
Green Synthesis of Sodium Cyanide Using Hydrogen Cyanide Extracted under Vacuum from Cassava (Manihot esculenta Crantz) Leaves



Abstract
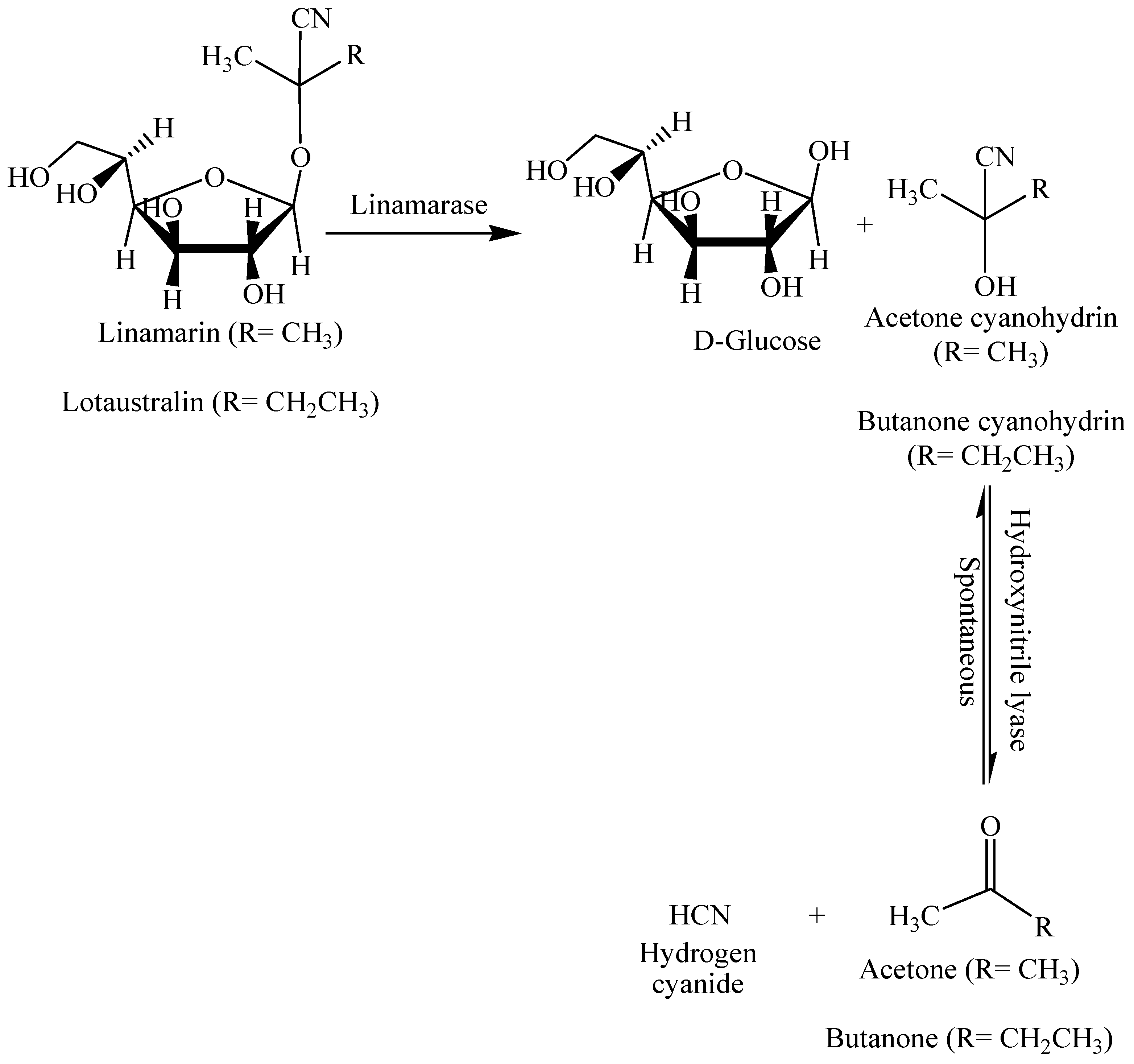
1. Introduction
1.1. Direct Synthesis of HCN
1.1.1. Andrussow Process
1.1.2. Methane–Ammonia Process or Blausaure Methane Anlage (BMA)
1.1.3. Shawinigan Process
1.2. Indirect Synthesis of HCN
Sohio Process
2. Materials and Methods
2.1. Materials
2.1.1. Sample Collection
2.1.2. Chemicals
2.2. Methods
2.2.1. Optimisation of Maceration Time and Temperature and HCN Recovery Time
2.2.2. Sample Preparation
2.2.3. Saturation of 3.6 mol/L NaOH Solution
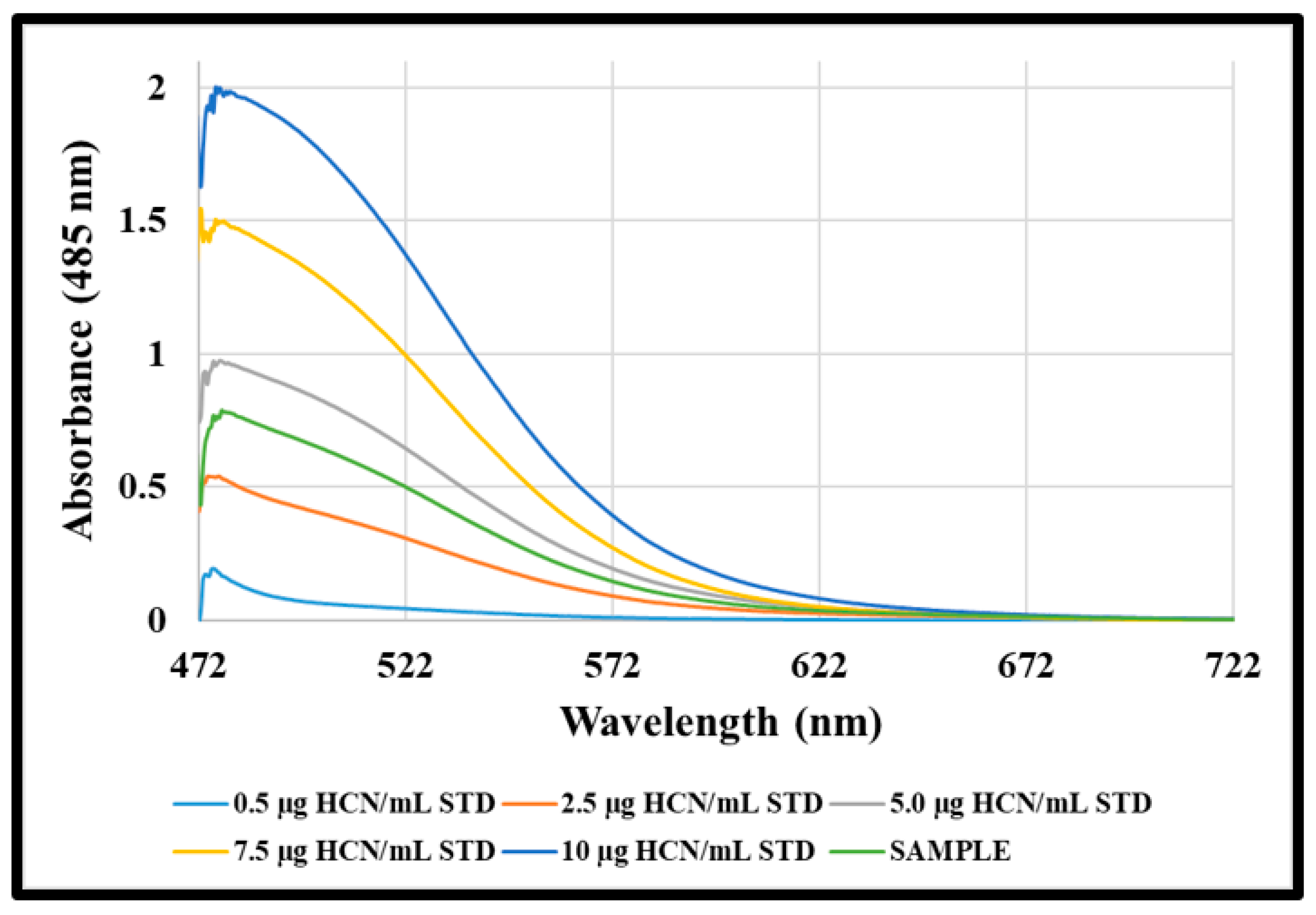
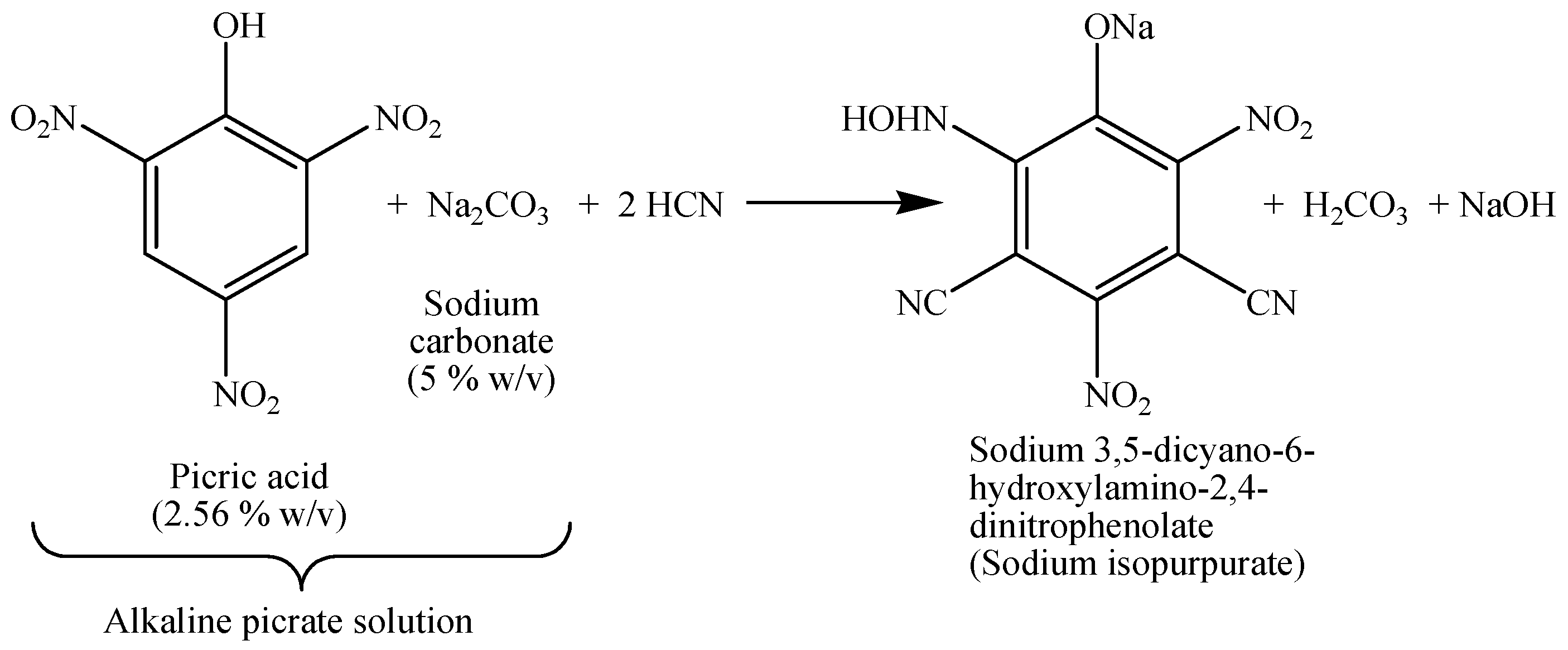
2.2.4. Quantification of NaCN Solution
2.2.5. Determination of Sodium Carbonate and Residual Sodium Hydroxide in Standard and Green-Sodium Cyanide Solutions
2.2.6. Drying of Green-Sodium Cyanide Solution
2.2.7. Structural Confirmation of Synthesised Sodium Cyanide Salt
- Attenuated total reflectance–Fourier transform infrared spectroscopy (ATR–FTIR)
- X-ray diffraction analysis (XRD)
- Scanning electron microscopy with energy-dispersive X-ray spectroscopy (SEM-EDS)
3. Results and Discussion
3.1. Optimisation of Maceration Time and Temperature and Recovery Time for Maximum Release of Hydrogen Cyanide from Cassava Leaves
3.2. Saturation of 3.6740 mol/L Absorbing Solution
3.3. Estimation of Sodium Carbonate and Residual Sodium Hydroxide in Standard and Green-Sodium Cyanide Salts
3.4. Structural Characterisation of Sodium Cyanide Salt
3.4.1. Attenuated Total Reflectance–Fourier Transform Infrared Spectroscopy (ATR–FTIR)
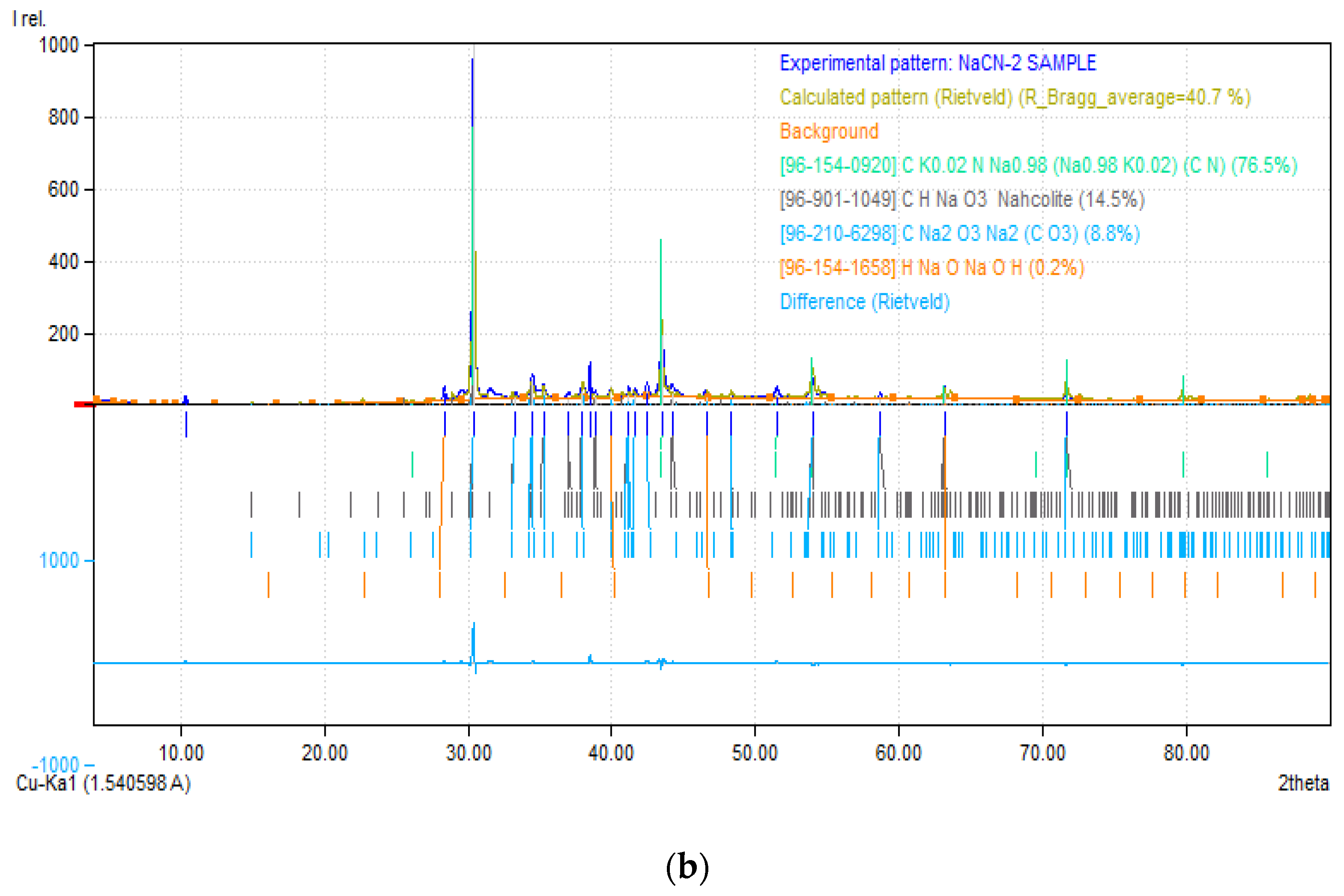
