
Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer



 and
and 
Abstract
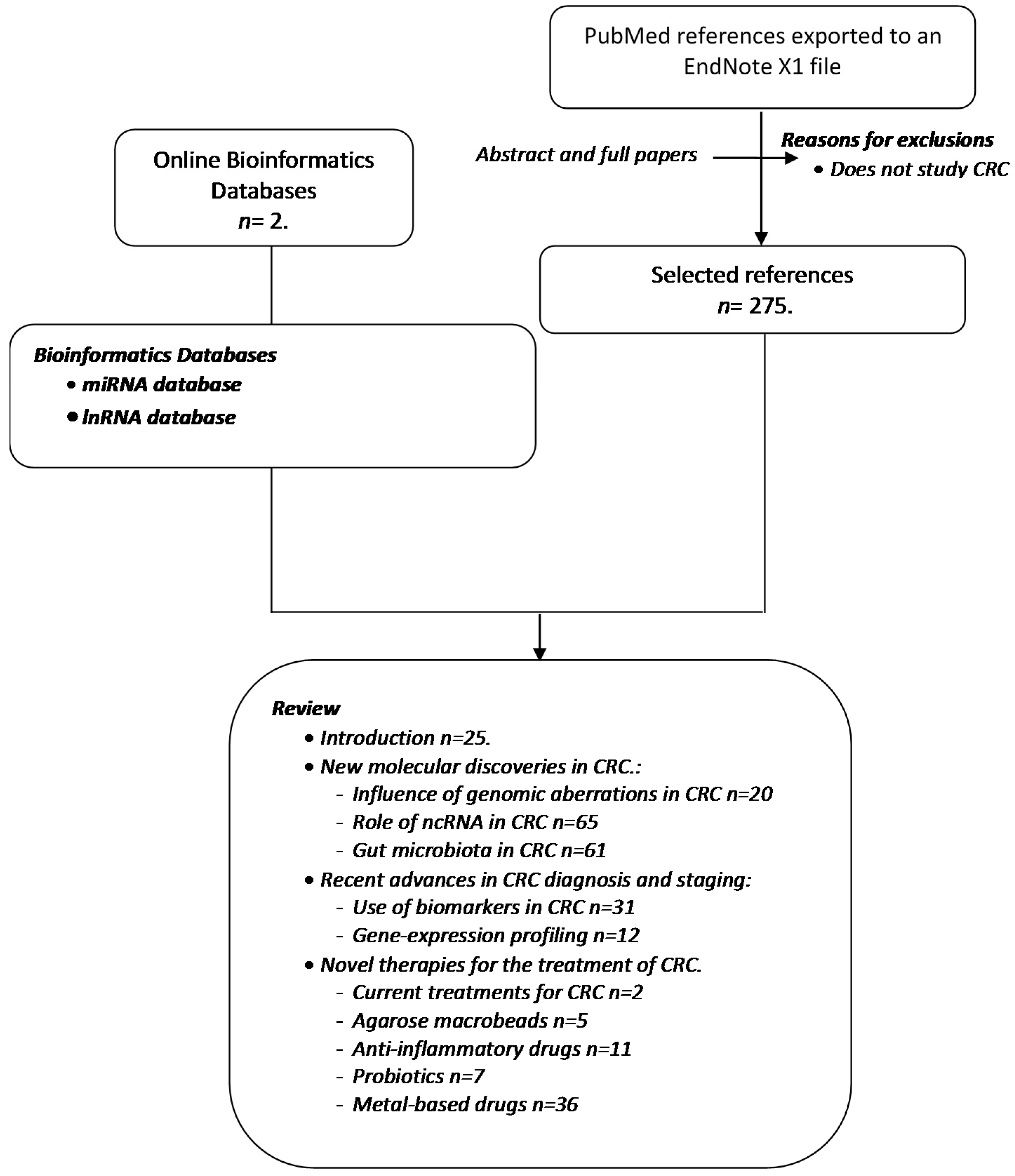
:1. Methodology
2. Introduction
2.1. Epidemiology
2.2. Aetiology
2.3. Risk Factors
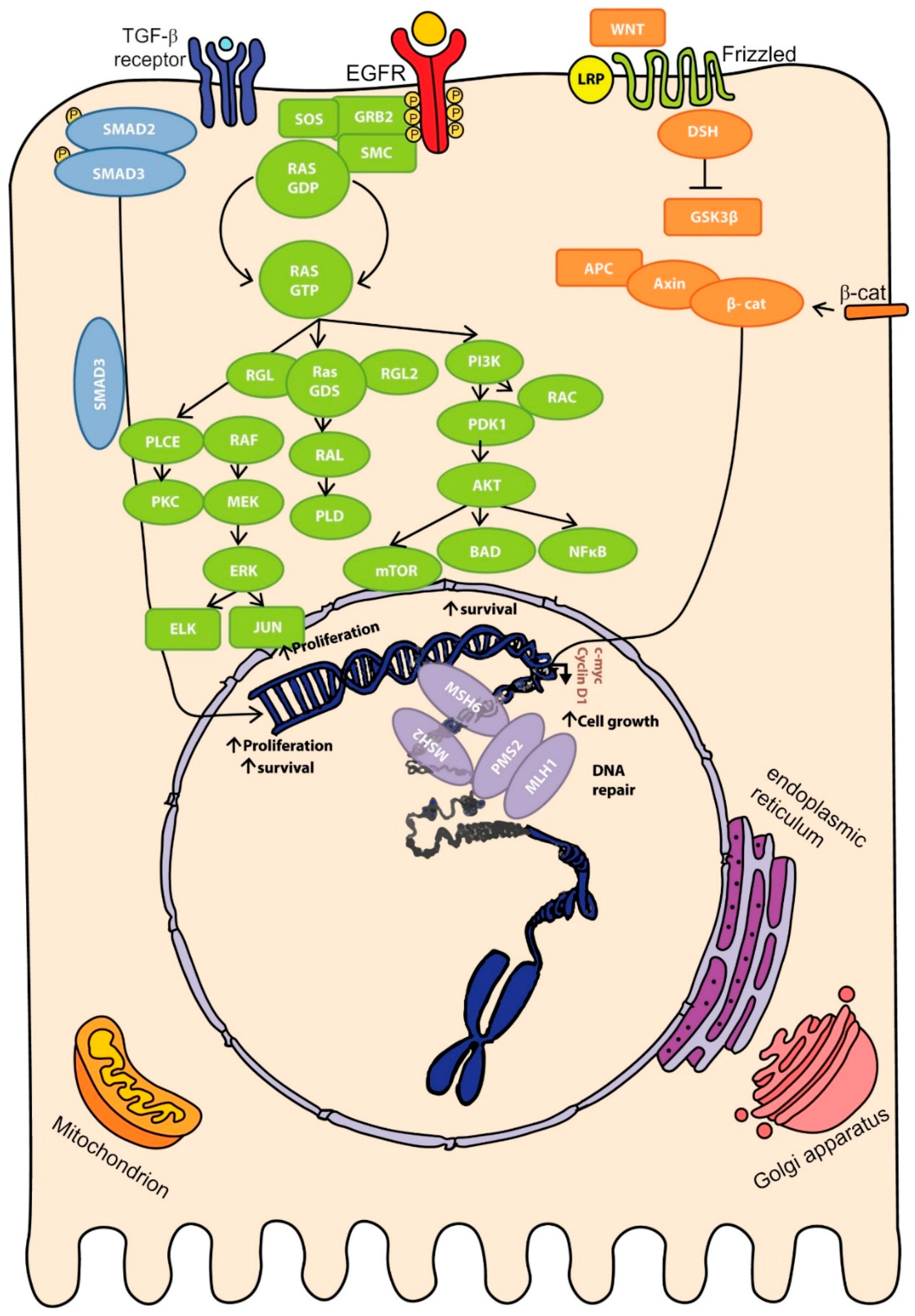
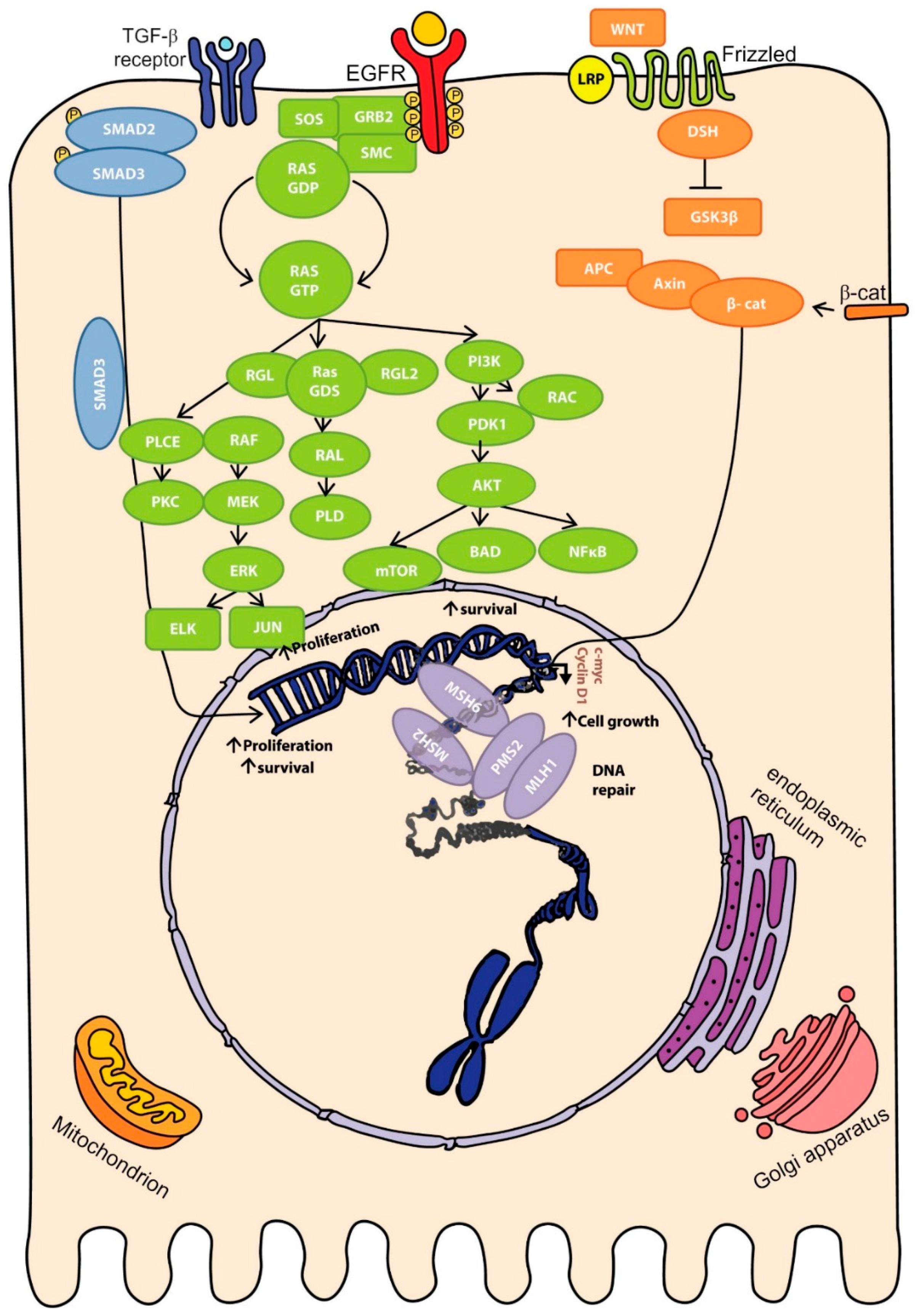
2.4. Molecular Pathways of Colorectal Cancer
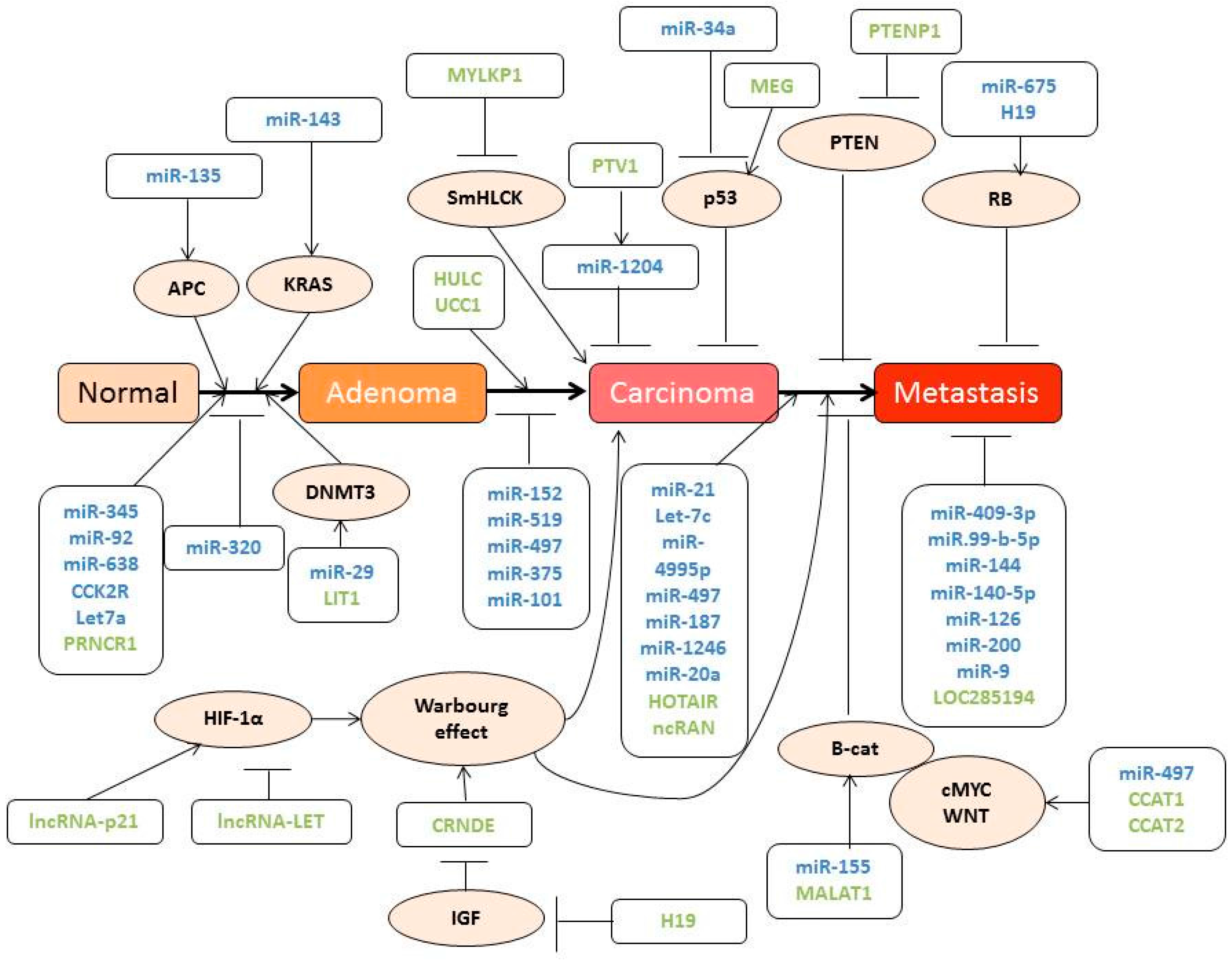
3. New Molecular Discoveries in Colorectal Cancer (CRC)
3.1. Influence of Genomic Aberrations on CRC Outcome
3.2. Role of ncRNA in Colon Carcinoma
3.3. Gut Microbiome in CRC
3.4. Dysbiosis and Colorectal Cancer: Breaking the Mutualism
3.4.1. Fusobacterium spp
3.4.2. Bacteroides Fragilis
3.4.3. Enteropathogenic Escherichia coli
3.5. Microbiome and Diet: A Possible Link with CRC
4. Recent Advances in CRC Diagnosis and Staging
4.1. Use of Biomarkers in CRC
4.2. Gene-Expression Profiling (GEP)
5. Novel Therapies for the Treatment of CRC
5.1. Current Treatments for CRC
5.2. Agarose Macrobeads
5.3. Anti-Inflammatory Drugs
5.4. Probiotics
5.5. Functional Foods
5.6. Metal-Based Drugs for CRC Treatment
5.6.1. Platinum
5.6.2. Gold
