |
| CPT1 | Carnitine palmitoyltransferase 1 |
| CREB | cAMP responsive element binding protein |
| FGF21 | Fibroblast growth factor 21 |
| GABA | γ amino butyric acid |
| HMGCL | 3-Hydroxy-3-methylglutaryl-CoA lyase |
| HMGCR | 3-Hydroxy-3-methylglutaryl-CoA reductase |
| HMGCS2 | 3-Hydroxy-3-methylglutaryl-CoA synthetase |
| HNF4 | Hepatocyte nuclear factor 4 |
| LCAD | Long chain acyl-CoA dehydrogenase |
| MCAD | Medium chain acyl-CoA dehydrogenase |
| MCT1 | Monocarboxylate transporter 1 |
| mTOR | Mammalian/mechanistic target of rapamycin |
| mTORC1 | mTOR complex 1 |
| NCoR | Nuclear coactivator repressor |
| NEFA | Nonesterified fatty acids |
| PD | Parkinson’s disease |
| PGC-1α | PPARγ coactivator 1α |
| PPAR | Peroxisome proliferator activated receptor |
| PPRE | Peroxisome proliferator response element |
| PUFA | Polyunsaturated fatty acid |
| ROR | Retinoid orphan receptor |
| SCOT | Succinyl-CoA:3-ketoacid-CoA transferase |
| TBI | Traumatic brain injury |
| TCA | Tricarboxylic acid cycle |
| TSC2 | Tuberous sclerosis complex 2 |
References
- Owen, O.E.; Morgan, A.P.; Kemp, H.G.; Sullivan, J.M.; Herrera, M.G.; Cahill, G.F., Jr. Brain metabolism during fasting. J. Clin. Investig. 1967, 46, 1589–1595. [Google Scholar] [CrossRef] [PubMed]
- Cahill, G.F., Jr. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Veech, R.L. The therapeutic implications of ketone bodies: The effects of ketone bodies in pathological conditions: Ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.W. Malonyl-coa: The regulator of fatty acid synthesis and oxidation. J. Clin. Investig. 2012, 122, 1958–1959. [Google Scholar] [CrossRef] [PubMed]
- Alberti, K.G.; Johnston, D.G.; Gill, A.; Barnes, A.J.; Orskov, H. Hormonal regulation of ketone-body metabolism in man. Biochem. Soc. Symp. 1978, 43, 163–182. [Google Scholar]
- McGarry, J.D.; Foster, D.W. Regulation of ketogenesis and clinical aspects of the ketotic state. Metabolism 1972, 21, 471–489. [Google Scholar] [CrossRef]
- Schade, D.S.; Eaton, R.P. Modulation of fatty acid metabolism by glucagon in man. II. Effects in insulin-deficient diabetics. Diabetes 1975, 24, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Keller, U.; Lustenberger, M.; Muller-Brand, J.; Gerber, P.P.; Stauffacher, W. Human ketone body production and utilization studied using tracer techniques: Regulation by free fatty acids, insulin, catecholamines, and thyroid hormones. Diabetes Metab. Rev. 1989, 5, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Bahnsen, M.; Burrin, J.M.; Johnston, D.G.; Pernet, A.; Walker, M.; Alberti, K.G. Mechanisms of catecholamine effects on ketogenesis. Am. J. Physiol. 1984, 247, E173–E180. [Google Scholar] [PubMed]
- Konig, B.; Koch, A.; Giggel, K.; Dordschbal, B.; Eder, K.; Stangl, G.I. Monocarboxylate transporter (MCT)-1 is up-regulated by PPARα. Biochim. Biophys. Acta 2008, 1780, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Schoonjans, K.; Staels, B.; Auwerx, J. Role of the peroxisome proliferator-activated receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression. J. Lipid Res. 1996, 37, 907–925. [Google Scholar] [PubMed]
- Qi, C.; Zhu, Y.; Reddy, J.K. Peroxisome proliferator-activated receptors, coactivators, and downstream targets. Cell Biochem. Biophys. 2000, 32, 187–204. [Google Scholar] [CrossRef]
- Varanasi, U.; Chu, R.; Huang, Q.; Castellon, R.; Yeldandi, A.V.; Reddy, J.K. Identification of a peroxisome proliferator-responsive element upstream of the human peroxisomal fatty acyl coenzyme a oxidase gene. J. Biol. Chem. 1996, 271, 2147–2155. [Google Scholar] [PubMed]
- Motojima, K. Differential effects of PPARα activators on induction of ectopic expression of tissue-specific fatty acid binding protein genes in the mouse liver. Int. J. Biochem. Cell Biol. 2000, 32, 1085–1092. [Google Scholar] [CrossRef]
- Kersten, S.; Seydoux, J.; Peters, J.M.; Gonzalez, F.J.; Desvergne, B.; Wahli, W. Peroxisome proliferator-activated receptor α mediates the adaptive response to fasting. J. Clin. Investig. 1999, 103, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of ppars in health and disease. Nature 2000, 405, 421–424. [Google Scholar] [PubMed]
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Cook, W.S.; Qi, C.; Yeldandi, A.V.; Reddy, J.K.; Rao, M.S. Defect in peroxisome proliferator-activated receptor α-inducible fatty acid oxidation determines the severity of hepatic steatosis in response to fasting. J. Biol. Chem. 2000, 275, 28918–28928. [Google Scholar] [CrossRef] [PubMed]
- Leone, T.C.; Weinheimer, C.J.; Kelly, D.P. A critical role for the peroxisome proliferator-activated receptor α (PPARα) in the cellular fasting response: The pparalpha-null mouse as a model of fatty acid oxidation disorders. Proc. Natl. Acad. Sci. USA 1999, 96, 7473–7478. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.C.; Gil-Gomez, G.; Hegardt, F.G.; Haro, D. Peroxisome proliferator-activated receptor mediates induction of the mitochondrial 3-hydroxy-3-methylglutaryl-COA synthase gene by fatty acids. J. Biol. Chem. 1994, 269, 18767–18772. [Google Scholar] [PubMed]
- Hegardt, F.G. Mitochondrial 3-hydroxy-3-methylglutaryl-coa synthase: A control enzyme in ketogenesis. Biochem. J. 1999, 338, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.M.; Miyata, K.S.; Cechetto, J.D.; Rachubinski, R.A.; Capone, J.P. A mitochondrial ketogenic enzyme regulates its gene expression by association with the nuclear hormone receptor PPARα. EMBO J. 1998, 17, 6972–6978. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Reddy, J.K. Transcription coactivators for peroxisome proliferator-activated receptors. Biochim. Biophys. Acta 2007, 1771, 936–951. [Google Scholar] [CrossRef] [PubMed]