3.4.2. X-ray Diffraction (XRD)
3.4.3. Scanning Electron Microscopy with Energy-Dispersive X-ray Spectroscopy (SEM-EDS)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- European Centre for Ecotoxicology and Toxicology of Chemicals. Cyanides of Hydrogen, Sodium and Potassium, and Acetone Cyanohydrin (CAS No. 74-90-8, 143-33-9, 151-50-8 and 75-86-5); ECETOC: Brussels, Belgium, 2007; pp. 13–31. [Google Scholar]
- Attahdaniel, B.E.; Ebisike, K.; Adeeyinwo, C.E.; Adetunji, A.R.; Olusunle, S.O.O.; Adewoye, O.O. Production of Sodium Cyanide from Cassava Wastes. Int. J. Sci. Technol. 2013, 2, 707–709. [Google Scholar]
- Gail, E.; Gos, S.; Kulzer, R.; Lorösch, J.; Rubo, A.; Sauer, M.; Kellens, R.; Reddy, J.; Steier, N.; Hasenpusch, W. Cyano Compounds, Inorganic. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2011; Volume 10, pp. 673–710. [Google Scholar]
- Deepa, H.A.; Raj, A.; Asha, P. Evaluation on Production and Economics of Acrylonitrile by Sohio Process. In Proceedings of the International Conference on Recent Trends in Engineering and Technologies, Mysuru, India, 13 March 2016. [Google Scholar]
- Attahdaniel, E.B.; Enwerem, P.O.; Lawrence, P.G.; Ofiwe, C.U.; Olusunle, S.O.O.; Adetunji, A.R. Green synthesis and characterization of sodium cyanide from cassava (Manihot esculenta CRANTZ). FTSTJ 2020, 5, 247–251. [Google Scholar]
- Davis, S.; Murray, J.; Katsiadaki, I. Cyanide in the Aquatic Environment and Its Metabolism by Fish; World Class Science for the Marine and Freshwater Environment, Ed.; Centre for Environment Fisheries & Aquaculture Science: Suffolk, UK, 2017; p. 72. [Google Scholar]