6. Discussion and Future Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Quek, X.C.; Thomson, D.W.; Maag, J.L.; Bartonicek, N.; Signal, B.; Clark, M.B.; Gloss, B.S.; Dinger, M.E. LncRNAdb v2. 0: Expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res. 2014, 43, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Amaral, P.P.; Clark, M.B.; Gascoigne, D.K.; Dinger, M.E.; Mattick, J.S. LncRNAdb: A reference database for long noncoding RNAs. Nucleic Acids Res. 2011, 39, 146–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, B.; Ding, Q.; Han, H.; Wu, D. miRCancer: A microRNA-cancer association database constructed by text mining on literature. Bioinformatics 2013, 29, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Ding, Q.; Wu, D. Text mining on big and complex biomedical literature. Big Data Anal. Bioinform. Healthc. 2014. [Google Scholar] [CrossRef]
- Xie, B.; Hochberg, R.; Ding, Q.; Wu, D. miRSAT & miRCDB: An Integrated microRNA Sequesce Analysis Tool and a Cancer-Associated microRNA Database. In Proceedings of the ISCA 2nd International Conference on Bioinformatics and Computational Biology, BICoB-2010, Sheraton Waikiki Hotel, Honolulu, HI, USA, 24–26 March 2010; pp. 159–164.
- Stewart, B.; Wild, C.P. (Eds.) World Cancer Report 2014; International Agency for Research on Cancer (IARC): Lyon, France, 2014.
