Insight into Oncogenic Viral Pathways as Drivers of Viral Cancers: Implication for Effective Therapy

 , , , , , , , and
, , , , , , , and
Abstract
:
1. Introduction
2. Basic Characteristics of Viral Cancers
3. Types of Viral Cancers
4. Possible Role of COVID-19 Infection in Cancer Progression
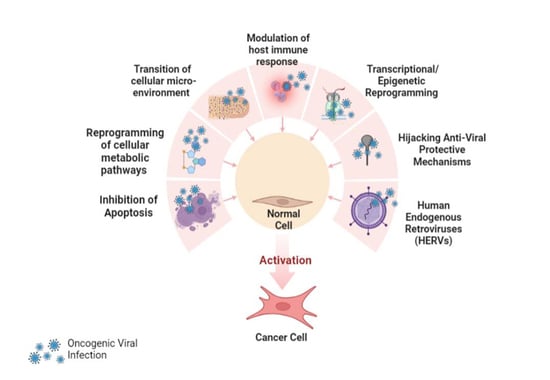
5. Multiple Mechanisms of Viral Carcinogenesis
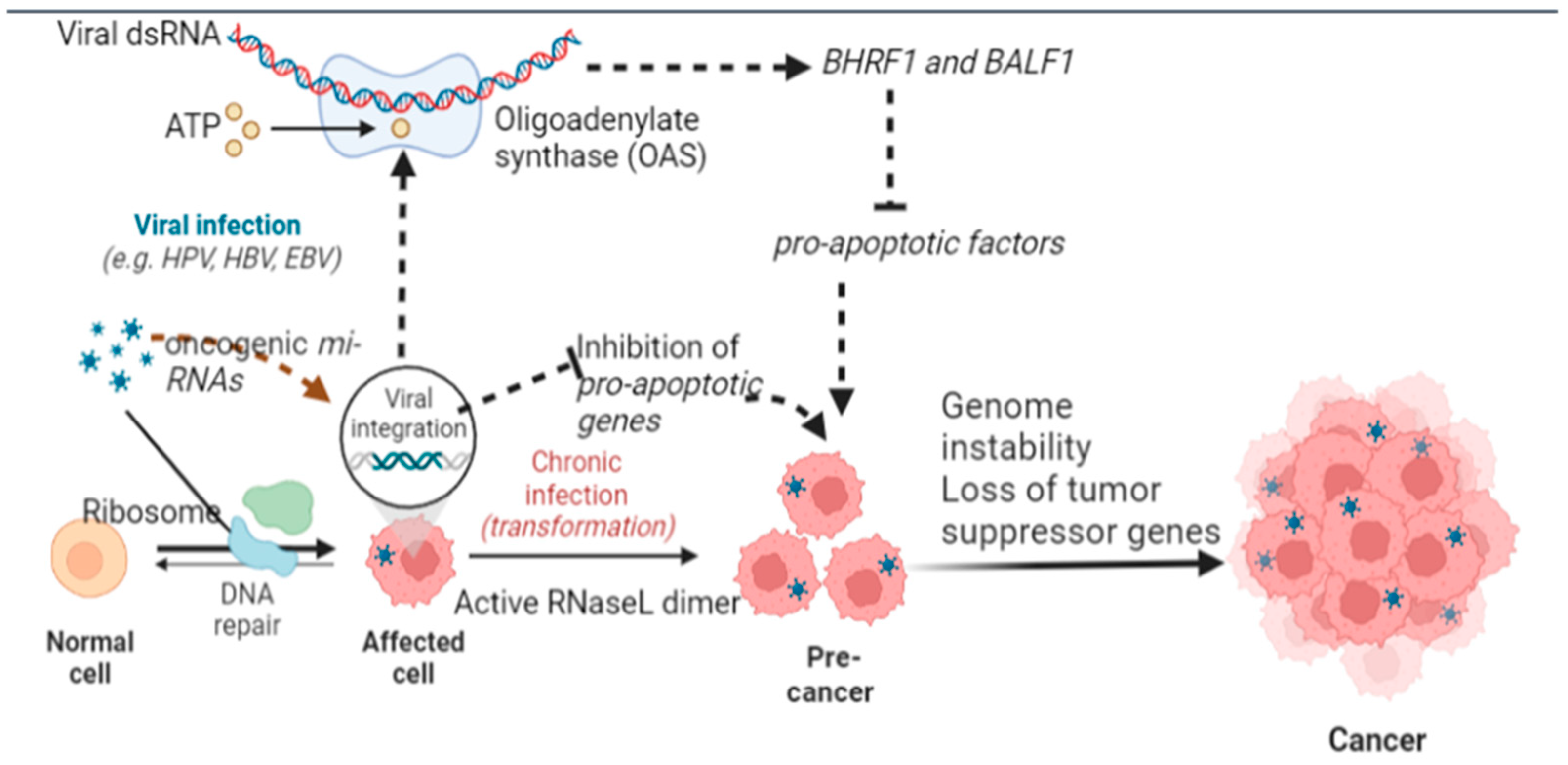
5.1. Inhibition of Apoptosis
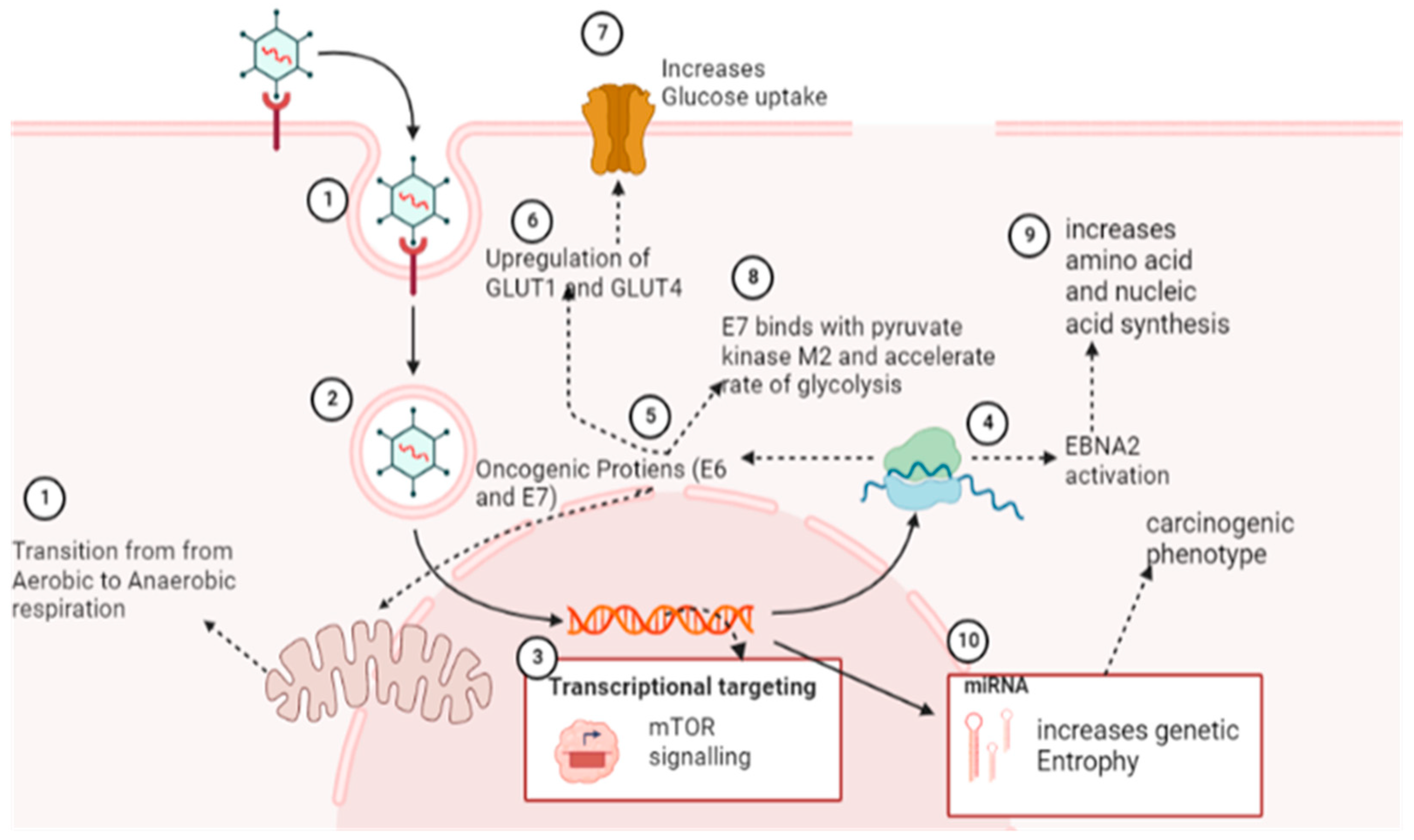
5.2. Reprogramming of Cellular Metabolic Pathways
5.3. Transition of Cellular Microenvironment
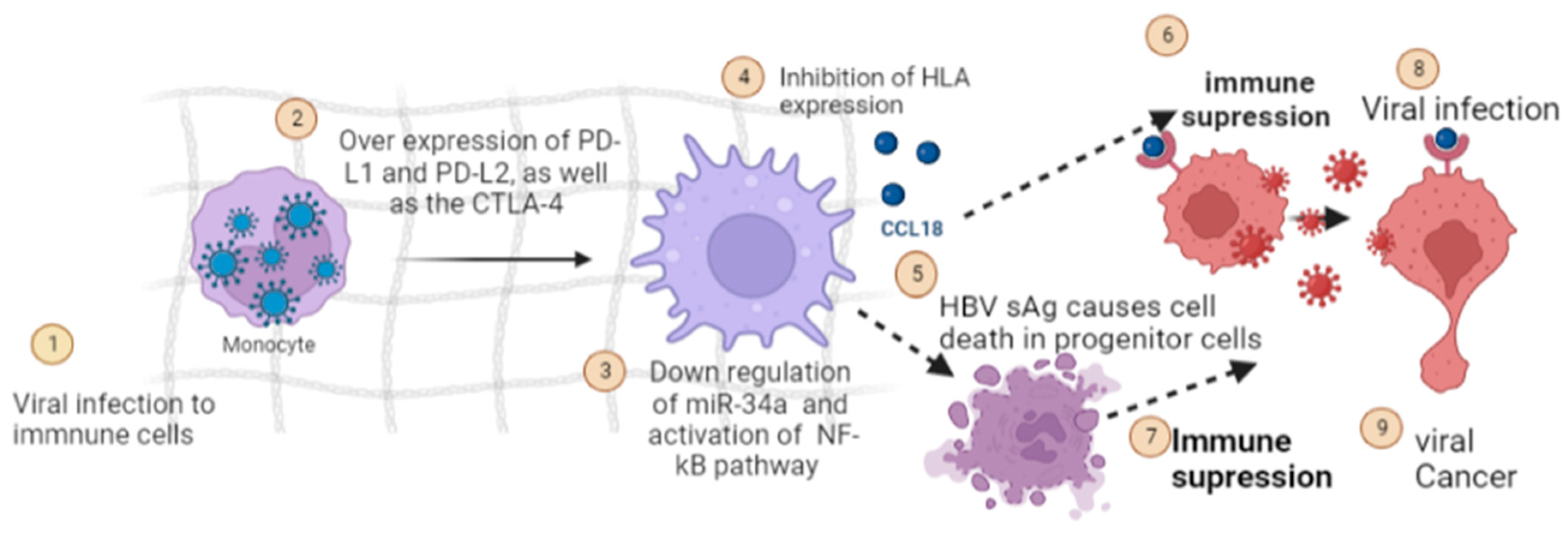
5.4. Modulation of Host Immune Response
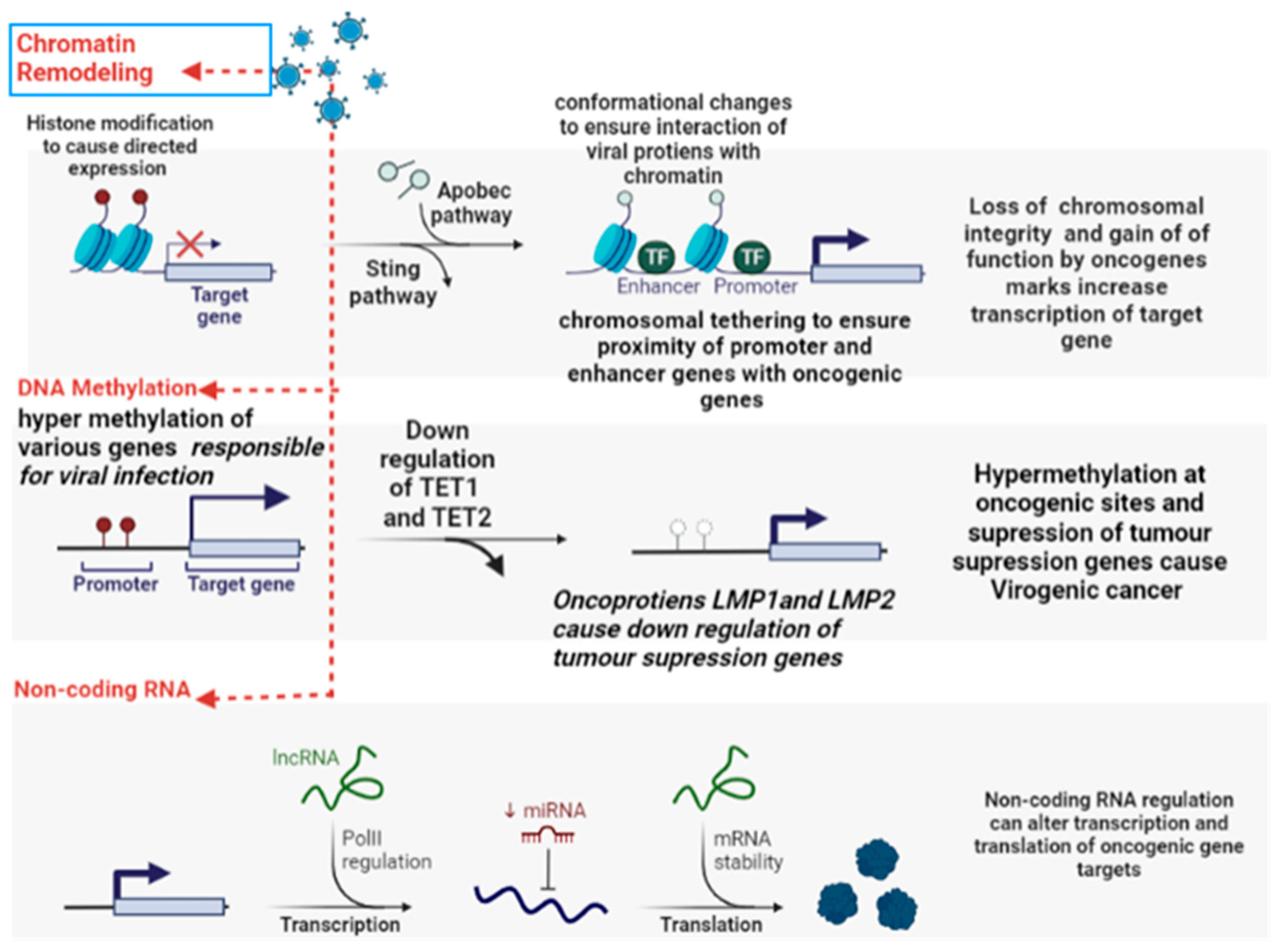
5.5. Transcriptional Reprogramming
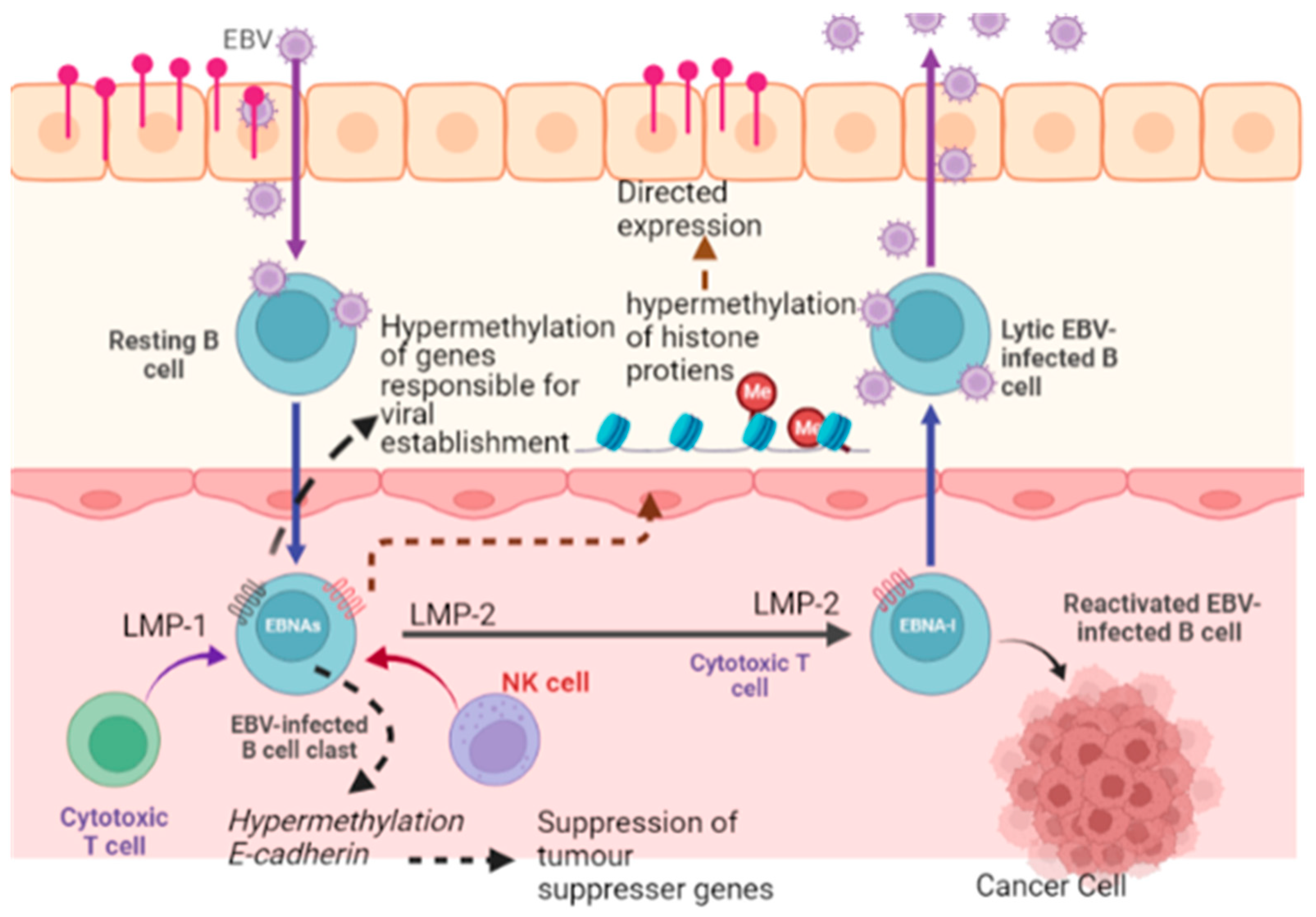
5.6. Epigenetic Reprogramming
5.7. Host Factors for Viral Oncogenesis
5.8. Hijacking Anti-Viral Protective Mechanisms
5.9. Role of Viral and Human miRNA in the Development of Cancer
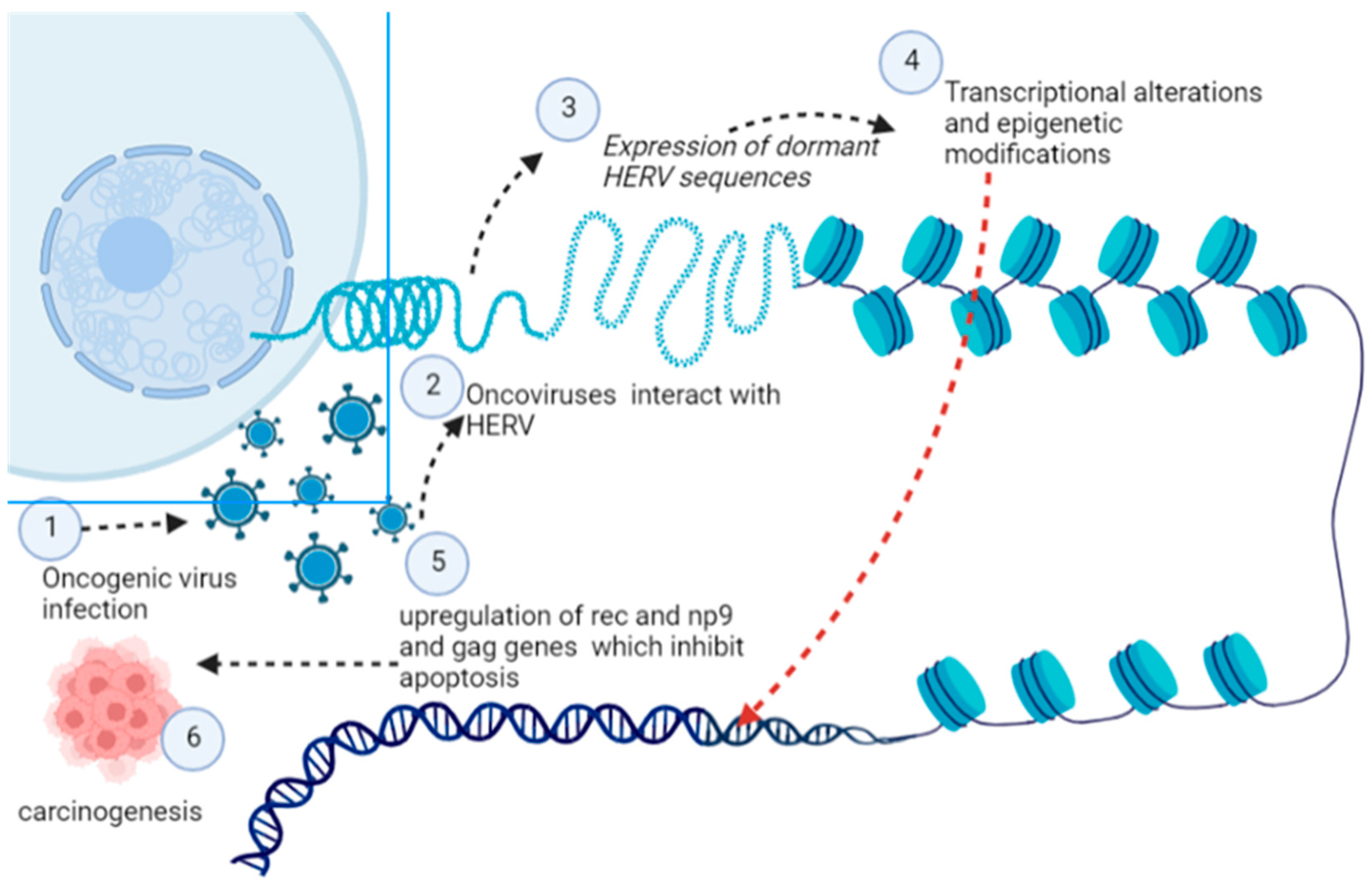
5.10. Human Endogenous Retroviruses (HERVs)
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- White, M.K.; Pagano, J.S.; Khalili, K. Viruses and human cancers: A long road of discovery of molecular paradigms. Clin. Microbiol. Rev. 2014, 27, 463–481. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R. Tumour-inducing viruses. Br. J. Hosp. Med. 2016, 77, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Kise, N.; Kinjyo, H.; Kondo, S.; Suzuki, M.; Tsukahara, N.; Murakami, A.; Kiyuna, A.; Agena, S.; Tanaka, K. Development of Antibodies against HPV-6 and HPV-11 for the Study of Laryngeal Papilloma. Viruses 2021, 13, 2024. [Google Scholar] [CrossRef] [PubMed]
- McEllin, B.; Searle, B.C.; DePledge, L.; Sun, G.; Cobbs, C.; Karimi, M. Detection of Human Papillomavirus Integration in Brain Metastases from Oropharyngeal Tumors by Targeted Sequencing. Viruses 2021, 13, 1536. [Google Scholar] [CrossRef] [PubMed]
- Mesri, E.A.; Feitelson, M.A.; Munger, K. Human viral oncogenesis: A cancer hallmarks analysis. Cell Host Microbe 2014, 15, 266–282. [Google Scholar] [CrossRef]
- Butler, M.D.; Griffin, K.; Brewster, C.D.; Kapuscinski, M.L.; Stenglein, M.D.; Tripp, D.W.; Quackenbush, S.L.; Fox, K.A. A Novel Retrovirus (Gunnison’s Prairie Dog Retrovirus) Associated with Thymic Lymphoma in Gunnison’s Prairie Dogs in Colorado, USA. Viruses 2020, 12, 606. [Google Scholar] [CrossRef] [PubMed]
- Wielgos, A.; Pietrzak, B.; Sikora, M.; Martirosian, G.; Suchonska, B.; Gozdowska, J.; Oldakowska-Jedynak, U.; Jabiry-Zieniewicz, Z.; Durlik, M.; Rudnicka, L. Human Papillomavirus (HPV) DNA Detection Using Self-Sampling Devices in Women Undergoing Long Term Immunosuppressive Therapy. Viruses 2020, 12, 962. [Google Scholar] [CrossRef]
- Oyervides-Muñoz, M.A.; Pérez-Maya, A.A.; Sánchez-Domínguez, C.N.; Berlanga-Garza, A.; Antonio-Macedo, M.; Valdéz-Chapa, L.D.; Cerda-Flores, R.M.; Trevino, V.; Barrera-Saldaña, H.A.; Garza-Rodríguez, M.L. Multiple HPV infections and viral load association in persistent cervical lesions in Mexican women. Viruses 2020, 12, 380. [Google Scholar] [CrossRef]
- Morgan, E.L.; Macdonald, A. Manipulation of JAK/STAT signalling by high-risk HPVs: Potential therapeutic targets for HPV-associated malignancies. Viruses 2020, 12, 977. [Google Scholar] [CrossRef]
- Santisteban-Espejo, A.; Perez-Requena, J.; Atienza-Cuevas, L.; Moran-Sanchez, J.; Fernandez-Valle, M.d.C.; Bernal-Florindo, I.; Romero-Garcia, R.; Garcia-Rojo, M. Prognostic Role of the Expression of Latent-Membrane Protein 1 of Epstein–Barr Virus in Classical Hodgkin Lymphoma. Viruses 2021, 13, 2523. [Google Scholar] [CrossRef]
- Nabi, S.U.; Rehman, M.U.; Arafah, A.; Taifa, S.; Khan, I.S.; Khan, A.; Rashid, S.; Jan, F.; Wani, H.A.; Ahmad, S.F. Treatment of Autism Spectrum Disorders by Mitochondrial-targeted Drug: Future of Neurological Diseases Therapeutics. Curr. Neuropharmacol. 2022. [Google Scholar] [CrossRef]
- Nabi, S.U.; Khan, A.; Siddiqui, E.M.; Rehman, M.U.; Alshahrani, S.; Arafah, A.; Mehan, S.; Alsaffar, R.M.; Alexiou, A.; Shen, B. Mechanisms of Mitochondrial Malfunction in Alzheimer’s Disease: New Therapeutic Hope. Oxidative Med. Cell. Longev. 2022, 2022, 4759963. [Google Scholar] [CrossRef]
- Krishna, G.; Soman Pillai, V.; Valiya Veettil, M. Upregulation of GLS1 isoforms KGA and GAC facilitates mitochondrial metabolism and cell proliferation in Epstein–Barr virus infected cells. Viruses 2020, 12, 811. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Choi, J.E.; Harms, P.W.; Chandrani, P. Merkel cell polyomavirus in Merkel cell carcinoma: Integration sites and involvement of the KMT2D tumor suppressor gene. Viruses 2020, 12, 966. [Google Scholar] [CrossRef] [PubMed]