- Kostiuk, M.A.; Keller, B.O.; Berthiaume, L.G. Palmitoylation of ketogenic enzyme HMGCs2 enhances its interaction with PPARα and transcription at the HMGCS2 PPRE. FASEB J. 2010, 24, 1914–1924. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.C.; Ortiz, J.A.; Hegardt, F.G.; Haro, D. Chicken ovalbumin upstream-promoter transcription factor (COUP-TF) could act as a transcriptional activator or repressor of the mitochondrial 3-hydroxy-3-methylglutaryl-COA synthase gene. Biochem. J. 1997, 326, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Nadal, A.; Marrero, P.F.; Haro, D. Down-regulation of the mitochondrial 3-hydroxy-3-methylglutaryl-COA synthase gene by insulin: The role of the forkhead transcription factor FKHRL1. Biochem. J. 2002, 366, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Wolfrum, C.; Asilmaz, E.; Luca, E.; Friedman, J.M.; Stoffel, M. FOXA2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature 2004, 432, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.C.; Ortiz, J.A.; Hegardt, F.G.; Haro, D. The hepatocyte nuclear factor 4 (HNF-4) represses the mitochondrial HMG-COA synthase gene. Biochem. Biophys. Res. Commun. 1998, 242, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Hirschey, M.D.; Shimazu, T.; Capra, J.A.; Pollard, K.S.; Verdin, E. SIRT1 and SIRT3 deacetylate homologous substrates: ACECS1,2 and HMGCS1,2. Aging 2011, 3, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.M.; Tubbs, P.K. Succinylation and inactivation of 3-hydroxy-3-methylglutaryl-COA synthase by succinyl-COA and its possible relevance to the control of ketogenesis. Biochem. J. 1985, 232, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Quant, P.A.; Tubbs, P.K.; Brand, M.D. Glucagon activates mitochondrial 3-hydroxy-3-methylglutaryl-COA synthase in vivo by decreasing the extent of succinylation of the enzyme. Eur. J. Biochem. 1990, 187, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Rardin, M.J.; He, W.; Nishida, Y.; Newman, J.C.; Carrico, C.; Danielson, S.R.; Guo, A.; Gut, P.; Sahu, A.K.; Li, B.; et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013, 18, 920–933. [Google Scholar] [CrossRef] [PubMed]
- Cleland, W.W. The kinetics of enzyme-catalyzed reactions with two or more substrates or products. I. Nomenclature and rate equations. Biochim. Biophys. Acta 1963, 67, 104–137. [Google Scholar] [CrossRef]
- Lopes-Cardozo, M.; van den Bergh, S.G. Ketogenesis in isolated rat liver mitochondria. I. Relationships with the citric acid cycle and with the mitochondrial energy state. Biochim. Biophys. Acta 1972, 283, 1–15. [Google Scholar] [CrossRef]
- Huth, W.; Jonas, R.; Wunderlich, I.; Seubert, W. On the mechanism of ketogenesis and its control. Purification, kinetic mechanism and regulation of different forms of mitochondrial acetoacetyl-COA thiolases from OX liver. Eur. J. Biochem. 1975, 59, 475–489. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Mannaerts, G.P.; Foster, D.W. A possible role for malonyl-COA in the regulation of hepatic fatty acid oxidation and ketogenesis. J. Clin. Investig. 1977, 60, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.K.; Ittmann, M.; Cooper, C. The role of leucine in ketogenesis in starved rats. Biochem. J. 1982, 204, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Noda, C.; Ichihara, A. Control of ketogenesis from amino acids. IV. Tissue specificity in oxidation of leucine, tyrosine, and lysine. J. Biochem. 1976, 80, 1159–1164. [Google Scholar] [PubMed]
- Williamson, J.R.; Walajtys-Rode, E.; Coll, K.E. Effects of branched chain α-ketoacids on the metabolism of isolated rat liver cells. I. Regulation of branched chain α-ketoacid metabolism. J. Biol. Chem. 1979, 254, 11511–11520. [Google Scholar] [PubMed]
- Brosnan, J.T.; Brosnan, M.E. Branched-chain amino acids: Enzyme and substrate regulation. J. Nutr. 2006, 136, 207S–211S. [Google Scholar] [PubMed]
- Liebich, H.M.; Forst, C. Hydroxycarboxylic and oxocarboxylic acids in urine: Products from branched-chain amino acid degradation and from ketogenesis. J. Chromatogr. 1984, 309, 225–242. [Google Scholar] [CrossRef]
- Siddiqui, R.A.; Williams, J.F. Tentative identification of the toxohormones of cancer cachexia: Roles of vasopressin, prostaglandin E2 and cachectin-TNF. Biochem. Int. 1990, 20, 787–797. [Google Scholar] [PubMed]
- Lundasen, T.; Hunt, M.C.; Nilsson, L.M.; Sanyal, S.; Angelin, B.; Alexson, S.E.; Rudling, M. PPARα is a key regulator of hepatic FGF21. Biochem. Biophys. Res. Commun. 2007, 360, 437–440. [Google Scholar] [CrossRef] [PubMed]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Erickson, A.; Moreau, R. The regulation of FGF21 gene expression by metabolic factors and nutrients. Horm. Mol. Biol. Clin. Investig. 2016. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Dutchak, P.; Zhao, G.; Ding, X.; Gautron, L.; Parameswara, V.; Li, Y.; Goetz, R.; Mohammadi, M.; Esser, V.; et al. Endocrine regulation of the fasting response by PPARα -mediated induction of fibroblast growth factor 21. Cell Metab. 2007, 5, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Inagaki, T.; Satapati, S.; Ding, X.; He, T.; Goetz, R.; Mohammadi, M.; Finck, B.N.; Mangelsdorf, D.J.; Kliewer, S.A.; et al. FGF21 induces PGC-1α and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc. Natl. Acad. Sci. USA 2009, 106, 10853–10858. [Google Scholar] [CrossRef] [PubMed]
- Lustig, Y.; Ruas, J.L.; Estall, J.L.; Lo, J.C.; Devarakonda, S.; Laznik, D.; Choi, J.H.; Ono, H.; Olsen, J.V.; Spiegelman, B.M. Separation of the gluconeogenic and mitochondrial functions of PGC-1α through s6 kinase. Genes Dev. 2011, 25, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.H.; Satoh, H.; Herzig, S.; Lee, C.H.; Hedrick, S.; Kulkarni, R.; Evans, R.M.; Olefsky, J.; Montminy, M. PGC-1 promotes insulin resistance in liver through PPARα-dependent induction of TRB-3. Nat. Med. 2004, 10, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Rhee, J.; Inoue, Y.; Yoon, J.C.; Puigserver, P.; Fan, M.; Gonzalez, F.J.; Spiegelman, B.M. Regulation of hepatic fasting response by PPARγ coactivator-1α (PGC-1): Requirement for hepatocyte nuclear factor 4α in gluconeogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 4012–4017. [Google Scholar] [CrossRef] [PubMed]