- Brody, H. Colorectal cancer. Nature 2015, 521, S1. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; Atkin, W.; Lenz, H.-J.; Lynch, H.T.; Minsky, B.; Nordlinger, B.; Starling, N. Colorectal cancer. Lancet 2010, 375, 1030–1047. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Lynch, H.T.; de la Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [PubMed]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R. Revised bethesda guidelines for hereditary nonpolyposis colorectal cancer (lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Stoffel, E.M.; Kastrinos, F. Familial colorectal cancer, beyond lynch syndrome. Clin. Gastroenterol. Hepatol. 2014, 12, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Levin, B.; Lieberman, D.A.; McFarland, B.; Smith, R.A.; Brooks, D.; Andrews, K.S.; Dash, C.; Giardiello, F.M.; Glick, S.; Levin, T.R. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: A joint guideline from the American cancer society, the US multi-society task force on colorectal cancer, and the American college of radiology. CA Cancer J. Clin. 2008, 58, 130–160. [Google Scholar] [CrossRef] [PubMed]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Canavan, C.; Abrams, K.R.; Mayberry, J. Meta-analysis: Colorectal and small bowel cancer risk in patients with crohn’s disease. Aliment. Pharmacol. Therap. 2006, 23, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Johns, L.E.; Houlston, R.S. A systematic review and meta-analysis of familial colorectal cancer risk. Am. J. Gastroenterol. 2001, 96, 2992–3003. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.J. ABC of colorectal cancer. Gastroenterology 2012, 143, 868–869. [Google Scholar] [CrossRef]
- Martinez-Useros, J.; Garcia-Foncillas, J. Obesity and colorectal cancer: Molecular features of adipose tissue. J. Transl. Med. 2016, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Willett, W.C. Diet and cancer: An evolving picture. JAMA 2005, 293, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Bastide, N.M.; Pierre, F.H.F.; Corpet, D.E. Heme iron from meat and risk of colorectal cancer: A meta-analysis and a review of the mechanisms involved. Cancer Prev. Res. 2011, 4, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, R.L.L.; Pierre, F.; Corpet, D.E. Processed meat and colorectal cancer: A review of epidemiologic and experimental evidence. Nutr. Cancer 2008, 60, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Pöschl, G.; Seitz, H.K. Alcohol and cancer. Alcohol Alcoholism 2004, 39, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Botteri, E.; Iodice, S.; Bagnardi, V.; Raimondi, S.; Lowenfels, A.B.; Maisonneuve, P. Smoking and colorectal cancer: A meta-analysis. JAMA 2008, 300, 2765–2778. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.J.; Boca, S.; Freedman, N.D.; Caporaso, N.E.; Huang, W.Y.; Sinha, R.; Sampson, J.N.; Moore, S.C. Metabolites of tobacco smoking and colorectal cancer risk. Carcinogenesis 2014, 35, 1516–1522. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.S.; Chen, T.Y.; Giovannucci, E. Cigarette smoking and colorectal cancer incidence and mortality: Systematic review and meta-analysis. Int. J. Cancer 2009, 124, 2406–2415. [Google Scholar] [CrossRef] [PubMed]
- Grady, W.M.; Carethers, J.M. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology 2008, 135, 1079–1099. [Google Scholar] [CrossRef] [PubMed]
- Pino, M.S.; Chung, D.C. The chromosomal instability pathway in colon cancer. Gastroenterology 2010, 138, 2059–2072. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef] [PubMed]
- Lao, V.V.; Grady, W.M. Epigenetics and colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 686–700. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Willett, C.G.; Chang, D.T.; Czito, B.G.; Meyer, J.; Wo, J. Cancer genome atlas network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Brocardo, M.; Henderson, B.R. APC shuttling to the membrane, nucleus and beyond. Trends Cell Boil. 2008, 18, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Herzig, D.O.; Tsikitis, V.L. Molecular markers for colon diagnosis, prognosis and targeted therapy. J. Surg. Oncol. 2015, 111, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Rennoll, S.; Yochum, G. Regulation of myc gene expression by aberrant Wnt/β-catenin signaling in colorectal cancer. World J. Biol. Chem. 2015, 6, 290. [Google Scholar] [CrossRef] [PubMed]
- Toon, C.W.; Chou, A.; Clarkson, A.; DeSilva, K.; Houang, M.; Chan, J.C.Y.; Sioson, L.L.; Jankova, L.; Gill, A.J. Immunohistochemistry for myc predicts survival in colorectal cancer. PLoS ONE 2014, 9, e87456. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guo, F.; Shi, X.; Zhang, L.; Zhang, A.; Jin, H.; He, Y. BRAF V600E mutation and KRAS codon 13 mutations predict poor survival in Chinese colorectal cancer patients. BMC Cancer 2014, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Qiu, T.; Zhi, W.; Shi, S.; Zou, S.; Ling, Y.; Shan, L.; Ying, J.; Lu, N. Colorectal carcinomas with KRAS codon 12 mutation are associated with more advanced tumor stages. BMC Cancer 2015, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Kawasaki, T.; Meyerhardt, J.A.; Loda, M.; Giovannucci, E.L.; Fuchs, C.S. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut 2009, 58, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.; Tran, B.; Ensor, J.; Gibbs, P.; Wong, H.L.; Wong, S.F.; Vilar, E.; Tie, J.; Broaddus, R.; Kopetz, S. Multicenter retrospective analysis of metastatic colorectal cancer (CRC) with high-level microsatellite instability (MSI-H). Ann. Oncol. 2014, 25, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Kadowaki, S.; Kakuta, M.; Takahashi, S.; Takahashi, A.; Arai, Y.; Nishimura, Y.; Yatsuoka, T.; Ooki, A.; Yamaguchi, K.; Matsuo, K. Prognostic value of KRAS and BRAF mutations in curatively resected colorectal cancer. World J. Gastroenterol. WJG 2015, 21, 1275. [Google Scholar] [CrossRef] [PubMed]
- Day, F.; Muranyi, A.; Singh, S.; Shanmugam, K.; Williams, D.; Byrne, D.; Pham, K.; Palmieri, M.; Tie, J.; Grogan, T. A mutant BRAF V600E-specific immunohistochemical assay: Correlation with molecular mutation status and clinical outcome in colorectal cancer. Target. Oncol. 2015, 10, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Cercek, A.; O’Reilly, E.M.; Reidy, D.L.; Kemeny, N.; Wolinsky, T.; Capanu, M.; Gollub, M.J.; Rosen, N.; Berger, M.F. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin. Cancer Res. 2015, 21, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Morkel, M.; Riemer, P.; Bläker, H.; Sers, C. Similar but different: Distinct roles for KRAS and BRAF oncogenes in colorectal cancer development and therapy resistance. Oncotarget 2015, 6, 20785. [Google Scholar] [CrossRef] [PubMed]
- Rosty, C.; Young, J.P.; Walsh, M.D.; Clendenning, M.; Sanderson, K.; Walters, R.J.; Parry, S.; Jenkins, M.A.; Win, A.K.; Southey, M.C. PIK3CA activating mutation in colorectal carcinoma: Associations with molecular features and survival. PLoS ONE 2013, 8, e65479. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Morikawa, T.; Lochhead, P.; Imamura, Y.; Kuchiba, A.; Yamauchi, M.; Nosho, K.; Qian, Z.R.; Nishihara, R.; Meyerhardt, J.A. Prognostic role of PIK3CA mutation in colorectal cancer: Cohort study and literature review. Clin. Cancer Res. 2012, 18, 2257–2268. [Google Scholar] [CrossRef] [PubMed]
- Atreya, C.E.; Sangale, Z.; Xu, N.; Matli, M.R.; Tikishvili, E.; Welbourn, W.; Stone, S.; Shokat, K.M.; Warren, R.S. PTEN expression is consistent in colorectal cancer primaries and metastases and associates with patient survival. Cancer Med. 2013, 2, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Sarli, L.; Bottarelli, L.; Bader, G.; Iusco, D.; Pizzi, S.; Costi, R.; Bertolani, M.; Roncoroni, L.; Bordi, C. Association between recurrence of sporadic colorectal cancer, high level of microsatellite instability, and loss of heterozygosity at chromosome 18q. Dis. Colon Rectum. 2004, 47, 1467–1482. [Google Scholar] [CrossRef] [PubMed]
- Popat, S.; Houlston, R.S. A systematic review and meta-analysis of the relationship between chromosome 18q genotype, DCC status and colorectal cancer prognosis. Eur. J. Cancer 2005, 41, 2060–2070. [Google Scholar] [CrossRef] [PubMed]
- Popat, S.; Zhao, D.; Chen, Z.; Pan, H.; Shao, Y.; Chandler, I.; Houlston, R.S. Relationship between chromosome 18q status and colorectal cancer prognosis: A prospective, blinded analysis of 280 patients. Anticancer Res. 2007, 27, 627–633. [Google Scholar] [PubMed]