- Rahman, R.; Gopinath, D.; Buajeeb, W.; Poomsawat, S.; Johnson, N.W. Potential Role of Epstein-Barr Virus in Oral Potentially Malignant Disorders and Oral Squamous Cell Carcinoma: A Scoping Review. Viruses 2022, 14, 801. [Google Scholar] [CrossRef]
- zur Hausen, H. Oncogenic DNA viruses. Oncogene 2001, 20, 7820–7823. [Google Scholar] [CrossRef] [PubMed]
- Bello-Perez, M.; Sola, I.; Novoa, B.; Klionsky, D.J.; Falco, A. Canonical and noncanonical autophagy as potential targets for COVID-19. Cells 2020, 9, 1619. [Google Scholar] [CrossRef]
- Cheerathodi, M.; Nkosi, D.; Cone, A.S.; York, S.B.; Meckes, D.G., Jr. Epstein-Barr virus LMP1 modulates the CD63 interactome. Viruses 2021, 13, 675. [Google Scholar] [CrossRef]
- Wright, R.J.; Hanson, H.A. A tipping point in cancer epidemiology: Embracing a life course exposomic framework. Trends Cancer 2022, 8, 280–282. [Google Scholar] [CrossRef]
- Aguayo, F.; Boccardo, E.; Corvalán, A.; Calaf, G.M.; Blanco, R. Interplay between Epstein-Barr virus infection and environmental xenobiotic exposure in cancer. Infect. Agents Cancer 2021, 16, 1–15. [Google Scholar] [CrossRef]
- Bhat, A.M.; Malik, H.; Singh, S.; Hussain, T.; Chaubhey, K.; Rehman, R.W.Y.; Qadri, S.I. Bio-prevalence and molecular diagnosis of Mycobacterium avium Subsp paratuberculosis infection in small ruminant population of Ganderbal district of Kashmir valley. J. Entomol. Zool. Stud. 2018, 6, 01–04. [Google Scholar]
- Kitsou, K.; Iliopoulou, M.; Spoulou, V.; Lagiou, P.; Magiorkinis, G. Viral causality of human cancer and potential roles of human endogenous retroviruses in the multi-omics era: An evolutionary epidemiology review. Front. Oncol. 2021, 11, 687631. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Hostager, B.S.; Bishop, G.A. Requirement for TRAF3 in signaling by LMP1 but not CD40 in B lymphocytes. J. Exp. Med. 2004, 199, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Li, L.; Yang, J.; Zhou, S.; Li, W.; Tang, M.; Shi, Y.; Yi, W.; Cao, Y. Latent membrane protein 1 encoded by Epstein–Barr virus induces telomerase activity via p16INK4A/Rb/E2F1 and JNK signaling pathways. J. Med. Virol. 2007, 79, 1153–1163. [Google Scholar] [CrossRef]
- Rassi, H.; Houshmand, M.; Hashemi, M.; Majidzadeh, A.; Akbari, M.H. Investigation of mitochondrial common deletion and BRCA mutations for detection of familial breast cancers in archival breast cancer materials. Int. J. Cancer Manag. 2009, 2, 77–83. [Google Scholar]
- Kumar, V.; Gabrilovich, D.I. Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunology 2014, 143, 512–519. [Google Scholar] [CrossRef]
- Hoang, P.H.; Landi, M.T. DNA Methylation in Lung Cancer: Mechanisms and Associations with Histological Subtypes, Molecular Alterations, and Major Epidemiological Factors. Cancers 2022, 14, 961. [Google Scholar] [CrossRef]
- Ojha, R.; Bhattacharyya, S.; Singh, S.K. Autophagy in cancer stem cells: A potential link between chemoresistance, recurrence, and metastasis. BioRes. Open Access 2015, 4, 97–108. [Google Scholar] [CrossRef]
- Cha, D.J.; Mengel, D.; Mustapic, M.; Liu, W.; Selkoe, D.J.; Kapogiannis, D.; Galasko, D.; Rissman, R.A.; Bennett, D.A.; Walsh, D.M. miR-212 and miR-132 are downregulated in neurally derived plasma exosomes of Alzheimer’s patients. Front. Neurosci. 2019, 13, 1208. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Ji, Y.G.; Cho, H.J.; Lee, D.H. Synergistic anti-cancer effects of AKT and SRC inhibition in human pancreatic cancer cells. Yonsei Med. J. 2018, 59, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.J.d.S.; Leão, A.H.F.F.; Erustes, A.G.; Morais, I.B.d.M.; Vrechi, T.A.d.M.; Zamarioli, L.d.S.; Pereira, C.A.S.; Marchioro, L.d.O.; Sperandio, L.P.; Lins, Í.V.F. Pharmacological Modulators of Autophagy as a Potential Strategy for the Treatment of COVID-19. Int. J. Mol. Sci. 2021, 22, 4067. [Google Scholar] [CrossRef] [PubMed]
- Kottaridi, C.; Resta, P.; Leventakou, D.; Gioti, K.; Zygouras, I.; Gouloumi, A.-R.; Sakagiannis, G.; Alzahrani, K.J.; Venetikou, M.S.; Anthouli-Anagnostopoulou, F. The T350G Variation of Human Papillomavirus 16 E6 Gene Prevails in Oropharyngeal Cancer from a Small Cohort of Greek Patients. Viruses 2022, 14, 1724. [Google Scholar] [CrossRef]
- Boyce, C.J.; Brown, G.D.; Moore, S.C. Money and happiness: Rank of income, not income, affects life satisfaction. Psychol. Sci. 2010, 21, 471–475. [Google Scholar] [CrossRef]