- Svensson, K.; Albert, V.; Cardel, B.; Salatino, S.; Handschin, C. Skeletal muscle PGC-1α modulates systemic ketone body homeostasis and ameliorates diabetic hyperketonemia in mice. FASEB J. 2016, 30, 1976–1986. [Google Scholar] [CrossRef] [PubMed]
- Galman, C.; Lundasen, T.; Kharitonenkov, A.; Bina, H.A.; Eriksson, M.; Hafstrom, I.; Dahlin, M.; Amark, P.; Angelin, B.; Rudling, M. The circulating metabolic regulator FGF21 is induced by prolonged fasting and PPARα activation in man. Cell Metab. 2008, 8, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Fazeli, P.K.; Lun, M.; Kim, S.M.; Bredella, M.A.; Wright, S.; Zhang, Y.; Lee, H.; Catana, C.; Klibanski, A.; Patwari, P.; et al. FGF21 and the late adaptive response to starvation in humans. J. Clin. Investig. 2015, 125, 4601–4611. [Google Scholar] [CrossRef] [PubMed]
- Corton, J.M.; Gillespie, J.G.; Hawley, S.A.; Hardie, D.G. 5-Aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 1995, 229, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Carling, D.; Clarke, P.R.; Zammit, V.A.; Hardie, D.G. Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-COA carboxylase kinase and 3-hydroxy-3-methylglutaryl-COA reductase kinase activities. Eur. J. Biochem. 1989, 186, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J. LKB1 and AMP-activated protein kinase control of mtor signalling and growth. Acta Physiol. 2009, 196, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mtor in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Nojima, H.; Tokunaga, C.; Eguchi, S.; Oshiro, N.; Hidayat, S.; Yoshino, K.; Hara, K.; Tanaka, N.; Avruch, J.; Yonezawa, K. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mtor substrates p70 S6 kinase and 4E-BP1 through their tor signaling (TOS) motif. J. Biol. Chem. 2003, 278, 15461–15464. [Google Scholar] [CrossRef] [PubMed]
- Schalm, S.S.; Fingar, D.C.; Sabatini, D.M.; Blenis, J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol. 2003, 13, 797–806. [Google Scholar] [CrossRef]
- Xu, K.; Liu, P.; Wei, W. Mtor signaling in tumorigenesis. Biochim. Biophys. Acta 2014, 1846, 638–654. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Sato, T.; Nakashima, A.; Guo, L.; Tamanoi, F. Specific activation of mtorc1 by RHEB G-protein in vitro involves enhanced recruitment of its substrate protein. J. Biol. Chem. 2009, 284, 12783–12791. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Peterson, T.R.; Laplante, M.; Oh, S.; Sabatini, D.M. MTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 2010, 468, 1100–1104. [Google Scholar] [CrossRef] [PubMed]
- Riggs, M.G.; Whittaker, R.G.; Neumann, J.R.; Ingram, V.M. N-butyrate causes histone modification in hela and friend erythroleukaemia cells. Nature 1977, 268, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.A.; Turner, B.M. Histone deacetylases: Complex transducers of nuclear signals. Semin. Cell Dev. Biol. 1999, 10, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Zupkovitz, G.; Tischler, J.; Posch, M.; Sadzak, I.; Ramsauer, K.; Egger, G.; Grausenburger, R.; Schweifer, N.; Chiocca, S.; Decker, T.; et al. Negative and positive regulation of gene expression by mouse histone deacetylase 1. Mol. Cell. Biol. 2006, 26, 7913–7928. [Google Scholar] [CrossRef] [PubMed]
- Cahill, G.F., Jr.; Herrera, M.G.; Morgan, A.P.; Soeldner, J.S.; Steinke, J.; Levy, P.L.; Reichard, G.A., Jr.; Kipnis, D.M. Hormone-fuel interrelationships during fasting. J. Clin. Investig. 1966, 45, 1751–1769. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.M.; Williamson, D.H. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol. Rev. 1980, 60, 143–187. [Google Scholar] [PubMed]
- Laffel, L. Ketone bodies: A review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 1999, 15, 412–426. [Google Scholar] [CrossRef]
- Rando, G.; Tan, C.K.; Khaled, N.; Montagner, A.; Leuenberger, N.; Bertrand-Michel, J.; Paramalingam, E.; Guillou, H.; Wahli, W. Glucocorticoid receptor-PPARα axis in fetal mouse liver prepares neonates for milk lipid catabolism. eLife 2016, 5, e11853. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gao, Z.; Zhang, J.; Ye, X.; Xu, A.; Ye, J.; Jia, W. Sodium butyrate stimulates expression of fibroblast growth factor 21 in liver by inhibition of histone deacetylase 3. Diabetes 2012, 61, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Tunaru, S.; Offermanns, S. Gpr109a, gpr109b and gpr81, a family of hydroxy-carboxylic acid receptors. Trends Pharmacol. Sci. 2009, 30, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Inoue, D.; Maeda, T.; Hara, T.; Ichimura, A.; Miyauchi, S.; Kobayashi, M.; Hirasawa, A.; Tsujimoto, G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc. Natl. Acad. Sci. USA 2011, 108, 8030–8035. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.P.; Liu, B.R.; Wang, J.F.; Xue, W.J.; Liu, H.M.; Zeng, Y.L.; Huang, B.X.; Li, S.N.; Lv, Q.K.; Wang, W.; et al. β-Hydroxybutyric acid inhibits growth hormone-releasing hormone synthesis and secretion through the GPR109A/Extracellular signal-regulated 1/2 signalling pathway in the hypothalamus. J. Neuroendocrinol. 2015, 27, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Muhammad, S.; Khan, M.A.; Chen, H.; Ridder, D.A.; Muller-Fielitz, H.; Pokorna, B.; Vollbrandt, T.; Stolting, I.; Nadrowitz, R.; et al. The β-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat. Commun. 2014, 5, 3944. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain: J. Neurol. 2016, 139, 1265–1281. [Google Scholar] [CrossRef] [PubMed]
- De Virgilio, A.; Greco, A.; Fabbrini, G.; Inghilleri, M.; Rizzo, M.I.; Gallo, A.; Conte, M.; Rosato, C.; Ciniglio Appiani, M.; de Vincentiis, M. Parkinson’s disease: Autoimmunity and neuroinflammation. Autoimmun. Rev. 2016, 15, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Sowers, J.L.; Johnson, K.M.; Conrad, C.; Patterson, J.T.; Sowers, L.C. The role of inflammation in brain cancer. Adv. Exp. Med. Biol. 2014, 816, 75–105. [Google Scholar] [PubMed]