- Munro, A.J.; Lain, S.; Lane, D.P. P53 abnormalities and outcomes in colorectal cancer: A systematic review. Br. J. Cancer 2005, 92, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Rooney, P.; Boonsong, A.; McKay, J.; Marsh, S.; Stevenson, D.; Murray, G.; Curran, S.; Haites, N.; Cassidy, J.; McLeod, H. Colorectal cancer genomics: Evidence for multiple genotypes which influence survival. Br. J. Cancer 2001, 85, 1492. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef] [PubMed]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. mir-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Sun, Y.; An, S.; Xin, S.; Ren, X.; Zhang, D.; Wu, P.; Liao, W.; Ding, Y.; Liang, L. MicroRNA-34a targets FMNL2 and E2F5 and suppresses the progression of colorectal cancer. Exp. Mol. Pathol. 2015, 99, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, H. The role of microRNAs in colorectal cancer. J. Genet. Genom. 2010, 37, 347–358. [Google Scholar] [CrossRef]
- Migliore, L.; Migheli, F.; Spisni, R.; Coppedè, F. Genetics, cytogenetics, and epigenetics of colorectal cancer. BioMed Res. Int. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.; Rasheed, S.; Nikolova, D.; Leupold, J.; Colburn, N.; Post, S.; Allgayer, H. microRNA-21 (mir-21) post-transcriptionally downregulates tumor suppressor PDCD4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008, 27, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.-T.; Wang, J.-L.; Du, W.; Hong, J.; Zhao, S.-L.; Wang, Y.-C.; Xiong, H.; Chen, H.-M.; Fang, J.-Y. MicroRNA 345, a methylation-sensitive microrna is involved in cell proliferation and invasion in human colorectal cancer. Carcinogenesis 2011, 32, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Xu, Y.; Wang, Z.; Chen, Y.; Yue, Z.; Gao, P.; Xing, C.; Xu, H. MicroRNA-148b suppresses cell growth by targeting cholecystokinin-2 receptor in colorectal cancer. Int. J. Cancer 2012, 131, 1042–1051. [Google Scholar] [CrossRef] [PubMed]
- Han, H.B.; Gu, J.; Zuo, H.J.; Chen, Z.G.; Zhao, W.; Li, M.; Ji, D.B.; Lu, Y.Y.; Zhang, Z.Q. LET-7c functions as a metastasis suppressor by targeting MMP11 and PBX3 in colorectal cancer. J. Pathol. 2012, 226, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, P.; Ma, Y.; Yang, J.; Moyer, M.P.; Shi, C.; Peng, J.; Qin, H. NIRF is frequently upregulated in colorectal cancer and its oncogenicity can be suppressed by LET-7a microRNA. Cancer Lett. 2012, 314, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Sun, L.; Chai, N.; Tang, S.; Jin, J.; Hu, H.; Nie, Y.; Wang, X.; Wu, K. microRNA-499-5p promotes cellular invasion and tumor metastasis in colorectal cancer by targeting FOXO4 and PDCD4. Carcinogenesis 2011, 32, 1798–1805. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhang, Z.; Wang, Y.; Li, C.; Gong, W.; Wang, X. MicroRNA-92a promotes colorectal cancer cell growth and migration by inhibiting KLF4. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2016, 23, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.L.; Bernardes, M.V.; Feitosa, M.R.; Peria, F.M.; Tirapelli, D.P.; Rocha, J.J.; Feres, O. Serological under expression of microRNA-21, microRNA-34a and microRNA-126 in colorectal cancer. Acta Cir. Bras. 2016, 31, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Vishnubalaji, R.; Hamam, R.; Yue, S.; Al-Obeed, O.; Kassem, M.; Liu, F.; Aldahmash, A.; Alajez, N. MicroRNA-320 suppresses colorectal cancer by targeting SOX4, FOXM1, and FOXQ1. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chang, J.; Wang, S.; Liu, X.; Peng, J.; Huang, D.; Sun, M.; Chen, Z.; Zhang, W.; Guo, W. miRNA-99b-5p suppresses liver metastasis of colorectal cancer by down-regulating mtor. Oncotarget 2015, 6, 24448–24462. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.R.; Lee, S.T.; Kim, S.L.; Liu, Y.C.; Lee, M.R.; Shin, J.H.; Seo, S.Y.; Kim, S.H.; Kim, I.H.; Lee, S.O. microRNA-9 suppresses cell migration and invasion through downregulation of TM4SF1 in colorectal cancer. Int. J. Oncol. 2016, 48, 2135–2143. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Toiyama, Y.; Kitajima, T.; Imaoka, H.; Hiro, J.; Saigusa, S.; Tanaka, K.; Inoue, Y.; Mohri, Y.; Toden, S. miRNA-503 promotes tumor progression and is associated with early recurrence and poor prognosis in human colorectal cancer. Oncology 2016, 90, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Bai, C.; Sun, Z. In microRNA-222 to enhance invasion and migration through MST3 in colorectal cancer and to predict for CRC patients progression. In Proceedings of the 2015 ASCO Annual Meeting Proceedings, Chicago, IL, USA, 29 May–2 June 2015; p. e22006.
- Zhao, L.-D.; Zheng, W.-W.; Wang, G.-X.; Kang, X.-C.; Qin, L.; Ji, J.-J.; Hao, S. Epigenetic silencing of mir-181b contributes to tumorigenicity in colorectal cancer by targeting RASSF1A. Int. J. Oncol. 2016, 48, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Yu, H.; Shi, X.; Xu, K.; Tang, Q.; Liang, B.; Hu, S.; Bao, Y.; Xu, J.; Cai, J. microRNA-497 inhibits invasion and metastasis of colorectal cancer cells by targeting vascular endothelial growth factor-A. Cell Prolif. 2016, 49, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jia, F.; Bai, P.; Liang, Y.; Sun, R.; Yuan, F.; Zhang, L.; Gao, L. Association between polymorphisms in long non-coding RNA PRNCR1 in 8q24 and risk of gastric cancer. Tumor Biol. 2016, 37, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Luo, Y.; Shao, Z.; Xu, L.; Liu, X.; Niu, Y.; Shi, J.; Sun, X.; Liu, Y.; Ding, Y. microRNA-187, a downstream effector of TGF-β pathway, suppresses SMAD-mediated epithelial-mesenchymal transition in colorectal cancer. Cancer Lett. 2016, 373, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Wang, J.; Zhang, J.; Cai, J.; Bai, Z.; Zhang, Z. ORAI1, a direct target of microRNA-519, promotes progression of colorectal cancer via AKT/GSK3Β signaling pathway. Digest. Dis. Sci. 2016, 61, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.C.; Li, T.; Han, Y.D.; Zhang, H.Y.; Lin, H.; Zhang, B. microRNA-155 enhances the activation of WNT/β-catenin signaling in colorectal carcinoma by suppressing HMG-box transcription factor 1. Mol. Med. Rep. 2016. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jiang, C.; Li, D.; Ge, X.; Shi, Z.; Li, C.; Liu, X.; Yin, Y.; Zhen, L.; Liu, L.-Z. MicroRNA-497 inhibits tumor growth and increases chemosensitivity to 5-fluorouracil treatment by targeting KSR1. Oncotarget 2015, 7, 2660–2671. [Google Scholar]
- Zaharie, F.; Muresan, M.-S.; Petrushev, B.; Berce, C.; Gafencu, G.-A.; Selicean, S.; Jurj, A.; Cojocneanu-Petric, R.; Lisencu, C.-I.; Pop, L.-A. Exosome-carried microRNA-375 inhibits cell progression and dissemination via Bcl-2 blocking in colon cancer. J. Gastrointest. Liver Dis. 2015, 24, 435–443. [Google Scholar]
- Wang, S.; Zeng, Y.; Zhou, J.M.; Nie, S.L.; Peng, Q.; Gong, J.; Huo, J.R. microRNA-1246 promotes growth and metastasis of colorectal cancer cells involving CCNG2 reduction. Mol. Med. Rep. 2016, 13, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Li, C.; Chai, B. miRNA-144 suppresses proliferation and migration of colorectal cancer cells through GSPT1. Biomed. Pharmacother. 2015, 74, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bian, Z.; Zhou, J.; Song, M.; Liu, Z.; Feng, Y.; Zhe, L.; Zhang, B.; Yin, Y.; Huang, Z. microRNA-638 inhibits cell proliferation by targeting phospholipase D1 in human gastric carcinoma. Protein Cell 2015, 6, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-B.; Yang, L.; Lu, P.-H.; Fu, X.-L.; Zhang, Y.; Zhu, Y.-Q.; Tian, Y. microRNA-101 down-regulates sphingosine kinase 1 in colorectal cancer cells. Biochem. Biophys. Res. Commun. 2015, 463, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Jing, C.; Shi, Y.; Miao, R.; Peng, L.; Kong, S.; Ma, Y.; Li, L. microRNA-20a enhances the epithelial-to-mesenchymal transition of colorectal cancer cells by modulating matrix metalloproteinases. Exp. Ther. Med. 2015, 10, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Weng, C.; Dong, H.; Li, S.; Chen, G.; Xu, Z. microRNA-409-3p suppresses colorectal cancer invasion and metastasis partly by targeting GAB1 expression. Int. J. Cancer 2015, 137, 2310–2322. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.V.; Mattick, J.S. The rise of regulatory RNA. Nat. Rev. Genet. 2014, 15, 423. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Wu, H.-J.; Hsu, J.-M.; Chang, S.-S.; LaBaff, A.M.; Li, C.-W.; Wang, Y.; Hsu, J.L.; Hung, M.-C. natures: Versatile master regulators of gene expression and crucial players in cancer. Am. J. Transl. Res. 2012, 4, 127–150. [Google Scholar] [PubMed]