- Zheng, X.; Huang, Y.; Li, K.; Luo, R.; Cai, M.; Yun, J. Immunosuppressive Tumor Microenvironment and Immunotherapy of Epstein–Barr Virus-Associated Malignancies. Viruses 2022, 14, 1017. [Google Scholar] [CrossRef] [PubMed]
- Brulois, K.; Jung, J.U. Interplay between Kaposi’s sarcoma-associated herpesvirus and the innate immune system. Cytokine Growth Factor Rev. 2014, 25, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Isaguliants, M.; Bayurova, E.; Avdoshina, D.; Kondrashova, A.; Chiodi, F.; Palefsky, J.M. Oncogenic effects of HIV-1 proteins, mechanisms behind. Cancers 2021, 13, 305. [Google Scholar] [CrossRef]
- Pal, A.; Kundu, R. Human papillomavirus E6 and E7: The cervical cancer hallmarks and targets for therapy. Front. Microbiol. 2020, 10, 3116. [Google Scholar] [CrossRef]
- Tempera, I.; Lieberman, P.M. Oncogenic Viruses as Entropic Drivers of Cancer Evolution. Front. Virol. 2021, 1, 753366. [Google Scholar] [CrossRef]
- Jiang, J.; Ying, H. Revealing the crosstalk between nasopharyngeal carcinoma and immune cells in the tumor microenvironment. J. Exp. Clin. Cancer Res. 2022, 41, 1–12. [Google Scholar] [CrossRef]
- Daniel, B.; Mukherjee, G.; Seshadri, L.; Vallikad, E.; Krishna, S. Changes in the physical state and expression of human papillomavirus type 16 in the progression of cervical intraepithelial neoplasia lesions analysed by PCR. J. Gen. Virol. 1995, 76, 2589–2593. [Google Scholar] [CrossRef]
- Kulmala, S.A.; Syrjänen, S.M.; Gyllensten, U.B.; Shabalova, I.P.; Petrovichev, N.; Tosi, P.; Syrjänen, K.J.; Johansson, B.C. Early integration of high copy HPV16 detectable in women with normal and low grade cervical cytology and histology. J. Clin. Pathol. 2006, 59, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Williams, V.M.; Filippova, M.; Soto, U.; Duerksen-Hughes, P.J. HPV-DNA integration and carcinogenesis: Putative roles for inflammation and oxidative stress. Future Virol. 2011, 6, 45–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckman, K.B.; Ames, B.N. Oxidative decay of DNA. J. Biol. Chem. 1997, 272, 19633–19636. [Google Scholar] [CrossRef]
- Kessis, T.D.; Connolly, D.C.; Hedrick, L.; Cho, K.R. Expression of HPV16 E6 or E7 increases integration of foreign DNA. Oncogene 1996, 13, 427–431. [Google Scholar] [PubMed]
- Sakakibara, N.; Mitra, R.; McBride, A.A. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol. 2011, 85, 8981–8995. [Google Scholar] [CrossRef]
- Kang, X.; Shu, X.; Hao, F.; Bai, L. Identification of Potential miRNAs-mRNAs Regulatory Network From CHB to HCC Based on Bioinformatic Analysis. J. Transl. Med. 2022, 17, 7. [Google Scholar]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef]
- Bossler, F.; Kuhn, B.J.; Günther, T.; Kraemer, S.J.; Khalkar, P.; Adrian, S.; Lohrey, C.; Holzer, A.; Shimobayashi, M.; Dürst, M. Repression of human papillomavirus oncogene expression under hypoxia is mediated by PI3K/mTORC2/AKT signaling. MBio 2019, 10, e02323-18. [Google Scholar] [CrossRef]
- Ohlenschläger, O.; Seiboth, T.; Zengerling, H.; Briese, L.; Marchanka, A.; Ramachandran, R.; Baum, M.; Korbas, M.; Meyer-Klaucke, W.; Dürst, M. Solution structure of the partially folded high-risk human papilloma virus 45 oncoprotein E7. Oncogene 2006, 25, 5953–5959. [Google Scholar] [CrossRef]
- Afzal, S.; Fiaz, K.; Noor, A.; Sindhu, A.S.; Hanif, A.; Bibi, A.; Asad, M.; Nawaz, S.; Zafar, S.; Ayub, S. Interrelated Oncogenic Viruses and Breast Cancer. Front. Mol. Biosci. 2022, 9, 781111. [Google Scholar] [CrossRef]
- Al Moustafa, A.-E.; Al-Antary, N.; Yasmeen, A. High-Risk Human Papillomavirus and Colorectal Carcinogenesis. In Human Papillomavirus-Research in a Global Perspective; IntechOpen: London, UK, 2016. [Google Scholar]
- McLaughlin-Drubin, M.E.; Park, D.; Munger, K. Tumor suppressor p16INK4A is necessary for survival of cervical carcinoma cell lines. Proc. Natl. Acad. Sci. USA 2013, 110, 16175–16180. [Google Scholar] [CrossRef]
- Zhang, Y.; Fan, S.; Meng, Q.; Ma, Y.; Katiyar, P.; Schlegel, R.; Rosen, E.M. BRCA1 interaction with human papillomavirus oncoproteins. J. Biol. Chem. 2005, 280, 33165–33177. [Google Scholar] [CrossRef]
- Al Moustafa, A.-E.; Al-Antary, N.; Aboulkassim, T.; Akil, N.; Batist, G.; Yasmeen, A. Co-prevalence of Epstein–Barr virus and high-risk human papillomaviruses in Syrian women with breast cancer. Hum. Vaccines Immunother. 2016, 12, 1936–1939. [Google Scholar] [CrossRef] [Green Version]
- Jácome-Santos, H.; da Silva e Silva, N.; Resende, R.G.; Costa Pinheiro, H.H.; Almeida Machado, L.F.; de Souza Silva, G.; de Oliveira Costa, F.; Brasil-Costa, I.; Amoras-Alves, A.C.B.; Mesquita, R.A. Simultaneous occurrence of Epstein-Barr virus (EBV) in periodontal pockets and in oral squamous cell carcinoma: A cross-sectional study. Clin. Oral Investig. 2022, 26, 2807–2815. [Google Scholar] [CrossRef]