- Taggart, A.K.; Kero, J.; Gan, X.; Cai, T.Q.; Cheng, K.; Ippolito, M.; Ren, N.; Kaplan, R.; Wu, K.; Wu, T.J.; et al. d-β-hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J. Biol. Chem. 2005, 280, 26649–26652. [Google Scholar] [CrossRef] [PubMed]
- Edmond, J.; Robbins, R.A.; Bergstrom, J.D.; Cole, R.A.; de Vellis, J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J. Neurosci. Res. 1987, 18, 551–561. [Google Scholar] [PubMed]
- Blazquez, C.; Sanchez, C.; Velasco, G.; Guzman, M. Role of carnitine palmitoyltransferase I in the control of ketogenesis in primary cultures of rat astrocytes. J. Neurochem. 1998, 71, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Ebert, D.; Haller, R.G.; Walton, M.E. Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J. Neurochem. 2003, 23, 5928–5935. [Google Scholar]
- Oldendorf, W.H. Carrier-mediated blood-brain barrier transport of short-chain monocarboxylic organic acids. Am. J. Physiol. 1973, 224, 1450–1453. [Google Scholar] [PubMed]
- Januszewicz, E.; Pajak, B.; Gajkowska, B.; Samluk, L.; Djavadian, R.L.; Hinton, B.T.; Nalecz, K.A. Organic cation/carnitine transporter OCTN3 is present in astrocytes and is up-regulated by peroxisome proliferators-activator receptor agonist. Int. J. Biochem. Cell Biol. 2009, 41, 2599–2609. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Connor, J.D.; Crawford, I.L. Permeability changes in the blood-brain barrier: Causes and consequences. CRC Crit. Rev. Toxicol. 1975, 3, 159–199. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Shinbo, S.; Asahi, A.; Imanaka, T. Very long chain fatty acid β-oxidation in astrocytes: Contribution of the ABCD1-dependent and -independent pathways. Biol. Pharm. Bull. 2012, 35, 1972–1979. [Google Scholar] [CrossRef] [PubMed]
- Guest, J.; Garg, M.; Bilgin, A.; Grant, R. Relationship between central and peripheral fatty acids in humans. Lipids Health Dis. 2013, 12, 79. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.W.; Hatch, G.M. Fatty acid transport into the brain: Of fatty acid fables and lipid tails. Prostaglandins Leukot. Essent. Fat. Acids 2011, 85, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Pelerin, H.; Jouin, M.; Lallemand, M.S.; Alessandri, J.M.; Cunnane, S.C.; Langelier, B.; Guesnet, P. Gene expression of fatty acid transport and binding proteins in the blood-brain barrier and the cerebral cortex of the rat: Differences across development and with different DHA brain status. Prostaglandins Leukot. Essent. Fat. Acids 2014, 91, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Pohl, J.; Ring, A.; Ehehalt, R.; Herrmann, T.; Stremmel, W. New concepts of cellular fatty acid uptake: Role of fatty acid transport proteins and of caveolae. Proc. Nutr. Soc. 2004, 63, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.N.; Ma, D.; Shui, G.; Wong, P.; Cazenave-Gassiot, A.; Zhang, X.; Wenk, M.R.; Goh, E.L.; Silver, D.L. MFSD2A is a transporter for the essential ω-3 fatty acid docosahexaenoic acid. Nature 2014, 509, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.W.; On, N.H.; Del Bigio, M.R.; Miller, D.W.; Hatch, G.M. Fatty acid transport protein expression in human brain and potential role in fatty acid transport across human brain microvessel endothelial cells. J. Neurochem. 2011, 117, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Croset, M.; Brossard, N.; Polette, A.; Lagarde, M. Characterization of plasma unsaturated lysophosphatidylcholines in human and rat. Biochem. J. 2000, 345, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, M.; Bernoud, N.; Brossard, N.; Lemaitre-Delaunay, D.; Thies, F.; Croset, M.; Lecerf, J. Lysophosphatidylcholine as a preferred carrier form of docosahexaenoic acid to the brain. J. Mol. Neurosci. 2001, 16, 201–204. [Google Scholar] [CrossRef]
- Lo Van, A.; Sakayori, N.; Hachem, M.; Belkouch, M.; Picq, M.; Lagarde, M.; Osumi, N.; Bernoud-Hubac, N. Mechanisms of DHA transport to the brain and potential therapy to neurodegenerative diseases. Biochimie 2016, 130, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, B.; Fenart, L.; Dehouck, M.P.; Pierce, A.; Torpier, G.; Cecchelli, R. A new function for the LDL receptor: Transcytosis of LDL across the blood-brain barrier. J. Cell Biol. 1997, 138, 877–889. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Bergersen, L.H.; Halestrap, A.P.; Pierre, K. Cellular and subcellular distribution of monocarboxylate transporters in cultured brain cells and in the adult brain. J. Neurosci. Res. 2005, 79, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Hugo, S.E.; Cruz-Garcia, L.; Karanth, S.; Anderson, R.M.; Stainier, D.Y.; Schlegel, A. A monocarboxylate transporter required for hepatocyte secretion of ketone bodies during fasting. Genes Dev. 2012, 26, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, P.; Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, C.; Woods, A.; de Ceballos, M.L.; Carling, D.; Guzman, M. The AMP-activated protein kinase is involved in the regulation of ketone body production by astrocytes. J. Neurochem. 1999, 73, 1674–1682. [Google Scholar] [CrossRef] [PubMed]
- Auestad, N.; Korsak, R.A.; Morrow, J.W.; Edmond, J. Fatty acid oxidation and ketogenesis by astrocytes in primary culture. J. Neurochem. 1991, 56, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Thevenet, J.; de Marchi, U.; Domingo, J.S.; Christinat, N.; Bultot, L.; Lefebvre, G.; Sakamoto, K.; Descombes, P.; Masoodi, M.; Wiederkehr, A. Medium-chain fatty acids inhibit mitochondrial metabolism in astrocytes promoting astrocyte-neuron lactate and ketone body shuttle systems. FASEB J. 2016, 30, 1913–1926. [Google Scholar] [CrossRef] [PubMed]