- Hamilton, M.J.; Young, M.D.; Sauer, S.; Martinez, E. The interplay of long non-coding RNAs and myc in cancer. AIMS Biophys. 2015, 2, 794–809. [Google Scholar] [PubMed]
- Gibb, E.A.; Brown, C.J.; Lam, W.L. The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 2011, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Tsang, W.P.; Ng, E.K.O.; Ng, S.S.M.; Jin, H.; Yu, J.; Sung, J.J.Y.; Kwok, T.T. Oncofetal H19-derived mir-675 regulates tumor suppressor RB in human colorectal cancer. Carcinogenesis 2010, 31, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Fellig, Y.; Ariel, I.; Ohana, P.; Schachter, P.; Sinelnikov, I.; Birman, T.; Ayesh, S.; Schneider, T.; de Groot, N.; Czerniak, A. H19 expression in hepatic metastases from a range of human carcinomas. J. Clin. Pathol. 2005, 58, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Ohana, P.; Schachter, P.; Ayesh, B.; Mizrahi, A.; Birman, T.; Schneider, T.; Matouk, I.; Ayesh, S.; Kuppen, P.J.K.; de Groot, N. Regulatory sequences of H19 and IGF2 genes in DNA-based therapy of colorectal rat liver metastases. J. Gene Med. 2005, 7, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L. Long non-coding RNA hotair reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Schorderet, P.; Duboule, D. Structural and functional differences in the long non-coding RNA hotair in mouse and human. PLoS Genet 2011, 7, e1002071. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Yang, M.H.; Tian, J.; Wang, X.Y.; Li, Z.G. Malat-1: A long non-coding RNA and its important 3′ end functional motif in colorectal cancer metastasis. Int. J. Oncol. 2011, 39, 169. [Google Scholar] [PubMed]
- Matouk, I.J.; Abbasi, I.; Hochberg, A.; Galun, E.; Dweik, H.; Akkawi, M. Highly upregulated in liver cancer noncoding RNA is overexpressed in hepatic colorectal metastasis. Eur. J. Gastroenterol. Hepatol. 2009, 21, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, Y.; Mehta, K.R.; Danila, D.C.; Scolavino, S.; Johnson, S.R.; Klibanski, A. A pituitary-derived MEG3 isoform functions as a growth suppressor in tumor cells. J. Clin. Endocrinol. Metab. 2003, 88, 5119–5126. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Xue, X.; Bi, J.; Zheng, L.; Zhi, K.; Gu, Y.; Fang, G. Long noncoding RNA CCAT1, which could be activated by c-myc, promotes the progression of gastric carcinoma. J. Cancer Res. Clin. Oncol. 2013, 139, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Cui, R.; Jeon, Y.-J.; Lee, J.-H.; Lee, J.H.; Sim, H.; Park, J.K.; Fadda, P.; Tili, E.; Nakanishi, H. Long–Range interaction and correlation between myc enhancer and oncogenic long noncoding RNA CARLO-5. Proc. Natl. Acad. Sci. USA 2014, 111, 4173–4178. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Spizzo, R.; Atlasi, Y.; Nicoloso, M.; Shimizu, M.; Redis, R.S.; Nishida, N.; Gafà, R.; Song, J.; Guo, Z. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res. 2013, 23, 1446–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, L.D.; Pedersen, S.K.; Brown, G.S.; Ho, T.; Kassir, Z.; Moynihan, A.T.; Vizgoft, E.K.; Dunne, R.; Pimlott, L.; Young, G.P. Colorectal neoplasia differentially expressed (CRNDE), a novel gene with elevated expression in colorectal adenomas and adenocarcinomas. Gene Cancer 2011, 2, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Huang, J.; Zhou, N.; Zhang, Z.; Zhang, A.; Lu, Z.; Wu, F.; Mo, Y.-Y. LncRNA loc285194 is a p53-regulated tumor suppressor. Nucleic Acids Res. 2013, 41, 4976–4987. [Google Scholar] [CrossRef] [PubMed]
- Pibouin, L.; Villaudy, J.; Ferbus, D.; Muleris, M.; Prospéri, M.-T.; Remvikos, Y.; Goubin, G.R. Cloning of the mRNA of overexpression in colon carcinoma-1: A sequence overexpressed in a subset of colon carcinomas. Cancer Genet. Cytogenet. 2002, 133, 55–60. [Google Scholar] [CrossRef]
- Yang, F.; Zhang, H.; Mei, Y.; Wu, M. Reciprocal regulation of HIF-1α and lincRNA-p21 modulates the Warburg effect. Mol. Cell 2014, 53, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Nakano, S.; Murakami, K.; Meguro, M.; Soejima, H.; Higashimoto, K.; Urano, T.; Kugoh, H.; Mukai, T.; Ikeguchi, M.; Oshimura, M. Expression profile of LIT1/KCNQ1OT1 and epigenetic status at the KVDMR1 in colorectal cancers. Cancer Sci. 2006, 97, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.J.; Ma, S.F.; Yourek, G.; Park, Y.-D.; Garcia, J.G.N. A transcribed pseudogene of MYLK promotes cell proliferation. FASEB J. 2011, 25, 2305–2312. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, I.; Moller, E.; Collin, A.; Mertens, F. The POU5F1P1 pseudogene encodes a putative protein similar to POU5F1 isoform 1. Oncol. Rep. 2008, 20, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, X.; Xie, X.; Zhao, L.; Chen, W. UCA1, a non-protein-coding RNA up-regulated in bladder carcinoma and embryo, influencing cell growth and promoting invasion. FEBS Lett. 2008, 582, 1919–1927. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Iyer, M.K.; Balbin, O.A.; Dhanasekaran, S.M.; Cao, Q.; Brenner, J.C.; Laxman, B.; Asangani, I.A.; Grasso, C.S.; Kominsky, H.D. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nat. Biotechnol. 2011, 29, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Chen, W.; Iyer, M.K.; Cao, Q.; Ma, T.; Han, S.; Sahu, A.; Malik, R.; Wilder-Romans, K.; Navone, N. PCAT-1, a long noncoding RNA, regulates BRCA2 and controls homologous recombination in cancer. Cancer Res. 2014, 74, 1651–1660. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Chen, Y.; Liao, X.; Liu, D.; Li, F.; Ruan, H.; Jia, W. Overexpression of long noncoding RNA PCAT-1 is a novel biomarker of poor prognosis in patients with colorectal cancer. Med. Oncol. 2013, 30, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Nakagawa, H.; Uemura, M.; Piao, L.; Ashikawa, K.; Hosono, N.; Takata, R.; Akamatsu, S.; Kawaguchi, T.; Morizono, T. Association of a novel long non-coding RNA in 8q24 with prostate cancer susceptibility. Cancer Sci. 2011, 102, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Sun, R.; Liang, Y.; Pan, X.; Li, Z.; Bai, P.; Zeng, X.; Zhang, D.; Zhang, L.; Gao, L. Association between polymorphisms in long non-coding RNA PRNCR1 in 8q24 and risk of colorectal cancer. J. Exp. Clin. Cancer Res. 2013, 32, 1. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Huo, X.S.; Yuan, S.X.; Zhang, L.; Zhou, W.P.; Wang, F.; Sun, S.H. Repression of the long noncoding RNA-LET by histone deacetylase 3 contributes to hypoxia-mediated metastasis. Mol. Cell 2013, 49, 1083–1096. [Google Scholar] [CrossRef] [PubMed]
- Qi, P.; Xu, M.D.; Ni, S.J.; Shen, X.H.; Wei, P.; Huang, D.; Tan, C.; Sheng, W.Q.; Zhou, X.Y.; Du, X. Down-regulation of NCRAN, a long non-coding RNA, contributes to colorectal cancer cell migration and invasion and predicts poor overall survival for colorectal cancer patients. Mol. Carcinog. 2015, 54, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Tseng, Y.-Y.; Moriarity, B.S.; Gong, W.; Akiyama, R.; Tiwari, A.; Kawakami, H.; Ronning, P.; Reuland, B.; Guenther, K.; Beadnell, T.C. PVT1 dependence in cancer with myc copy-number increase. Nature 2014, 512, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Sawada, G.; Kurashige, J.; Uchi, R.; Matsumura, T.; Ueo, H.; Takano, Y.; Eguchi, H.; Sudo, T.; Sugimachi, K. Amplification of PVT1 is involved in poor prognosis via apoptosis inhibition in colorectal cancers. Br. J. Cancer 2014, 110, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.E.; Ahmed, N.C.; Vos, P.W.; Bonnerup, C.; Atkins, J.N.; Casey, M.; Nuovo, G.J.; Naziri, W.; Wiley, J.E.; Mota, H. Diagnostic microRNA markers to screen for sporadic human colon cancer in stool: I. Proof of principle. Cancer Genom. Proteom. 2013, 10, 93–113. [Google Scholar]
- Yang, X.; Zhong, J.; Ji, Y.; Li, J.; Jian, Y.; Zhang, J.; Yang, W. The expression and clinical significance of microRNAs in colorectal cancer detecting. Tumor Biol. 2015, 36, 2675–2684. [Google Scholar] [CrossRef] [PubMed]
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the human microbiome. Nutr. Rev. 2012, 70, S38–S44. [Google Scholar] [CrossRef] [PubMed]
- McNally, L.; Brown, S.P. Building the microbiome in health and disease: Niche construction and social conflict in bacteria. Phil. Trans. R. Soc. B 2015. [Google Scholar] [CrossRef] [PubMed]