- Jiménez-Santos, M.J.; García-Martín, S.; Fustero-Torre, C.; Di Domenico, T.; Gómez-López, G.; Al-Shahrour, F. A bioinformatics roadmap for therapy selection in cancer genomics. Mol. Oncol. 2022, 16, 3881–3908. [Google Scholar] [CrossRef]
- Shah, N.N.; Dar, K.A.; Quibtiya, S.; Azad, A.M.U.D.; Mushtaq, M.; Bashir, S.M.; Rather, M.A.; Ali, S.I.; Sheikh, W.M.; Nabi, S.U. Repurposing of Mycobacterium indicus pranii for the severe form of COVID-19 patients in India: A cohort study. J. Med. Virol. 2022, 94, 1906–1919. [Google Scholar] [CrossRef]
- Shah, N.N.; Khan, Z.; Ahad, H.; Elderdery, A.Y.; Alomary, M.N.; Atwah, B.; Alhindi, Z.; Alsugoor, M.H.; Elkhalifa, A.M.; Nabi, S. Mucormycosis an added burden to Covid-19 Patients: An in-depth Systematic Review. J. Infect. Public Health 2022, 15, 1299–1314. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Nabi, S.U.; Rather, M.A.; Kalwar, Q.; Ali, S.I.; Sheikh, W.M.; Ganai, A.; Bashir, S.M. An update on emerging therapeutics to combat COVID-19. Basic Clin. Pharmacol. Toxicol. 2021, 129, 104–129. [Google Scholar] [CrossRef] [PubMed]
- Zalpoor, H.; Akbari, A.; Samei, A.; Forghaniesfidvajani, R.; Kamali, M.; Afzalnia, A.; Manshouri, S.; Heidari, F.; Pornour, M.; Khoshmirsafa, M. The roles of Eph receptors, neuropilin-1, P2X7, and CD147 in COVID-19-associated neurodegenerative diseases: Inflammasome and JaK inhibitors as potential promising therapies. Cell. Mol. Biol. Lett. 2022, 27, 1–21. [Google Scholar] [CrossRef]
- Zalpoor, H.; Akbari, A.; Nabi-Afjadi, M. Ephrin (Eph) receptor and downstream signaling pathways: A promising potential targeted therapy for COVID-19 and associated cancers and diseases. Hum. Cell 2022, 35, 952–954. [Google Scholar] [CrossRef] [PubMed]
- Zalpoor, H.; Bakhtiyari, M.; Liaghat, M.; Nabi-Afjadi, M.; Ganjalikhani-Hakemi, M. Quercetin potential effects against SARS-CoV-2 infection and COVID-19-associated cancer progression by inhibiting mTOR and hypoxia-inducible factor-1α (HIF-1α). Phytother. Res. 2022, 36, 2679–2682. [Google Scholar] [CrossRef]
- Zalpoor, H.; Shapourian, H.; Akbari, A.; Shahveh, S.; Haghshenas, L. Increased neuropilin-1 expression by COVID-19: A possible cause of long-term neurological complications and progression of primary brain tumors. Hum. Cell 2022, 35, 1301–1303. [Google Scholar] [CrossRef]
- Miller, K.; McGrath, M.E.; Hu, Z.; Ariannejad, S.; Weston, S.; Frieman, M.; Jackson, W.T. Coronavirus interactions with the cellular autophagy machinery. Autophagy 2020, 16, 2131–2139. [Google Scholar] [CrossRef]
- Akkoc, Y.; Peker, N.; Akcay, A.; Gozuacik, D. Autophagy and cancer dormancy. Front. Oncol. 2021, 11, 627023. [Google Scholar] [CrossRef]
- Mehri, F.; Rahbar, A.H.; Ghane, E.T.; Souri, B.; Esfahani, M. Changes in oxidative markers in COVID-19 patients. Arch. Med. Res. 2021, 52, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Assiri, A.; Al-Tawfiq, J.A.; Al-Rabeeah, A.A.; Al-Rabiah, F.A.; Al-Hajjar, S.; Al-Barrak, A.; Flemban, H.; Al-Nassir, W.N.; Balkhy, H.H.; Al-Hakeem, R.F. Epidemiological, demographic, and clinical characteristics of 47 cases of Middle East respiratory syndrome coronavirus disease from Saudi Arabia: A descriptive study. Lancet Infect. Dis. 2013, 13, 752–761. [Google Scholar] [CrossRef]
- Peiris, J.S.M.; Chu, C.-M.; Cheng, V.C.-C.; Chan, K.; Hung, I.; Poon, L.L.; Law, K.-I.; Tang, B.; Hon, T.; Chan, C. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: A prospective study. Lancet 2003, 361, 1767–1772. [Google Scholar] [CrossRef]
- Kim, Y.; Lee, S.; Chu, C.; Choe, S.; Hong, S.; Shin, Y. The characteristics of Middle Eastern respiratory syndrome coronavirus transmission dynamics in South Korea. Osong Public Health Res. Perspect. 2016, 7, 49–55. [Google Scholar] [CrossRef]
- Saad, M.; Omrani, A.S.; Baig, K.; Bahloul, A.; Elzein, F.; Matin, M.A.; Selim, M.A.; Al Mutairi, M.; Al Nakhli, D.; Al Aidaroos, A.Y. Clinical aspects and outcomes of 70 patients with Middle East respiratory syndrome coronavirus infection: A single-center experience in Saudi Arabia. Int. J. Infect. Dis. 2014, 29, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.G.; Macleod, K.F. Autophagy, cancer stem cells and drug resistance. J. Pathol. 2019, 247, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; Hait, N.C. Autophagy-mediated tumor cell survival and progression of breast cancer metastasis to the brain. J. Cancer 2021, 12, 954. [Google Scholar] [CrossRef]