- Panou, M.; Gkelis, S. Cyano-assassins: Widespread cyanogenic production from cyanobacteria. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bhalla, T.C.; Kumar, V.; Kumar, V. Microbial Remediation of Cyanides. In Bioremediation Current Research and Applications, 1st ed.; Ashok, D., Rathoure, K., Eds.; IK International: Delhi, India, 2017; p. 23. [Google Scholar]
- Ubwa, S.T.; Otache, M.A.; Igbum, G.O.; Shambe, T. Determination of Cyanide Content in Three Sweet Cassava Cultivars in Three Local Government Areas of Benue State, Nigeria. Food Sci. Nutr. 2015, 6, 1078–1085. [Google Scholar] [CrossRef]
- Zuk, M.; Pelc, K.; Szperlik, J.; Sawula, A.; Szopa, J. Metabolism of the Cyanogenic Glucosides in Developing Flax: Metabolic Analysis, and Expression Pattern of Genes. Metabolites 2020, 10, 288. [Google Scholar] [CrossRef] [PubMed]
- Bayitse, R.; Tornyie, F.; Bjerre, A.-B. Cassava cultivation, processing and potential uses in Ghana. In Handbook on Cassava; Klein, C., Ed.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2017; p. 21. [Google Scholar]
- Omotayo, A.R.; EL-Ishaq, A.; Babatunde, A.S. Assessment of cyanide content in white, light yellow and deep yellow cassava Grit (Gari) sold in Damaturu Metropolis. Am. J. Food Sci. Health 2015, 1, 5. [Google Scholar]
- Nyirenda, K.K. Toxicity Potential of Cyanogenic Glycosides in Edible Plants. In Medical Toxicology; Erkekoglu, P., Ogawa, T., Eds.; IntechOpen: London, UK, 2021; pp. 1–19. [Google Scholar]
- Shackelford, G.E.; Haddaway, N.R.; Usieta, H.O.; Pypers, P.; Petrovan, S.O.; Sutherland, W.J. Cassava farming practices and their agricultural and environmental impacts: A systematic map protocol. Environ. Evid. 2018, 7, 30. [Google Scholar] [CrossRef]