- Scarpellini, E.; Ianiro, G.; Attili, F.; Bassanelli, C.; de Santis, A.; Gasbarrini, A. The human gut microbiota and virome: Potential therapeutic implications. Dig. Liver Dis. 2011, 47, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Stearns, J.C.; Lynch, M.D.J.; Senadheera, D.B.; Tenenbaum, H.C.; Goldberg, M.B.; Cvitkovitch, D.G.; Croitoru, K.; Moreno-Hagelsieb, G.; Neufeld, J.D. Bacterial biogeography of the human digestive tract. Sci. Rep. 2011, 1. [Google Scholar] [CrossRef] [PubMed]
- Rajilić-Stojanović, M.; Smidt, H.; de Vos, W.M. Diversity of the human gastrointestinal tract microbiota revisited. Environ. Microbial. 2007, 9, 2125–2136. [Google Scholar] [CrossRef] [PubMed]
- Matamoros, S.; Gras-Leguen, C.; Le Vacon, F.; Potel, G.; de La Cochetiere, M.-F. Development of intestinal microbiota in infants and its impact on health. Trends Microbial. 2013, 21, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.; Ghosh, T.S.; Mande, S.S. Global investigation of composition and interaction networks in gut microbiomes of individuals belonging to diverse geographies and age-groups. Gut Pathog. 2016, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Engen, P.A.; Green, S.J.; Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. The gastrointestinal microbiome: Alcohol effects on the composition of intestinal microbiota. Alcohol Res. Curr. Rev. 2015, 37, 223. [Google Scholar]
- Xu, Z.; Knight, R. Dietary effects on human gut microbiome diversity. Br. J. Nutr. 2015, 113, S1–S5. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef] [PubMed]
- Davenport, E.R.; Mizrahi-Man, O.; Michelini, K.; Barreiro, L.B.; Ober, C.; Gilad, Y. Seasonal variation in human gut microbiome composition. PLoS ONE 2014, 9, e90731. [Google Scholar] [CrossRef] [PubMed]
- Sankar, S.A.; Lagier, J.-C.; Pontarotti, P.; Raoult, D.; Fournier, P.-E. The human gut microbiome, a taxonomic conundrum. Syst. Appl. Microbial. 2015, 38, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.J.; Cookson, W.O.; Moffatt, M.F. Sequencing the human microbiome in health and disease. Hum. Mol. Genet. 2013, 22, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Garza, D.R.; Dutilh, B.E. From cultured to uncultured genome sequences: Metagenomics and modeling microbial ecosystems. Cell. Mol. Life Sci. 2015, 72, 4287–4308. [Google Scholar] [CrossRef] [PubMed]
- Geuking, M.B.; Köller, Y.; Rupp, S.; McCoy, K.D. The interplay between the gut microbiota and the immune system. Gut Microb. 2014, 5, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Kasper, D.L. Microbiota-stimulated immune mechanisms to maintain gut homeostasis. Curr. Opin. Immunol. 2010, 22, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Alden, N.; Lee, K. Pathways and functions of gut microbiota metabolism impacting host physiology. Curr. Opin. Biotechnol. 2015, 36, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-J.; Li, S.; Gan, R.-Y.; Zhou, T.; Xu, D.-P.; Li, H.-B. Impacts of gut bacteria on human health and diseases. Int. J. Mol. Sci. 2015, 16, 7493–7519. [Google Scholar] [CrossRef] [PubMed]
- Serino, M.; Blasco-Baque, V.; Nicolas, S.; Burcelin, R. Far from the eyes, close to the heart: Dysbiosis of gut microbiota and cardiovascular consequences. Curr. Cardiol. Rep. 2014, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-G.; Udayanga, K.G.S.; Totsuka, N.; Weinberg, J.B.; Núñez, G.; Shibuya, A. Gut dysbiosis promotes M2 macrophage polarization and allergic airway inflammation via fungi-induced PGE2. Cell Host Microb. 2014, 15, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Iebba, V.; Totino, V.; Gagliardi, A.; Santangelo, F.; Cacciotti, F.; Trancassini, M.; Mancini, C.; Cicerone, C.; Corazziari, E.; Pantanella, F. Eubiosis and dysbiosis: The two sides of the microbiota. New Microbial. 2016, 39, 1–12. [Google Scholar]
- Schippa, S.; Conte, M.P. Dysbiotic events in gut microbiota: Impact on human health. Nutrients 2014, 6, 5786–5805. [Google Scholar] [CrossRef] [PubMed]
- Nagao-Kitamoto, H.; Kitamoto, S.; Kuffa, P.; Kamada, N. Pathogenic role of the gut microbiota in gastrointestinal diseases. Intest. Res. 2016, 14, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Peloquin, J.M.; Nguyen, D.D. The microbiota and inflammatory bowel disease: Insights from animal models. Anaerobe 2013, 24, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Tomasello, G.; Tralongo, P.; Damiani, P.; Sinagra, E.; di Trapani, B.; Zeenny, M.N.; Hussein, I.H.; Jurjus, A.; Leone, A. Dismicrobism in inflammatory bowel disease and colorectal cancer: Changes in response of colocytes. World J. Gastroenterol. 2014, 20, 18121–18130. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Yang, X.; Zhang, R.; Li, J.; Xiao, X.; Hu, Y.; Chen, Y.; Yang, F.; Lu, N.; Wang, Z. Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb. Ecol. 2013, 66, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Sheflin, A.M.; Whitney, A.K.; Weir, T.L. Cancer-promoting effects of microbial dysbiosis. Curr. Oncol. Rep. 2014, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cai, G.; Qiu, Y.; Fei, N.; Zhang, M.; Pang, X.; Jia, W.; Cai, S.; Zhao, L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012, 6, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, J.; Lange, B.; Frick, J.-S.; Sauer, H.; Zimmermann, K.; Schwiertz, A.; Rusch, K.; Klosterhalfen, S.; Enck, P. A vegan or vegetarian diet substantially alters the human colonic faecal microbiota. Eur. J. Clin. Nutr. 2012, 66, 53–60. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, R.J.; Ulrich, C.M.; Goode, E.L.; Brhane, Y.; Muir, K.; Chan, A.T.; Le Marchand, L.; Schildkraut, J.; Witte, J.S.; Eeles, R. Cross cancer genomic investigation of inflammation pathway for five common cancers: Lung, ovary, prostate, breast, and colorectal cancer. J. Natl. Cancer Inst. 2015, 107, 246. [Google Scholar] [CrossRef] [PubMed]
- Chai, E.Z.P.; Siveen, K.S.; Shanmugam, M.K.; Arfuso, F.; Sethi, G. Analysis of the intricate relationship between chronic inflammation and cancer. Biochem. J. 2015, 468, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.M.; Wolff, R.K.; Valeri, N.; Khan, M.; Robinson, D.; Paone, A.; Bowman, E.D.; Lundgreen, A.; Caan, B.; Potter, J. An analysis of genetic factors related to risk of inflammatory bowel disease and colon cancer. Cancer Epidemiol. 2015, 38, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, I.; Amiot, A.; Le Baleur, Y.; Levy, M.; Auriault, M.-L.; van Nhieu, J.T.; Delchier, J.C. Microbial dysbiosis and colon carcinogenesis: Could colon cancer be considered a bacteria-related disease? Ther. Adv. Gastroenterol. 2013, 6, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Borges-Canha, M.; Portela-Cidade, J.P.; Dinis-Ribeiro, M.; Leite-Moreira, A.F.; Pimentel-Nunes, P. Role of colonic microbiota in colorectal carcinogenesis: A systematic review. Rev. Esp. Enferm. Dig. 2015, 107, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Candela, M.; Turroni, S.; Biagi, E.; Carbonero, F.; Rampelli, S.; Fiorentini, C.; Brigidi, P. Inflammation and colorectal cancer, when microbiota-host mutualism breaks. World J. Gastroenterol. 2014, 20, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Zackular, J.P.; Baxter, N.T.; Iverson, K.D.; Sadler, W.D.; Petrosino, J.F.; Chen, G.Y.; Schloss, P.D. The gut microbiome modulates colon tumorigenesis. MBio 2013, 4, e00692-13. [Google Scholar] [CrossRef] [PubMed]
- Nosho, K.; Sukawa, Y.; Adachi, Y.; Ito, M.; Mitsuhashi, K.; Kurihara, H.; Kanno, S.; Yamamoto, I.; Ishigami, K.; Igarashi, H. Association of fusobacterium nucleatum with immunity and molecular alterations in colorectal cancer. World J. Gastroenterol. 2016, 22, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microb. 2014, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-Y.; Ge, Q.-X.; Cao, J.; Zhou, Y.-J.; Du, Y.-L.; Shen, B.; Wan, Y.-J.Y.; Nie, Y.-Q. Association of fusobacterium nucleatum infection with colorectal cancer in Chinese patients. World J. Gastroenterol. 2016, 22, 3227. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.N.; Araujo-Perez, F.; Azcarate-Peril, A.; Yeh, J.J.; Sandler, R.S.; Keku, T.O. Fusobacterium is associated with colorectal adenomas. PLoS ONE 2013, 8, e53653. [Google Scholar] [CrossRef] [PubMed]
- Bashir, A.; Miskeen, A.Y.; Hazari, Y.M.; Asrafuzzaman, S.; Fazili, K.M. Fusobacterium nucleatum, inflammation, and immunity: The fire within human gut. Tumor Biol. 2016, 37, 2805–2810. [Google Scholar] [CrossRef] [PubMed]
- Sears, C.L.; Geis, A.L.; Housseau, F. Bacteroides fragilis subverts mucosal biology: From symbiont to colon carcinogenesis. J. Clin. Investing. 2014, 124, 4166–4172. [Google Scholar] [CrossRef] [PubMed]