- Capparelli, R.; Iannelli, D. Epigenetics and Helicobacter pylori. Int. J. Mol. Sci. 2022, 23, 1759. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Rickinson, A. The global landscape of EBV-associated tumors. Front. Oncol. 2019, 6, 713. [Google Scholar] [CrossRef] [Green Version]
- Shannon-Lowe, C.; Rickinson, A.B.; Bell, A.I. Epstein–Barr virus-associated lymphomas. Philos. Trans. R. Soc. B: Biol. Sci. 2017, 372, 20160271. [Google Scholar] [CrossRef]
- Yan, B.; Wang, C.; Chakravorty, S.; Zhang, Z.; Kadadi, S.D.; Zhuang, Y.; Sirit, I.; Hu, Y.; Jung, M.; Sahoo, S. A comprehensive single cell data analysis of in lymphoblastoid cells reveals the role of Super-enhancers in maintaining EBV latency. J. Med. Virol. 2023, 95, e28362. [Google Scholar] [CrossRef] [PubMed]
- Al-Salam, S.; Awwad, A.; Sudhadevi, M.; Daoud, S.; Nagelkerke, N.J.; Castella, A.; Chong, S.; Alashari, M. Epstein-Barr virus infection correlates with the expression of COX-2, p16INK4A and p53 in classic Hodgkin lymphoma. Int. J. Clin. Exp. Pathol. 2013, 6, 2765. [Google Scholar] [PubMed]
- zur Hausen, H. Papillomaviruses in the causation of human cancers—A brief historical account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef]
- Akram, M.; Iqbal, M.; Daniyal, M.; Khan, A.U. Awareness and current knowledge of breast cancer. Biol. Res. 2017, 50, 1–23. [Google Scholar] [CrossRef]
- Kane, G.C. It’s a Network, Not an Encyclopedia: A Social Network Perspective on Wikipedia Collaboration. In Academy of Management Proceedings; Academy of Management USA: Boston, MA, USA, 2009; pp. 1–6. [Google Scholar]
- Retamal-Morales, G.; Mehnert, M.; Schwabe, R.; Tischler, D.; Zapata, C.; Chávez, R.; Schlömann, M.; Levicán, G. Detection of arsenic-binding siderophores in arsenic-tolerating Actinobacteria by a modified CAS assay. Ecotoxicol. Environ. Saf. 2018, 157, 176–181. [Google Scholar] [CrossRef]
- Liu, L.; Oza, S.; Hogan, D.; Perin, J.; Rudan, I.; Lawn, J.E.; Cousens, S.; Mathers, C.; Black, R.E. Global, regional, and national causes of child mortality in 2000–13, with projections to inform post-2015 priorities: An updated systematic analysis. Lancet 2015, 385, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Talapin, D.V.; Murray, C.B. PbSe nanocrystal solids for n-and p-channel thin film field-effect transistors. Science 2005, 310, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, L.; Fujito, N.; Sugimoto, R.; Inoue, I. Detection of Ancient Viruses and Long-Term Viral Evolution. Viruses 2022, 14, 1336. [Google Scholar] [CrossRef]
- Yang, X.; Jiang, Z.; Li, Y.; Zhang, Y.; Han, Y.; Gao, L. Non-coding RNAs regulating epithelial-mesenchymal transition: Research progress in liver disease. Biomed. Pharmacother. 2022, 150, 112972. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Golovkina, T. Common threads in persistent viral infections. J. Virol. 2010, 84, 4116–4123. [Google Scholar] [CrossRef]
- Levican, J.; Acevedo, M.; León, O.; Gaggero, A.; Aguayo, F. Role of BK human polyomavirus in cancer. Infect. Agents Cancer 2018, 13, 1–8. [Google Scholar] [CrossRef]
- Rizzo, G.E.M.; Cabibbo, G.; Craxì, A. Hepatitis B Virus-Associated Hepatocellular Carcinoma. Viruses 2022, 14, 986. [Google Scholar] [CrossRef]
- Amemiya, T.; Yamaguchi, T. Oscillations and Dynamic Symbiosis in Cellular Metabolism in Cancer. Front. Oncol. 2022, 12, 783908. [Google Scholar] [CrossRef]
- Liu, X.; Zhu, C.; Wang, Y.; Wei, F.; Cai, Q. KSHV reprogramming of host energy metabolism for pathogenesis. Front. Cell. Infect. Microbiol. 2021, 11, 621156. [Google Scholar] [CrossRef]
- Barillari, G.; Bei, R.; Manzari, V.; Modesti, A. Infection by High-Risk Human Papillomaviruses, Epithelial-to-Mesenchymal Transition and Squamous Pre-Malignant or Malignant Lesions of the Uterine Cervix: A Series of Chained Events? Int. J. Mol. Sci. 2021, 22, 13543. [Google Scholar] [CrossRef]
- Vazifehmand, R.; Ali, D.S.; Othman, Z.; Chau, D.-M.; Stanslas, J.; Shafa, M.; Sekawi, Z. The evaluation expression of non-coding RNAs in response to HSV-G47∆ oncolytic virus infection in glioblastoma multiforme cancer stem cells. J. NeuroVirol. 2022, 28, 566–582. [Google Scholar] [CrossRef] [PubMed]
- Cluster, G. Genetic Organization and Hypoxic. J. Virol 2006, 80, 7037. [Google Scholar]
- Wang, V.; Davis, D.A.; Deleage, C.; Brands, C.; Choi, H.S.; Haque, M.; Yarchoan, R. Induction of Kaposi’s sarcoma-associated herpesvirus-encoded thymidine kinase (ORF21) by X-box binding protein 1. J. Virol. 2020, 94, e01555-19. [Google Scholar] [CrossRef]