- Murin, R.; Hamprecht, B. Metabolic and regulatory roles of leucine in neural cells. Neurochem. Res. 2008, 33, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Cullingford, T.E.; Dolphin, C.T.; Sato, H. The peroxisome proliferator-activated receptor α-selective activator ciprofibrate upregulates expression of genes encoding fatty acid oxidation and ketogenesis enzymes in rat brain. Neuropharmacology 2002, 42, 724–730. [Google Scholar] [CrossRef]
- Barros, L.F.; Deitmer, J.W. Glucose and lactate supply to the synapse. Brain Res. Rev. 2010, 63, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Sibson, N.R.; Dhankhar, A.; Mason, G.F.; Rothman, D.L.; Behar, K.L.; Shulman, R.G. Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc. Natl. Acad. Sci. USA 1998, 95, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Bonvento, G.; Chatton, J.Y.; Pierre, K.; Magistretti, P.J. Role of neuron-glia interaction in the regulation of brain glucose utilization. Diabetes Nutr. Metab. 2002, 15, 268–273. [Google Scholar] [PubMed]
- Veech, R.L.; Chance, B.; Kashiwaya, Y.; Lardy, H.A.; Cahill, G.F., Jr. Ketone bodies, potential therapeutic uses. IUBMB Life 2001, 51, 241–247. [Google Scholar] [PubMed]
- Cahill, G.F., Jr.; Veech, R.L. Ketoacids? Good medicine? Trans. Am. Clin. Climatol. Assoc. 2003, 114, 149–161. [Google Scholar] [PubMed]
- Yudkoff, M.; Daikhin, Y.; Melo, T.M.; Nissim, I.; Sonnewald, U.; Nissim, I. The ketogenic diet and brain metabolism of amino acids: Relationship to the anticonvulsant effect. Annu. Rev. Nutr. 2007, 27, 415–430. [Google Scholar] [CrossRef] [PubMed]
- Peterman, M.G. Ketogenic diet in the treatment of epilepsy. J. Am. Med. Assoc. 1925, 84, 1979–1983. [Google Scholar] [CrossRef]
- Barborka, C.J. Epilepsy in adults: Results of treatment by ketogenic diet in one hundred cases. Arch. Neurol. Psychiatr. 1930, 23, 904–914. [Google Scholar] [CrossRef]
- Wibisono, C.; Rowe, N.; Beavis, E.; Kepreotes, H.; Mackie, F.E.; Lawson, J.A.; Cardamone, M. Ten-year single-center experience of the ketogenic diet: Factors influencing efficacy, tolerability, and compliance. J. Pediatr. 2015, 166, 1030–1036.e1. [Google Scholar] [CrossRef] [PubMed]
- Gulati, S.; Chakrabarty, B. Dietary therapy in childhood epilepsy, an overview. Int. J. Epilepsy 2014, 1, 27–35. [Google Scholar] [CrossRef]
- Cullingford, T.E. The ketogenic diet; fatty acids, fatty acid-activated receptors and neurological disorders. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Lampen, A.; Carlberg, C.; Nau, H. Peroxisome proliferator-activated receptor delta is a specific sensor for teratogenic valproic acid derivatives. Eur. J. Pharmacol. 2001, 431, 25–33. [Google Scholar] [CrossRef]
- Puligheddu, M.; Pillolla, G.; Melis, M.; Lecca, S.; Marrosu, F.; de Montis, M.G.; Scheggi, S.; Carta, G.; Murru, E.; Aroni, S.; et al. PPARα agonists as novel antiepileptic drugs: Preclinical findings. PLoS ONE 2013, 8, e64541. [Google Scholar] [CrossRef] [PubMed]
- Phiel, C.J.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef] [PubMed]
- Gottlicher, M.; Minucci, S.; Zhu, P.; Kramer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Lo Coco, F.; Nervi, C.; Pelicci, P.G.; et al. Valproic acid defines a novel class of hdac inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef] [PubMed]
- Gurvich, N.; Tsygankova, O.M.; Meinkoth, J.L.; Klein, P.S. Histone deacetylase is a target of valproic acid-mediated cellular differentiation. Cancer Res. 2004, 64, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Eyal, S.; Yagen, B.; Sobol, E.; Altschuler, Y.; Shmuel, M.; Bialer, M. The activity of antiepileptic drugs as histone deacetylase inhibitors. Epilepsia 2004, 45, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Landgrave-Gomez, J.; Mercado-Gomez, O.F.; Vazquez-Garcia, M.; Rodriguez-Molina, V.; Cordova-Davalos, L.; Arriaga-Avila, V.; Miranda-Martinez, A.; Guevara-Guzman, R. Anticonvulsant effect of time-restricted feeding in a pilocarpine-induced seizure model: Metabolic and epigenetic implications. Front. Cell. Neurosci. 2016, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Czapp, M.; Loscher, W. Increase in antiepileptic efficacy during prolonged treatment with valproic acid: Role of inhibition of histone deacetylases? Epilepsy Res. 2008, 81, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Gasior, M.; Rogawski, M.A.; Hartman, A.L. Neuroprotective and disease-modifying effects of the ketogenic diet. Behav. Pharmacol. 2006, 17, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Stafstrom, C.E.; Rho, J.M. The ketogenic diet as a treatment paradigm for diverse neurological disorders. Front. Pharmacol. 2012, 3, 59. [Google Scholar] [CrossRef] [PubMed]
- Reger, M.A.; Henderson, S.T.; Hale, C.; Cholerton, B.; Baker, L.D.; Watson, G.S.; Hyde, K.; Chapman, D.; Craft, S. Effects of β-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 2004, 25, 311–314. [Google Scholar] [CrossRef]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 2009, 6, 31. [Google Scholar] [CrossRef] [PubMed]
- Vanitallie, T.B.; Nonas, C.; di Rocco, A.; Boyar, K.; Hyams, K.; Heymsfield, S.B. Treatment of Parkinson disease with diet-induced hyperketonemia: A feasibility study. Neurology 2005, 64, 728–730. [Google Scholar] [CrossRef] [PubMed]
- Prins, M.L.; Fujima, L.S.; Hovda, D.A. Age-dependent reduction of cortical contusion volume by ketones after traumatic brain injury. J. Neurosci. Res. 2005, 82, 413–420. [Google Scholar] [CrossRef] [PubMed]