- Spencer, D.S.C.; Ezedinma, C. Cassava cultivation in sub-Saharan Africa. In Achieving Sustainable Cultivation of Cassava Volume 1, 1st ed.; Burleigh Dodds Science Publishing: Cambridge, UK, 2017; pp. 123–148. [Google Scholar]
- Otekunrin, O.A.; Sawicka, B. Cassava, a 21st Century Staple Crop: How can Nigeria Harness Its Enormous Trade Potentials? Acta Sci. Agric. 2019, 3, 194–202. [Google Scholar] [CrossRef]
- Mudombi, C.R. An Ex-Ante Economic Evaluation of Genetically Modified Cassava in South Africa. Master’s Thesis, University of Pretoria, Pretoria, South Africa, 2010. [Google Scholar]
- Mabasa, K.G. Epidemiology of Cassava Mosaic Disease and Molecular Characterization of Cassava Mosaic Viruses and Their Associated Whitefly (Bemisia Tabaci) Vector in South Africa. Master’s Thesis, University of the Witwatersrand, Johannesburg, South Africa, 2007. [Google Scholar]
- Makwarela, M.; Rey, C. Cassava Biotechnology, a southern African perspective. Biotechnol. Mol. Biol. Rev. 2006, 1, 10. [Google Scholar]
- Cuvaca, I.B.; Eash, N.S.; Zivanovic, S.; Lambert, D.M.; Walker, F.; Rustrick, B. Cassava (Manihot esculenta Crantz) Tuber Quality as Measured by Starch and Cyanide (HCN) Affected by Nitrogen, Phosphorus, and Potassium Fertilizer Rates. J. Agric. Sci. 2015, 7, 36–49. [Google Scholar] [CrossRef][Green Version]
- Srihawong, W.; Kongsil, P.; Petchpoung, K.; Sarobol, E. Effect of genotype, age and soil moisture on cyanogenic glycosides content and root yield in cassava (Manihot esculenta Crantz). Agric. Nat. Resour. 2015, 49, 13. [Google Scholar]