- McNamara, R.P.; Eason, A.B.; Zhou, Y.; Bigi, R.; Griffith, J.D.; Costantini, L.M.; Rudek, M.A.; Anders, N.M.; Damania, B.A.; Dittmer, D.P. Exosome-Encased Nucleic Acid Scaffold Chemotherapeutic Agents for Superior Anti-Tumor and Anti-Angiogenesis Activity. ACS Bio Med. Chem. Au 2022, 2, 140–149. [Google Scholar] [CrossRef]
- Guo, W.; Qiao, T.; Dong, B.; Li, T.; Liu, Q.; Xu, X. The Effect of Hypoxia-Induced Exosomes on Anti-Tumor Immunity and Its Implication for Immunotherapy. Front. Immunol. 2022, 13, 3214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, H.; Liu, J.; Tao, T.; Zeng, Z.; Wang, M. RGS1 and related genes as potential targets for immunotherapy in cervical cancer: Computational biology and experimental validation. J. Transl. Med. 2022, 20, 1–17. [Google Scholar] [CrossRef]
- Nabi, S.U.; Ali, S.I.; Rather, M.A.; Sheikh, W.M.; Altaf, M.; Singh, H.; Mumtaz, P.T.; Mishra, N.C.; Nazir, S.U.; Bashir, S.M. Organoids: A new approach in toxicity testing of nanotherapeutics. J. Appl. Toxicol. 2022, 42, 52–72. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Jiao, P.; Wang, Z.; Chen, M.; Du, H.; Xu, L.; Xu, J.; Dai, Y.; Wu, F.-g.; Zhang, Y. Endoplasmic reticulum stress promotes the release of exosomal PD-L1 from head and neck cancer cells and facilitates M2 macrophage polarization. Cell Commun. Signal. 2022, 20, 1–13. [Google Scholar] [CrossRef]
- Meraviglia-Crivelli, D.; Zheleva, A.; Barainka, M.; Moreno, B.; Villanueva, H.; Pastor, F. Therapeutic Strategies to Enhance Tumor Antigenicity: Making the Tumor Detectable by the Immune System. Biomedicines 2022, 10, 1842. [Google Scholar] [CrossRef] [PubMed]
- Harley, J.B.; Chen, X.; Pujato, M.; Miller, D.; Maddox, A.; Forney, C.; Magnusen, A.F.; Lynch, A.; Chetal, K.; Yukawa, M. Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat. Genet. 2018, 50, 699–707. [Google Scholar] [CrossRef]
- Mrozek-Gorska, P.; Buschle, A.; Pich, D.; Schwarzmayr, T.; Fechtner, R.; Scialdone, A.; Hammerschmidt, W. Epstein–Barr virus reprograms human B lymphocytes immediately in the prelatent phase of infection. Proc. Natl. Acad. Sci. USA 2019, 116, 16046–16055. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.-S.; Kieff, E. Epstein–Barr virus latent genes. Exp. Mol. Med. 2015, 47, e131. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I. Herpesvirus latency. J. Clin. Investig. 2020, 130, 3361–3369. [Google Scholar] [CrossRef]
- Giudice, A.; D’Arena, G.; Crispo, A.; Tecce, M.F.; Nocerino, F.; Grimaldi, M.; Rotondo, E.; D’Ursi, A.M.; Scrima, M.; Galdiero, M. Role of viral miRNAs and epigenetic modifications in epstein-barr virus-associated gastric carcinogenesis. Oxidative Med. Cell. Longev. 2016, 2016, 6021934. [Google Scholar] [CrossRef] [PubMed]
- Nash, A.; Ryan, E.J. The oncogenic gamma herpesviruses Epstein-Barr virus (EBV) and Kaposi’s sarcoma-associated herpesvirus (KSHV) hijack retinoic acid-inducible gene I (RIG-I) facilitating both viral and tumour immune evasion. Tumour Virus Res. 2022, 14, 200246. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Okabe, A.; Kaneda, A. Epigenetic contribution to tumorigenesis of host cells by Epstein-Barr virus infection. Chiba Medical. J. 2022, 98, 1–7. [Google Scholar]
- Kapusta, A.; Feschotte, C. Volatile evolution of long noncoding RNA repertoires: Mechanisms and biological implications. Trends Genet. 2014, 30, 439–452. [Google Scholar] [CrossRef]
- Krump, N.A.; You, J. Molecular mechanisms of viral oncogenesis in humans. Nat. Rev. Microbiol. 2018, 16, 684–698. [Google Scholar] [CrossRef]
- Fierti, A.O.; Yakass, M.B.; Okertchiri, E.A.; Adadey, S.M.; Quaye, O. The Role of Epstein-Barr Virus in Modulating Key Tumor Suppressor Genes in Associated Malignancies: Epigenetics, Transcriptional, and Post-Translational Modifications. Biomolecules 2022, 12, 127. [Google Scholar] [CrossRef]
- Moody, C.A. Regulation of the Innate Immune Response during the Human Papillomavirus Life Cycle. Viruses 2022, 14, 1797. [Google Scholar] [CrossRef]
- Grillo, M.J.; Jones, K.F.; Carpenter, M.A.; Harris, R.S.; Harki, D.A. The current toolbox for APOBEC drug discovery. Trends Pharmacol. Sci. 2022, 43, 362–377. [Google Scholar] [CrossRef]
- Zhao, J.; Luo, Z. Discovery of Raf Family Is a Milestone in Deciphering the Ras-Mediated Intracellular Signaling Pathway. Int. J. Mol. Sci. 2022, 23, 5158. [Google Scholar] [CrossRef] [PubMed]
- Chitcharoen, S.; Sivapornnukul, P.; Payungporn, S. Revolutionized virome research using systems microbiology approaches. Exp. Biol. Med. 2022, 247, 1135–1147. [Google Scholar] [CrossRef]
- Soliman, S.H.A.; Orlacchio, A.; Verginelli, F. Viral Manipulation of the Host Epigenome as a Driver of Virus-Induced Oncogenesis. Microorganisms 2021, 9, 1179. [Google Scholar] [CrossRef] [PubMed]