- White, H.; Venkatesh, B. Clinical review: Ketones and brain injury. Crit. Care 2011, 15, 219. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Suzuki, M.; Sato, K.; Dohi, S.; Sato, T.; Matsuura, A.; Hiraide, A. Effect of beta-hydroxybutyrate, a cerebral function improving agent, on cerebral hypoxia, anoxia and ischemia in mice and rats. Jpn. J. Pharmacol. 2001, 87, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Prins, M.L.; Matsumoto, J.H. The collective therapeutic potential of cerebral ketone metabolism in traumatic brain injury. J. Lipid Res. 2014, 55, 2450–2457. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Prins, M.L.; Samii, M.; Hovda, D.A. Cerebral metabolic response to traumatic brain injury sustained early in development: A 2-deoxy-d-glucose autoradiographic study. J. Neurotrauma 2000, 17, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Prins, M.L.; Hovda, D.A. Mapping cerebral glucose metabolism during spatial learning: Interactions of development and traumatic brain injury. J. Neurotrauma 2001, 18, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, A.; Hovda, D.A.; Kawamata, T.; Katayama, Y.; Becker, D.P. Dynamic changes in local cerebral glucose utilization following cerebral conclusion in rats: Evidence of a hyper- and subsequent hypometabolic state. Brain Res. 1991, 561, 106–119. [Google Scholar] [CrossRef]
- Robertson, C.L.; Scafidi, S.; McKenna, M.C.; Fiskum, G. Mitochondrial mechanisms of cell death and neuroprotection in pediatric ischemic and traumatic brain injury. Exp. Neurol. 2009, 218, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.; Blazquez, C. Ketone body synthesis in the brain: Possible neuroprotective effects. Prostaglandins Leukot. Essent. Fat. Acids 2004, 70, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Maalouf, M.; Rho, J.M.; Mattson, M.P. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res. Rev. 2009, 59, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.L.; Heal, D.J.; Martin, K.F. KTX 0101: A potential metabolic approach to cytoprotection in major surgery and neurological disorders. CNS Drug Rev. 2005, 11, 113–140. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, J.; Morales, L.; Barreto, G.E. Metabolic and inflammatory adaptation of reactive astrocytes: Role of PPARs. Mol. Neurobiol. 2016, in press. [Google Scholar]
- Cullingford, T.E.; Bhakoo, K.; Peuchen, S.; Dolphin, C.T.; Patel, R.; Clark, J.B. Distribution of mRNAs encoding the peroxisome proliferator-activated receptor α, β, and γ and the retinoid X receptor α, β, and γ in rat central nervous system. J. Neurochem. 1998, 70, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Cimini, A.; Benedetti, E.; Cristiano, L.; Sebastiani, P.; D’Amico, M.A.; D’Angelo, B.; di Loreto, S. Expression of peroxisome proliferator-activated receptors (PPARs) and retinoic acid receptors (RXRS) in rat cortical neurons. Neuroscience 2005, 130, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Cristiano, L.; Bernardo, A.; Ceru, M.P. Peroxisome proliferator-activated receptors (PPARs) and peroxisomes in rat cortical and cerebellar astrocytes. J. Neurocytol. 2001, 30, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Drew, P.D.; Xu, J.; Storer, P.D.; Chavis, J.A.; Racke, M.K. Peroxisome proliferator-activated receptor agonist regulation of glial activation: Relevance to CNS inflammatory disorders. Neurochem. Int. 2006, 49, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chavis, J.A.; Racke, M.K.; Drew, P.D. Peroxisome proliferator-activated receptor-α and retinoid X receptor agonists inhibit inflammatory responses of astrocytes. J. Neuroimmunol. 2006, 176, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Racke, M.K.; Drew, P.D. Peroxisome proliferator-activated receptor-α agonist fenofibrate regulates IL-12 family cytokine expression in the CNS: Relevance to multiple sclerosis. J. Neurochem. 2007, 103, 1801–1810. [Google Scholar] [CrossRef] [PubMed]
- Paukkeri, E.L.; Leppanen, T.; Sareila, O.; Vuolteenaho, K.; Kankaanranta, H.; Moilanen, E. PPARα agonists inhibit nitric oxide production by enhancing iNOS degradation in LPS-treated macrophages. Br. J. Pharmacol. 2007, 152, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Cuzzocrea, S.; Bruscoli, S.; Mazzon, E.; Crisafulli, C.; Donato, V.; Di Paola, R.; Velardi, E.; Esposito, E.; Nocentini, G.; Riccardi, C. Peroxisome proliferator-activated receptor-α contributes to the anti-inflammatory activity of glucocorticoids. Mol. Pharmacol. 2008, 73, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Koenig, W.; Habib, A.; Merval, R.; Lebret, M.; Torra, I.P.; Delerive, P.; Fadel, A.; Chinetti, G.; Fruchart, J.C.; et al. Activation of human aortic smooth-muscle cells is inhibited by PPARα but not by PPARγ activators. Nature 1998, 393, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Delerive, P.; de Bosscher, K.; Vanden Berghe, W.; Fruchart, J.C.; Haegeman, G.; Staels, B. DNA binding-independent induction of IκBα gene transcription by PPARα. Mol. Endocrinol. 2002, 16, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Dubrac, S.; Stoitzner, P.; Pirkebner, D.; Elentner, A.; Schoonjans, K.; Auwerx, J.; Saeland, S.; Hengster, P.; Fritsch, P.; Romani, N.; et al. Peroxisome proliferator-activated receptor-α activation inhibits langerhans cell function. J. Immunol. 2007, 178, 4362–4372. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Chaudhary, A.; Sethi, S. Oxidized ω-3 fatty acids inhibit NF-κB activation via a PPARα-dependent pathway. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1621–1627. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Bujo, H.; Unoki, H.; Saito, Y. Effect of PPARα activation of macrophages on the secretion of inflammatory cytokines in cultured adipocytes. Eur. J. Pharmacol. 2007, 561, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Narala, V.R.; Adapala, R.K.; Suresh, M.V.; Brock, T.G.; Peters-Golden, M.; Reddy, R.C. Leukotriene B4 is a physiologically relevant endogenous peroxisome proliferator-activated receptor-α agonist. J. Biol. Chem. 2010, 285, 22067–22074. [Google Scholar] [CrossRef] [PubMed]
- Devchand, P.R.; Keller, H.; Peters, J.M.; Vazquez, M.; Gonzalez, F.J.; Wahli, W. The PPARα-leukotriene B4 pathway to inflammation control. Nature 1996, 384, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.F.; Hsu, M.H.; Savas, U.; Griffin, K.J. Regulation of p450 4a expression by peroxisome proliferator activated receptors. Toxicology 2002, 181–182, 203–206. [Google Scholar] [CrossRef]