- Brito, V.H.S.; Ramalho, R.T.; Rabacow, A.P.M.; Moreno, S.E.; Cereda, M.P. Colorimetric method for free and potential cyanide analysis of cassava tissue. Gene Conserve 2009, 8, 1–11. [Google Scholar]
- Oshima, H.; Ueno, E.; Saito, I.; Matsumoto, H. Quantitative Determination of Cyanide in Foods by Spectrophotometry using Picric Acid Test Strips. Jpn. J. Food Chem. 2003, 10, 96–100. [Google Scholar]
- Asterion, A. Carbonates in the Plating Bath. Available online: https://asterionstc.com/2016/06/carbonates-plating-bath/ (accessed on 22 July 2021).
- Dusica, I.; Bojana, K.; Tea, B.; Radmilo, C.; Djuro, V.; Jovanka, L.; Slavica, S. Effect of microwave heating on content of cyanogenic glycosides in linseed. Ratar. Povrt. 2012, 49, 63–68. [Google Scholar] [CrossRef]
- Castada, H.Z.; Liu, J.; Ann Barringer, S.; Huang, X. Cyanogenesis in Macadamia and Direct Analysis of Hydrogen Cyanide in Macadamia Flowers, Leaves, Husks, and Nuts Using Selected Ion Flow Tube-Mass Spectrometry. Foods 2020, 9, 174. [Google Scholar] [CrossRef] [PubMed]
- Nandiyanto, A.B.D.; Oktiani, R.; Ragadhita, R. How to Read and Interpret FTIR Spectroscope of Organic Material. Indones. J. Sci. Technol. 2019, 4, 97–118. [Google Scholar] [CrossRef]
- Tesleva, E.P.; Gil, L.B.; Solovyan, A.V. Thermodeformational Behavior of Cubic Crystals of Sodium Cyanide. In Proceedings of the VII International Scientific Practical Conference “Innovative Technologies in Engineering”, Yurga, Russia, 19–21 May 2016. [Google Scholar]




























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Initial HCN concentration (mg/kg) * | ||||||
|---|---|---|---|---|---|---|
| Run 1 | Run 2 | Run 3 | Run 4 | |||
| 581.40 | 784.86 | 414.01 | 317.96 | |||