- Ginefra, P.; Carrasco Hope, H.; Spagna, M.; Zecchillo, A.; Vannini, N. Ionic Regulation of T-Cell Function and Anti-Tumour Immunity. Int. J. Mol. Sci. 2021, 22, 13668. [Google Scholar] [CrossRef]
- Goon, P.; Schürmann, M.; Oppel, F.; Shao, S.; Schleyer, S.; Pfeiffer, C.J.; Todt, I.; Brasch, F.; Scholtz, L.-U.; Göerner, M. Viral and Clinical Oncology of Head and Neck Cancers. Curr. Oncol. Rep. 2022, 24, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Yasunaga, J.I.; Matsuoka, M. Oncogenic spiral by infectious pathogens: Cooperation of multiple factors in cancer development. Cancer Sci. 2018, 109, 24–32. [Google Scholar] [CrossRef]
- Tong, L.; Stow, N.D. Analysis of herpes simplex virus type 1 DNA packaging signal mutations in the context of the viral genome. J. Virol. 2010, 84, 321–329. [Google Scholar] [CrossRef]
- Cao, Z.; Sugimura, N.; Burgermeister, E.; Ebert, M.P.; Zuo, T.; Lan, P. The gut virome: A new microbiome component in health and disease. EBioMedicine 2022, 81, 104113. [Google Scholar] [CrossRef]
- Yang, X.; Da, M.; Zhang, W.; Qi, Q.; Zhang, C.; Han, S. Role of Lactobacillus in cervical cancer. Cancer Manag. Res. 2018, 10, 1219. [Google Scholar] [CrossRef]
- Lei, V.; Petty, A.J.; Atwater, A.R.; Wolfe, S.A.; MacLeod, A.S. Skin viral infections: Host antiviral innate immunity and viral immune evasion. Front. Immunol. 2020, 11, 593901. [Google Scholar] [CrossRef]
- Guzman, N.D. Characterization and miRNA Analysis of Cancer Cell-Secreted Microvesicles; The Ohio State University: Columbus, OH, USA, 2012. [Google Scholar]
- Židovec Lepej, S.; Matulić, M.; Gršković, P.; Pavlica, M.; Radmanić, L.; Korać, P. miRNAs: EBV mechanism for escaping host’s immune response and supporting tumorigenesis. Pathogens 2020, 9, 353. [Google Scholar] [CrossRef] [PubMed]
- Zamai, L. Unveiling human non-random genome editing mechanisms activated in response to chronic environmental changes: I. Where might these mechanisms come from and what might they have led to? Cells 2020, 9, 2362. [Google Scholar] [CrossRef]
- Yu, H.; Wang, Y.; Wang, D.; Yi, Y.; Liu, Z.; Wu, M.; Wu, Y.; Zhang, Q. Landscape of the epigenetic regulation in wound healing. Front. Physiol. 2022, 13, 949498. [Google Scholar] [CrossRef] [PubMed]
- Hurst, T.P.; Magiorkinis, G. Epigenetic control of human endogenous retrovirus expression: Focus on regulation of long-terminal repeats (LTRs). Viruses 2017, 9, 130. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, R.; Uldrick, T.S. HIV-associated cancers and related diseases. N. Engl. J. Med. 2018, 378, 1029–1041. [Google Scholar] [CrossRef]
- Curty, G.; Marston, J.L.; de Mulder Rougvie, M.; Leal, F.E.; Nixon, D.F.; Soares, M.A. Human endogenous retrovirus K in cancer: A potential biomarker and immunotherapeutic target. Viruses 2020, 12, 726. [Google Scholar] [CrossRef]
- Kristensen, M.K.; Christensen, T. Regulation of the expression of human endogenous retroviruses: Elements in fetal development and a possible role in the development of cancer and neurological diseases. Apmis 2021, 129, 241–253. [Google Scholar] [CrossRef]
- Yau, H.L.; Ettayebi, I.; De Carvalho, D.D. The cancer epigenome: Exploiting its vulnerabilities for immunotherapy. Trends Cell Biol. 2019, 29, 31–43. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus | Oncogenic Cancer-Associated | Oncogenic Viral Products | Mechanism Involved | References |
|---|---|---|---|---|
| EBV | Hodgkin’s lymphoma, Gastric Cancer, Lymphoepithelioma | circular RNAs (circRNAs), EBV circular BamHI A rightward transcripts (circBARTs), EBV latent membrane protein 1 (EBV LMP1) | Various genes and signaling pathways influence the development of EBV-related neoplasms. Activate oncogenes such as Bcl-2 and MYC and signaling pathways, including NF-B, JNK, JAK/STAT, and PI3K/Akt, and deactivate tumor suppressors such as p53, p27kip1, p21WAF1/CIP1, p16INK4A, p73, PRDM1, DICE1, and p27kip1. Methylation modification, evasion of apoptosis, alterations in the transcription of DNA methyltransferases, suppression of anti-tumor genes (p16 and p53) | [11,23,24,25,26,27] |
| HHV-8 | Kaposi’s sarcoma, Multicentre Castleman’s Disease, Primary Effusion Lymphoma | cirRNAs, vIRF4 viral locus (circvIRF4), RNase-R resistant Polyadenylated Nuclear (circPAN), non-coding RNA, Latency Associated Nuclear Antigens | Alteration in Notch pathways and hypermethylation followedby slower hypomethylation, upregulation of Protocadherin Beta-5 (PCDHB5), | [23,28,29,30] |