- Pace, E.; Ferraro, M.; Mody, C.H.; Melis, M.R.; Scafidi, V.; Bonanno, A.; Profita, M.; Giarratano, A.; Gjomarkaj, M. Pleural mesothelial cells express both BLT2 and PPARα and mount an integrated response to pleural leukotriene B4. J. Immunol. 2008, 181, 7292–7299. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Esposito, G.; Blasio, A.; Valenza, M.; Arietti, P.; Steardo, L., Jr.; Carnuccio, R.; de Filippis, D.; Petrosino, S.; Iuvone, T.; et al. Palmitoylethanolamide counteracts reactive astrogliosis induced by β-amyloid peptide. J. Cell. Mol. Med. 2011, 15, 2664–2674. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, C.; Valenza, M.; Stecca, C.; Esposito, G.; Carratu, M.R.; Steardo, L. Palmitoylethanolamide exerts neuroprotective effects in mixed neuroglial cultures and organotypic hippocampal slices via peroxisome proliferator-activated receptor-α. J. Neuroinflamm. 2012, 9, 49. [Google Scholar] [CrossRef] [PubMed]
- Deplanque, D.; Gele, P.; Petrault, O.; Six, I.; Furman, C.; Bouly, M.; Nion, S.; Dupuis, B.; Leys, D.; Fruchart, J.C.; et al. Peroxisome proliferator-activated receptor-α activation as a mechanism of preventive neuroprotection induced by chronic fenofibrate treatment. J. Neurosci. 2003, 23, 6264–6271. [Google Scholar] [PubMed]
- Xuan, A.G.; Chen, Y.; Long, D.H.; Zhang, M.; Ji, W.D.; Zhang, W.J.; Liu, J.H.; Hong, L.P.; He, X.S.; Chen, W.L. PPARα agonist fenofibrate ameliorates learning and memory deficits in rats following global cerebral ischemia. Mol. Neurobiol. 2015, 52, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Maurer, G.D.; Brucker, D.P.; Bahr, O.; Harter, P.N.; Hattingen, E.; Walenta, S.; Mueller-Klieser, W.; Steinbach, J.P.; Rieger, J. Differential utilization of ketone bodies by neurons and glioma cell lines: A rationale for ketogenic diet as experimental glioma therapy. BMC Cancer 2011, 11, 315. [Google Scholar] [CrossRef] [PubMed]
- Fredericks, M.; Ramsey, R.B. 3-Oxo acid coenzyme a transferase activity in brain and tumors of the nervous system. J. Neurochem. 1978, 31, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J.; Brennan, R.A. Loss of acetoacetate coenzyme a transferase activity in tumours of peripheral tissues. Br. J. Cancer 1983, 47, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J.; Brennan, R.A. Metabolic substrate utilization by a tumour cell line which induces cachexia in vivo. Br. J. Cancer 1986, 54, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Poduslo, S.E. Induction of ketone body enzymes in glial cells. Arch. Biochem. Biophys. 1989, 272, 318–322. [Google Scholar] [CrossRef]
- Roeder, L.M.; Poduslo, S.E.; Tildon, J.T. Utilization of ketone bodies and glucose by established neural cell lines. J. Neurosci. Res. 1982, 8, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, L.A.; Spennetta, T.; Elson, C.; Shrago, E. Utilization and preferred metabolic pathway of ketone bodies for lipid synthesis by isolated rat hepatoma cells. Am. J. Physiol. 1995, 269, C22–C27. [Google Scholar] [PubMed]
- De Feyter, H.M.; Behar, K.L.; Rao, J.U.; Madden-Hennessey, K.; Ip, K.L.; Hyder, F.; Drewes, L.R.; Geschwind, J.F.; de Graaf, R.A.; Rothman, D.L. A ketogenic diet increases transport and oxidation of ketone bodies in RG2 and 9l gliomas without affecting tumor growth. Neuro-oncology 2016, 18, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.T.; Olson, L.K.; Schwartz, K.A. Ketolytic and glycolytic enzymatic expression profiles in malignant gliomas: Implication for ketogenic diet therapy. Nutr. Metab. 2013, 10, 47. [Google Scholar] [CrossRef] [PubMed]
- Eloqayli, H.; Melo, T.M.; Haukvik, A.; Sonnewald, U. [2,4-(13)c]β-hydroxybutyrate metabolism in astrocytes and C6 glioblastoma cells. Neurochem. Res. 2011, 36, 1566–1573. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.S.; Russell, J.J.; Gershman, H. Ketone-body metabolism in glioma and neuroblastoma cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7214–7218. [Google Scholar] [CrossRef] [PubMed]
- Saraon, P.; Cretu, D.; Musrap, N.; Karagiannis, G.S.; Batruch, I.; Drabovich, A.P.; van der Kwast, T.; Mizokami, A.; Morrissey, C.; Jarvi, K.; et al. Quantitative proteomics reveals that enzymes of the ketogenic pathway are associated with prostate cancer progression. Mol. Cell. Proteom. 2013, 12, 1589–1601. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Lin, Z.; Lamb, R.; Hulit, J.; Howell, A.; Sotgia, F.; Rubin, E.; Lisanti, M.P. Ethanol exposure induces the cancer-associated fibroblast phenotype and lethal tumor metabolism: Implications for breast cancer prevention. Cell Cycle 2013, 12, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Dueregger, A.; Schopf, B.; Eder, T.; Hofer, J.; Gnaiger, E.; Aufinger, A.; Kenner, L.; Perktold, B.; Ramoner, R.; Klocker, H.; et al. Differential utilization of dietary fatty acids in benign and malignant cells of the prostate. PLoS ONE 2015, 10, e0135704. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Li, T.; Wang, L.; Zhang, L.; Yan, R.; Li, K.; Xing, S.; Wu, G.; Hu, L.; Jia, W.; et al. Hepatocellular carcinoma redirects to ketolysis for progression under nutrition deprivation stress. Cell Res. 2016, 26, 1112–1130. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.L.; Gao, H.W.; Chiang, C.P.; Wang, W.M.; Huang, S.M.; Ku, C.F.; Liu, G.Y.; Hung, H.C. Human mitochondrial NADP(+)-dependent malic enzyme participates in cutaneous melanoma progression and invasion. J. Investig. Dermatol. 2015, 135, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Marcondes, S.; Turko, I.V.; Murad, F. Nitration of succinyl-COA:3-oxoacid COA-transferase in rats after endotoxin administration. Proc. Natl. Acad. Sci. USA 2001, 98, 7146–7151. [Google Scholar] [CrossRef] [PubMed]
- Turko, I.V.; Marcondes, S.; Murad, F. Diabetes-associated nitration of tyrosine and inactivation of succinyl-COA:3-oxoacid COA-transferase. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H2289–H2294. [Google Scholar] [PubMed]
- Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Lisanti, M.P.; Sotgia, F. Ketone bodies and two-compartment tumor metabolism: Stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle 2012, 11, 3956–3963. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.; Sotnikov, A.V.; Mangian, H.J.; Zhou, J.R.; Visek, W.J.; Clinton, S.K. Energy intake and prostate tumor growth, angiogenesis, and vascular endothelial growth factor expression. J. Natl. Cancer Inst. 1999, 91, 512–523. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzo, M.S.; Baljinnyam, E.; Vatner, D.E.; Abarzua, P.; Vatner, S.F.; Rabson, A.B. Caloric restriction reduces growth of mammary tumors and metastases. Carcinogenesis 2011, 32, 1381–1387. [Google Scholar] [CrossRef] [PubMed]
- Phoenix, K.N.; Vumbaca, F.; Fox, M.M.; Evans, R.; Claffey, K.P. Dietary energy availability affects primary and metastatic breast cancer and metformin efficacy. Breast Cancer Res. Treat. 2010, 123, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.J.; McGinley, J.N.; Spoelstra, N.S.; Jiang, W.; Zhu, Z.; Wolfe, P. Effect of dietary energy restriction on vascular density during mammary carcinogenesis. Cancer Res. 2004, 64, 5643–5650. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.; Kaemmerer, U.; Illert, B.; Muehling, B.; Pfetzer, N.; Wittig, R.; Voelker, H.U.; Thiede, A.; Coy, J.F. Growth of human gastric cancer cells in nude mice is delayed by a ketogenic diet supplemented with ω-3 fatty acids and medium-chain triglycerides. BMC Cancer 2008, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Flores, R.; Poff, A.M.; D′Agostino, D.P.; Mukherjee, P. Metabolic therapy: A new paradigm for managing malignant brain cancer. Cancer Lett. 2015, 356, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Woolf, E.C.; Scheck, A.C. The ketogenic diet for the treatment of malignant glioma. J. Lipid Res. 2015, 56, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Magee, B.A.; Potezny, N.; Rofe, A.M.; Conyers, R.A. The inhibition of malignant cell growth by ketone bodies. Aust. J. Exp. Biol. Med. Sci. 1979, 57, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Skinner, R.; Trujillo, A.; Ma, X.; Beierle, E.A. Ketone bodies inhibit the viability of human neuroblastoma cells. J. Pediatr. Surg. 2009, 44, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Stemmer, K.; Zani, F.; Habegger, K.M.; Neff, C.; Kotzbeck, P.; Bauer, M.; Yalamanchilli, S.; Azad, A.; Lehti, M.; Martins, P.J.; et al. FGF21 is not required for glucose homeostasis, ketosis or tumour suppression associated with ketogenic diets in mice. Diabetologia 2015, 58, 2414–2423. [Google Scholar] [CrossRef] [PubMed]
- Abdelwahab, M.G.; Fenton, K.E.; Preul, M.C.; Rho, J.M.; Lynch, A.; Stafford, P.; Scheck, A.C. The ketogenic diet is an effective adjuvant to radiation therapy for the treatment of malignant glioma. PLoS ONE 2012, 7, e36197. [Google Scholar] [CrossRef] [PubMed]
- Stafford, P.; Abdelwahab, M.G.; Kim, D.Y.; Preul, M.C.; Rho, J.M.; Scheck, A.C. The ketogenic diet reverses gene expression patterns and reduces reactive oxygen species levels when used as an adjuvant therapy for glioma. Nutr. Metab. 2010, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Mukherjee, P.; Kiebish, M.A.; Markis, W.T.; Mantis, J.G.; Seyfried, T.N. The calorically restricted ketogenic diet, an effective alternative therapy for malignant brain cancer. Nutr. Metab. 2007, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.; El-Abbadi, M.M.; Kasperzyk, J.L.; Ranes, M.K.; Seyfried, T.N. Dietary restriction reduces angiogenesis and growth in an orthotopic mouse brain tumour model. Br. J. Cancer 2002, 86, 1615–1621. [Google Scholar] [CrossRef] [PubMed]
- Nebeling, L.C.; Lerner, E. Implementing a ketogenic diet based on medium-chain triglyceride oil in pediatric patients with cancer. J. Am. Diet. Assoc. 1995, 95, 693–697. [Google Scholar] [CrossRef]
- Nebeling, L.C.; Miraldi, F.; Shurin, S.B.; Lerner, E. Effects of a ketogenic diet on tumor metabolism and nutritional status in pediatric oncology patients: Two case reports. J. Am. Coll. Nutr. 1995, 14, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Zuccoli, G.; Marcello, N.; Pisanello, A.; Servadei, F.; Vaccaro, S.; Mukherjee, P.; Seyfried, T.N. Metabolic management of glioblastoma multiforme using standard therapy together with a restricted ketogenic diet: Case report. Nutr. Metab. 2010, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.B.; Fan, J.; Lin, R.; Elf, S.; Ji, Q.; Zhao, L.; Jin, L.; Seo, J.H.; Shan, C.; Arbiser, J.L.; et al. Metabolic rewiring by oncogenic BRAF V600E links ketogenesis pathway to BRAF-MEK1 signaling. Mol. Cell 2015, 59, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Grabacka, M.M.; Wilk, A.; Antonczyk, A.; Banks, P.; Walczyk-Tytko, E.; Dean, M.; Pierzchalska, M.; Reiss, K. Fenofibrate induces ketone body production in melanoma and glioblastoma cells. Front. Endocrinol. 2016, 7, 5. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabacka, M.; Pierzchalska, M.; Dean, M.; Reiss, K. Regulation of Ketone Body Metabolism and the Role of PPARα. Int. J. Mol. Sci. 2016, 17, 2093. https://doi.org/10.3390/ijms17122093
Grabacka M, Pierzchalska M, Dean M, Reiss K. Regulation of Ketone Body Metabolism and the Role of PPARα. International Journal of Molecular Sciences. 2016; 17(12):2093. https://doi.org/10.3390/ijms17122093
Chicago/Turabian StyleGrabacka, Maja, Malgorzata Pierzchalska, Matthew Dean, and Krzysztof Reiss. 2016. "Regulation of Ketone Body Metabolism and the Role of PPARα" International Journal of Molecular Sciences 17, no. 12: 2093. https://doi.org/10.3390/ijms17122093
APA StyleGrabacka, M., Pierzchalska, M., Dean, M., & Reiss, K. (2016). Regulation of Ketone Body Metabolism and the Role of PPARα. International Journal of Molecular Sciences, 17(12), 2093. https://doi.org/10.3390/ijms17122093