| Final/recovered HCN concentration (mg/kg) | ||||||
| Run 1 | Run 2 | Run 3 | Run 4 | |||
| 30 min extraction | 574.19 | 756.18 | 406.24 | 309.98 | Average % recovered | % RSD |
| Recovery (%) | 98.76 | 96.35 | 98.12 | 97.49 | 97.68 | 1.05 |
| 45 min extraction | 571.22 | 766.26 | 403.56 | 317.17 | Average % recovered | % RSD |
| Recovery (%) | 98.25 | 97.63 | 97.47 | 99.75 | 98.28 | 1.06 |
| 60 min extraction | 568.38 | 761.29 | 394.72 | 305.62 | Average % recovered | % RSD |
| Recovery (%) | 97.76 | 97.00 | 95.34 | 96.12 | 96.55 | 1.09 |
| Sample Aliquot (mL) | Burette Reading (mL) | Titrant Used for NaOH and Half of Na2CO3 (mL) | Titrant Used for NaOH and Na2CO3 (mL) | Titrant Used for HCO3− (mL) | Titrant Used for Na2CO3 (mL) | Titrant Used for NaOH (mL) | ||
|---|---|---|---|---|---|---|---|---|
| Initial Volume | 1st Endpoint | 2nd Endpoint | ||||||
| V1 | V2 | V3 | V4 = (V2 − V1) | V5 = (V3 − V1) | V6 = (V5 − V4) | Va = 2V6 | Vb = (V5 − 2V6) | |
| Titration of 2.6408 mol/L control NaCN solution | ||||||||
| 25 | 0.00 | 4.47 ± 0.01 | 5.00 ± 0.01 | 4.47 ± 0.01 | 5.00 ± 0.01 | 0.53 ± 0.00 | 1.05 ± 0.01 | 3.95 ± 0.01 |
| Titration of 2.6408 mol/L green-NaCN solution | ||||||||
| 25 | 0.00 | 17.52 ± 0.02 | 20.25 ± 0.02 | 17.52 ± 0.02 | 20.25 ± 0.02 | 2.74 ± 0.01 | 5.47 ± 0.02 | 14.78 ± 0.02 |
| Estimation of Na2CO3 and residual NaOH | ||||||||
| Na2CO3 | Residual NaOH | |||||||
| Control NaCN solution | Green-NaCN solution | Control NaCN solution | Green-NaCN solution | |||||
| Molarity (mol/L) | 0.05746 | 0.2165 | 0.4323 | 1.1699 | ||||
| Strength (g/L) | 6.0902 | 22.9468 | 17.2920 | 46.7960 | ||||
| Percentage (% w/v) | 0.61 | 2.29 | 1.73 | 4.68 | ||||