- Boleij, A.; Hechenbleikner, E.M.; Goodwin, A.C.; Badani, R.; Stein, E.M.; Lazarev, M.G.; Ellis, B.; Carroll, K.C.; Albesiano, E.; Wick, E.C. The bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin. Infect. Dis. 2014. [CrossRef] [PubMed]
- Kohoutova, D.; Smajs, D.; Moravkova, P.; Cyrany, J.; Moravkova, M.; Forstlova, M.; Cihak, M.; Rejchrt, S.; Bures, J. Escherichia coli strains of phylogenetic group B2 and D and bacteriocin production are associated with advanced colorectal neoplasia. BMC Infect. Dis. 2014, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Allen-Vercoe, E.; Jobin, C. Fusobacterium and enterobacteriaceae: Important players for CRC? Immunol. Lett. 2014, 162, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, M.; Buc, E.; Sauvanet, P.; Darcha, C.; Dubois, D.; Pereira, B.; Déchelotte, P.; Bonnet, R.; Pezet, D.; Darfeuille-Michaud, A. Colonization of the human gut by E. Coli and colorectal cancer risk. Clin. Cancer Res. 2014, 20, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.A.; Manan, A.A.; Chow, K.Y.; Cornain, S.F.; Devi, C.R.; Triningsih, F.X.; Laudico, A.; Mapua, C.A.; Mirasol-Lumague, M.R.; Noorwati, S. Cancer epidemiology and control in peninsular and island South-east Asia-past, present and future. Asian Pac. J. Cancer Prev. 2010, 11, 81–98. [Google Scholar] [PubMed]
- Farazi, P.A. Cancer trends and risk factors in Cyprus. ecancermedicalscience 2014, 8. [Google Scholar] [CrossRef]
- Koifman, S.; Koifman, R.J. Environment and cancer in Brazil: An overview from a public health perspective. Rev. Mutat. Res. 2003, 544, 305–311. [Google Scholar] [CrossRef]
- Okreglicka, K. Health effects of changes in the structure of dietary macronutrients intake in western societies. Rocz. Panstw. Zakl. Hig. 2015, 66, 97–105. [Google Scholar] [PubMed]
- Aykan, N.F. Red meat and colorectal cancer. Oncol. Rev. 2015, 9, 288. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Coelho, D.; Blachier, F. Review of the association between meat consumption and risk of colorectal cancer. Nutr. Res. 2013, 33, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Carbonero, F.; Zoetendal, E.G.; DeLany, J.P.; Wang, M.; Newton, K.; Gaskins, H.R.; O’Keefe, S.J.D. Diet, microbiota, and microbial metabolites in colon cancer risk in rural africans and African americans. Am. J. Clin. Nutr. 2013, 98, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, S.J.D.; Li, J.V.; Lahti, L.; Ou, J.; Carbonero, F.; Mohammed, K.; Posma, J.M.; Kinross, J.; Wahl, E.; Ruder, E. Fat, fibre and cancer risk in African americans and rural africans. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Fung, K.Y.C.; Cosgrove, L.; Lockett, T.; Head, R.; Topping, D.L. A review of the potential mechanisms for the lowering of colorectal oncogenesis by butyrate. Br. J. Nutr. 2012, 108, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Hofmanova, J.; Hyrslova Vaculova, A.; Kozubik, A. Regulation of the metabolism of polyunsaturated fatty acids and butyrate in colon cancer cells. Curr. Pharm. Biotechnol. 2013, 14, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Holley, D.; Collins, L.B.; Montgomery, S.A.; Whitmore, A.C.; Hillhouse, A.; Curry, K.P.; Renner, S.W.; Greenwalt, A.; Ryan, E.P. A gnotobiotic mouse model demonstrates that dietary fiber protects against colorectal tumorigenesis in a microbiota- and butyrate-dependent manner. Cancer Discov. 2014, 4, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Diamandis, E.P. Cancer biomarkers: Can we turn recent failures into success? J. Natl. Cancer Inst. 2010, 102, 1462–1467. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, J.A.; Weinstein, J.N. Biomarkers in cancer staging, prognosis and treatment selection. Nat. Rev. Cancer 2005, 5, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Coghlin, C.; Murray, G.I. Biomarkers of colorectal cancer: Recent advances and future challenges. Proteom. Clin. Appl. 2015, 9, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Bacher, J.W.; Flanagan, L.A.; Smalley, R.L.; Nassif, N.A.; Burgart, L.J.; Halberg, R.B.; Megid, W.M.A.; Thibodeau, S.N. Development of a fluorescent multiplex assay for detection of MSI-high tumors. Dis. Mark. 2004, 20, 237–250. [Google Scholar] [CrossRef]
- Geiersbach, K.B.; Samowitz, W.S. Microsatellite instability and colorectal cancer. Arch. Pathol. Lab. Med. 2011, 135, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Iacopetta, B.; Watanabe, T. Predictive value of microsatellite instability for benefit from adjuvant fluorouracil chemotherapy in colorectal cancer. Gut 2006, 55, 1671–1672. [Google Scholar] [PubMed]
- De Roock, W.; Claes, B.; Bernasconi, D.; de Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol. 2010, 11, 753–762. [Google Scholar] [CrossRef]
- Aprile, G.; Macerelli, M.; De Maglio, G.; Pizzolitto, S.; Fasola, G. The relevance of BRAF and extended ras mutational analyses for metastatic colorectal cancer patients. OA Mol. Oncol. 2013, 1, 7. [Google Scholar] [CrossRef]
- Kislitsin, D.; Lerner, A.; Rennert, G.; Lev, Z. KRAS mutations in sporadic colorectal tumors in Israel: Unusual high frequency of codon 13 mutations and evidence for nonhomogeneous representation of mutation subtypes. Dig. Dis. Sci. 2002, 47, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Fransén, K.; Klintenas, M.; Osterstrom, A.; Dimberg, J.; Monstein, H.-J.; Soderkvist, P. Mutation analysis of the BRAF, ARAF and RAF-1 genes in human colorectal adenocarcinomas. Carcinogenesis 2004, 25, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.; Cunningham, D. Using predictive biomarkers to select patients with advanced colorectal cancer for treatment with epidermal growth factor receptor antibodies. J. Clin. Oncol. 2008, 26, 5668–5670. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Martini, M.; Molinari, F.; Veronese, S.; Nichelatti, M.; Artale, S.; di Nicolantonio, F.; Saletti, P.; de Dosso, S.; Mazzucchelli, L. PIK3CA mutations in colorectal cancer are associated with clinical resistance to egfr-targeted monoclonal antibodies. Cancer Res. 2009, 69, 1851–1857. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, J.M.D.; Loukola, A.; Aaltonen, L.A.; Mortensen, N.J.M.; Bodmer, W.F. The role of hypermethylation of thehmlh1 promoter region in hnpcc versus MSI+ sporadic colorectal cancers. J. Med. Genet. 2000, 37, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Pons, M.; Cruz-Correa, M. Colorectal cancer biomarkers: Where are we now? BioMed Res. Int. 2015, 2015, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Umetani, N.; Kim, J.; Hiramatsu, S.; Reber, H.A.; Hines, O.J.; Bilchik, A.J.; Hoon, D.S.B. Increased integrity of free circulating DNA in sera of patients with colorectal or periampullary cancer: Direct quantitative PCR for alu repeats. Clin. Chem. 2006, 52, 1062–1069. [Google Scholar] [CrossRef] [PubMed]
- Barrier, A.; Boelle, P.-Y.; Roser, F.O.; Gregg, J.; Tse, C.; Brault, D.; Lacaine, F.O.; Houry, S.; Huguier, M.; Franc, B. Stage II colon cancer prognosis prediction by tumor gene expression profiling. J. Clin. Oncol. 2006, 24, 4685–4691. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jatkoe, T.; Zhang, Y.; Mutch, M.G.; Talantov, D.; Jiang, J.; McLeod, H.L.; Atkins, D. Gene expression profiles and molecular markers to predict recurrence of dukes’ B colon cancer. J. Clin. Oncol. 2004, 22, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- Takai, T.; Kanaoka, S.; Yoshida, K.-I.; Hamaya, Y.; Ikuma, M.; Miura, N.; Sugimura, H.; Kajimura, M.; Hishida, A. Fecal cyclooxygenase 2 plus matrix metalloproteinase 7 mRNA assays as a marker for colorectal cancer screening. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1888–1893. [Google Scholar] [CrossRef] [PubMed]
- Link, A.; Balaguer, F.; Shen, Y.; Nagasaka, T.; Lozano, J.J.; Boland, C.R.; Goel, A. Fecal microRNAs as novel biomarkers for colon cancer screening. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1766–1774. [Google Scholar] [CrossRef] [PubMed]