| HTLV-1 | leukemia/ lymphoma | Tax proteins, HTLV-1 Tax protein, translocation of methylcytosine dioxygenase genes | proviral integrations on chromatin loops, disruption of transcriptional pathways, inactivation of p53, and hypermethylation at oncogenic promoter regions. | [1,29,31,32] |
| HBV | Pancreatic Carcinoma, Hepatocellular carcinoma, | short mRNAs, latent membrane protein 1 (EBV LMP1), Latency Associated Nuclear Antigens | chromosomal instability, upregulation of Small Protein of the HBV Surface Antigen, enhanced cell migration, Methylation mechanisms | [13,33,34,35] |
| HIV-1 | Kaposi’s sarcoma, skin Carcinoma, Hepatocellular carcinoma, cancer of the conjunctiva | Intrinsically Disordered Proteins (IDPs) | Suppression of p16 expression, immune dysregulation, and immune evasion, there are five viral proteins: transactivator of transcription, accessory protein negative factor Nef, matrix protein p17, and envelope protein gp120. Reverse transcriptase RT and Tat. When secreted from HIV-1-infected cells, Gp120, Nef, p17, Tat, and RT produce oxidative stress and are carcinogenic | [33,36,37,38,39] |
| HPV | Cervical Cancer, oral Cancer, tonsillar carcinoma, penile Cancer | endogenous protein E6 and E7, retinoblastoma protein, HPV6, 11, 16, and 18, Dedicator of cytokinesis-8 (DOCK-8), Alpha-7 genotypes of HPV | Inactivation of p53, DNA methylation, methylation modifications, and Integration serves as a precursor to the passage from LSIL to HSIL, Oxidative stress resulting in DNA damage | [26,28,40,41,42,43,44,45] |
| HCV | Non-Hodgkin’s lymphoma, Gallbladder carcinoma, Thyroid carcinoma | E1 and E2 membrane proteins, non-coding RNA, histone modification, transcriptional alteration | Immune evasion, signal transducer and activator of transcription 3 (STAT-3) activation, suppression of DNA methyltransferases activity, suppression of tumor suppression genes, Genomic hypomethylation of apoptosis genes. | [17,19,46] |
| MCPyV | T-cell leukemia/lymphoma and Merkel Cell Carcinoma | large T antigen (LT), Full-length LT, Truncated LT antigen mutation (tLT), small T antigen (ST) | cell immortalization, cell Transformation, immune evasion, Onco-suppressive proteins (Rb), downregulation of Toll-like Receptor-9 (TLR-9) | [17,27,47,48] |
| HPV E6 | Cervical Cancer Breast Cancer Colorectal Cancer | cellular proteins(p21 and pRb) deregulates expression of p53 and BCL2 E6 stimulates cell proliferation independently from E7 through its C-terminal PDZ-ligand domain | E6 degrades p53, targets c-myc oncogene (a marker protein for several cancer forms including cervical cancer), Inactivates p53, and releases repression of BCL2Mediates suprabasal cell proliferation and disrupts normal cell adhesion to contribute to the development of metastatic tumors | [37,49,50,51] |
| HPV E7 | Cervical Cancer Breast cancer Colorectal Cancer | Interacts with DREAM (dimerization partner, RB-like, E2F4, and MuyB) BRCA1 gene and dissociates the pRB-E2F complex by binding to pRB | Inhibits retinoblastoma protein (pRb)epigenetic derepression through KDM6B (H3K27-specific) Demethylase 6B triggers the expression of p16INK4A, targets c-myc oncogene, lowers p53, and increases BCL2 levelExpresses proteins necessary for DNA replication | [25,37,52,53,54] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elkhalifa, A.M.E.; Nabi, S.U.; Shah, O.S.; Bashir, S.M.; Muzaffer, U.; Ali, S.I.; Wani, I.A.; Alzerwi, N.A.N.; Elderdery, A.Y.; Alanazi, A.; et al. Insight into Oncogenic Viral Pathways as Drivers of Viral Cancers: Implication for Effective Therapy. Curr. Oncol. 2023, 30, 1924-1944. https://doi.org/10.3390/curroncol30020150
Elkhalifa AME, Nabi SU, Shah OS, Bashir SM, Muzaffer U, Ali SI, Wani IA, Alzerwi NAN, Elderdery AY, Alanazi A, et al. Insight into Oncogenic Viral Pathways as Drivers of Viral Cancers: Implication for Effective Therapy. Current Oncology. 2023; 30(2):1924-1944. https://doi.org/10.3390/curroncol30020150
Chicago/Turabian StyleElkhalifa, Ahmed M. E., Showkat Ul Nabi, Ovais Shabir Shah, Showkeen Muzamil Bashir, Umar Muzaffer, Sofi Imtiyaz Ali, Imtiyaz Ahmad Wani, Nasser A. N. Alzerwi, Abozer Y. Elderdery, Awadh Alanazi, and et al. 2023. "Insight into Oncogenic Viral Pathways as Drivers of Viral Cancers: Implication for Effective Therapy" Current Oncology 30, no. 2: 1924-1944. https://doi.org/10.3390/curroncol30020150
APA StyleElkhalifa, A. M. E., Nabi, S. U., Shah, O. S., Bashir, S. M., Muzaffer, U., Ali, S. I., Wani, I. A., Alzerwi, N. A. N., Elderdery, A. Y., Alanazi, A., Alenazy, F. O., & Alharbi, A. H. A. (2023). Insight into Oncogenic Viral Pathways as Drivers of Viral Cancers: Implication for Effective Therapy. Current Oncology, 30(2), 1924-1944. https://doi.org/10.3390/curroncol30020150