| Cassava Sample | Method Used to Extract Cyanogenic Glucosides | Reference | ||||||
|---|---|---|---|---|---|---|---|---|
| Fresh Leaves | Fresh Peels | Fresh Tuber Tissue | Fresh Whole Tuber Tissue | Dried Leaves | Dried Peels | Dried Whole Tuber | ||
| 5.68 | 5.50 | 5.90 | 5.27 | 4.61 | 4.27 | 5.11 | Acid hydrolysis | [2] |
| - | 10.08 | 10.06 | 9.46 | - | - | - | Acid hydrolysis | [5] |
| - | 5.50 | 5.86 | 3.92 | - | - | - | Direct hydrolysis (Deionised water) | |
| 0.21 | - | - | - | - | - | - | Direct hydrolysis (Deionised water) | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monga, I.; Paul, V.; Muniyasamy, S.; Zinyemba, O. Green Synthesis of Sodium Cyanide Using Hydrogen Cyanide Extracted under Vacuum from Cassava (Manihot esculenta Crantz) Leaves. Sustain. Chem. 2022, 3, 312-333. https://doi.org/10.3390/suschem3030020
Monga I, Paul V, Muniyasamy S, Zinyemba O. Green Synthesis of Sodium Cyanide Using Hydrogen Cyanide Extracted under Vacuum from Cassava (Manihot esculenta Crantz) Leaves. Sustainable Chemistry. 2022; 3(3):312-333. https://doi.org/10.3390/suschem3030020
Chicago/Turabian StyleMonga, Ilunga, Vimla Paul, Sudhakar Muniyasamy, and Orpah Zinyemba. 2022. "Green Synthesis of Sodium Cyanide Using Hydrogen Cyanide Extracted under Vacuum from Cassava (Manihot esculenta Crantz) Leaves" Sustainable Chemistry 3, no. 3: 312-333. https://doi.org/10.3390/suschem3030020
APA StyleMonga, I., Paul, V., Muniyasamy, S., & Zinyemba, O. (2022). Green Synthesis of Sodium Cyanide Using Hydrogen Cyanide Extracted under Vacuum from Cassava (Manihot esculenta Crantz) Leaves. Sustainable Chemistry, 3(3), 312-333. https://doi.org/10.3390/suschem3030020