- Khambata-Ford, S.; Garrett, C.R.; Meropol, N.J.; Basik, M.; Harbison, C.T.; Wu, S.; Wong, T.W.; Huang, X.; Takimoto, C.H.; Godwin, A.K. Expression of epiregulin and amphiregulin and KRAS mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J. Clin. Oncol. 2007, 25, 3230–3237. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, B.; de Roock, W.; Piessevaux, H.; van Oirbeek, R.; Biesmans, B.; de Schutter, J.; Fieuws, S.; Vandesompele, J.; Peeters, M.; van Laethem, J.-L. Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J. Clin. Oncol. 2009, 27, 5068–5074. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.B.; Dutta, D.; Watson, D.; Maddala, T.; Munneke, B.M.; Shak, S.; Rowinsky, E.K.; Xu, L.A.; Harbison, C.T.; Clark, E.A. Tumour gene expression predicts response to cetuximab in patients with kras wild-type metastatic colorectal cancer. Br. J. Cancer 2011, 104, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Von Roon, A.C.; Karamountzos, L.; Purkayastha, S.; Reese, G.E.; Darzi, A.W.; Teare, J.P.; Paraskeva, P.; Tekkis, P.P. Diagnostic precision of fecal calprotectin for inflammatory bowel disease and colorectal malignancy. Am. J. Gastroenterol. 2007, 102, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Xing, P.X.; Young, G.P.; Ho, D.; Sinatra, M.A.; Hoj, P.B.; McKenzie, I.F.C. A new approach to fecal occult blood testing based on the detection of haptoglobin. Cancer 1996, 78, 48–56. [Google Scholar] [CrossRef]
- Quaye, I.K. Haptoglobin, inflammation and disease. Trans. R. Soc. Trop. Med. Hyg. 2008, 102, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Hundt, S.; Haug, U.; Brenner, H. Blood markers for early detection of colorectal cancer: A systematic review. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1935–1953. [Google Scholar] [CrossRef] [PubMed]
- Holten-Andersen, M.N.; Christensen, I.J.; Nielsen, H.; Stephens, R.W.; Jensen, V.; Nielsen, O.H.; Sørensen, S.; Overgaard, J.; Lilja, H.; Harris, A. Total levels of tissue inhibitor of metalloproteinases 1 in plasma yield high diagnostic sensitivity and specificity in patients with colon cancer. Clin. Cancer Res. 2002, 8, 156–164. [Google Scholar] [PubMed]
- Sastre, J.; Maestro, M.L.; Puente, J.; Veganzones, S.; Alfonso, R.; Rafael, S.; Garcia-Saenz, J.A.; Vidaurreta, M.; Martin, M.; Arroyo, M. Circulating tumor cells in colorectal cancer: Correlation with clinical and pathological variables. Ann. Oncol. 2008, 19, 935–938. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.J.; Punt, C.J.A.; Iannotti, N.; Saidman, B.H.; Sabbath, K.D.; Gabrail, N.Y.; Picus, J.; Morse, M.A.; Mitchell, E.; Miller, M.C. Prognostic significance of circulating tumor cells in patients with metastatic colorectal cancer. Ann. Oncol. 2009, 20, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Uen, Y.-H.; Lu, C.-Y.; Tsai, H.-L.; Yu, F.-J.; Huang, M.-Y.; Cheng, T.-L.; Lin, S.-R.; Wang, J.-Y. Persistent presence of postoperative circulating tumor cells is a poor prognostic factor for patients with stage I–III colorectal cancer after curative resection. Ann. Surg. Oncol. 2008, 15, 2120–2128. [Google Scholar] [CrossRef] [PubMed]
- Bertucci, F.; Salas, S.; Eysteries, S.; Nasser, V.; Finetti, P.; Ginestier, C.; Charafe-Jauffret, E.; Loriod, B.; Bachelart, L.C.; Montfort, J.M. Gene expression profiling of colon cancer by DNA microarrays and correlation with histoclinical parameters. Oncogene 2004, 23, 1377–1391. [Google Scholar] [CrossRef] [PubMed]
- Marshall, K.W.; Mohr, S.; Khettabi, F.E.; Nossova, N.; Chao, S.; Bao, W.; Ma, J.; Li, X.-J.; Liew, C.-C. A blood-based biomarker panel for stratifying current risk for colorectal cancer. Int. J. Cancer 2010, 126, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Yip, K.-T.; Das, P.K.; Suria, D.; Lim, C.-R.; Ng, G.-H.; Liew, C.-C. A case-controlled validation study of a blood-based seven-gene biomarker panel for colorectal cancer in Malaysia. J. Exp. Clin. Cancer Res. 2010, 29, 1. [Google Scholar] [CrossRef] [PubMed]
- Yothers, G.; O’Connell, M.J.; Lee, M.; Lopatin, M.; Clark-Langone, K.M.; Millward, C.; Paik, S.; Sharif, S.; Shak, S.; Wolmark, N. Validation of the 12-gene colon cancer recurrence score in NSABP C-07 as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin (FU/LV) and FU/LV plus oxaliplatin. J. Clin. Oncol. 2013, 31, 4512–4519. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, G.; Renfro, L.A.; Behrens, R.J.; Lopatin, M.; Chao, C.; Soori, G.S.; Dakhil, S.R.; Mowat, R.B.; Kuebler, J.P.; Kim, G. Prospective multicenter study of the impact of oncotype DX colon cancer assay results on treatment recommendations in stage II colon cancer patients. Oncologist 2014, 19, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Salazar, R.; Roepman, P.; Capella, G.; Moreno, V.; Simon, I.; Dreezen, C.; Lopez-Doriga, A.; Santos, C.; Marijnen, C.; Westerga, J. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J. Clin. Oncol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Maak, M.; Simon, I.; Nitsche, U.; Roepman, P.; Snel, M.; Glas, A.M.; Schuster, T.; Keller, G.; Zeestraten, E.; Goossens, I.S. Independent validation of a prognostic genomic signature (coloprint) for patients with stage II colon cancer. Ann. Surg. 2013, 257, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, R.D.; Bylesjo, M.; Kerr, P.; Davison, T.; Black, J.M.; Kay, E.W.; Holt, R.J.; Proutski, V.; Ahdesmaki, M.; Farztdinov, V. Development and independent validation of a prognostic assay for stage II colon cancer using formalin-fixed paraffin-embedded tissue. J. Clin. Oncol. 2011, 29, 4620–4626. [Google Scholar] [CrossRef] [PubMed]
- Sveen, A.; Nesbakken, A.; Agesen, T.H.; Guren, M.G.; Tveit, K.M.; Skotheim, R.I.; Lothe, R.A. Anticipating the clinical use of prognostic gene expression-based tests for colon cancer stage II and III: Is godot finally arriving? Clin. Cancer Res. 2013, 19, 6669–6677. [Google Scholar] [CrossRef] [PubMed]
- Lenehan, P.F.; Boardman, L.A.; Riegert-Johnson, D.; de Petris, G.; Fry, D.W.; Ohrnberger, J.; Heyman, E.R.; Gerard, B.; Almal, A.A.; Worzel, W.P. Generation and external validation of a tumor-derived 5-gene prognostic signature for recurrence of lymph node-negative, invasive colorectal carcinoma. Cancer 2012, 118, 5234–5244. [Google Scholar] [CrossRef] [PubMed]
- Black, E.R.; Falzon, L.; Aronson, N. Gene Expression Profiling for Predicting Outcomes in Stage II Colon Cancer; Report No.: 12(13)-EHC143-EF; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2012.
- Carlson, M.R. Previstage (TM) GCC colorectal cancer staging test: A new molecular test to identify lymph node metastases and provide more accurate information about the stage of patients with colorectal cancer. Mol. Diagn. Ther. 2009, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Nordlinger, B.; Cervantes, A.; Group, E.G.W. Advanced colorectal cancer: ESMO clinical practice guidelines for treatment. Ann. Oncol. 2010, 21, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Cervantes, A.; Nordlinger, B.; Arnold, D. Metastatic colorectal cancer: Esmo clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Venook, A. Critical evaluation of current treatments in metastatic colorectal cancer. Oncologist 2005, 10, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Prehn, R.T. The inhibition of tumor growth by tumor mass. Cancer Res. 1991, 51, 2–4. [Google Scholar] [PubMed]
- Brown, L.M.; Malkinson, A.M.; Rannels, D.E.; Rannels, S.R. Compensatory lung growth after partial pneumonectomy enhances lung tumorigenesis induced by 3-methylcholanthrene. Cancer Res. 1999, 59, 5089–5092. [Google Scholar] [PubMed]
- Smith, B.H.; Gazda, L.S.; Conn, B.L.; Jain, K.; Asina, S.; Levine, D.M.; Parker, T.S.; Laramore, M.A.; Martis, P.C.; Vinerean, H.V. Hydrophilic agarose macrobead cultures select for outgrowth of carcinoma cell populations that can restrict tumor growth. Cancer Res. 2011, 71, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Asina, S.; Jain, K.; Rubin, A.L.; Smith, B.; Stenzel, K. Cancer-Cell Proliferation-Suppressing Material Produced by Cancer Cells Restricted by Entrapment. Google Patents U.S. 6303151 B1, 16 October 2001. [Google Scholar]
- Ocean, A.J.; Parikh, T.; Berman, N.; Escalon, J.; Shah, M.A.; Andrada, Z.; Akahoho, E.; Pogoda, J.M.; Stoms, G.B.; Escobia, V.B. Phase I/II trial of intraperitoneal implantation of agarose-agarose macrobeads (MB) containing mouse renal adenocarcinoma cells (RENCA) in patients (PTS) with advanced colorectal cancer (CRC). In Proceedings of the 2013 ASCO Annual Meeting, Chicago, IL, USA, 31 May–4 June 2013; p. e14517.
- Janakiram, N.B.; Rao, C.V. The role of inflammation in colon cancer. Adv. Exp. Med. Biol. 2014, 816, 25–52. [Google Scholar] [PubMed]
- Suh, O.; Mettlin, C.; Petrelli, N.J. Aspirin use, cancer, and polyps of the large bowel. Cancer 1993, 72, 1171–1177. [Google Scholar] [CrossRef]
- Rigau, J.; Piqué, J.M.; Rubio, E.; Planas, R.N.; Tarrech, J.M.; Bordas, J.M. Effects of long-term sulindac therapy on colonic polyposis. Ann. Intern. Med. 1991, 115, 952–954. [Google Scholar] [CrossRef] [PubMed]
- Suh, N.; Reddy, B.S.; DeCastro, A.; Paul, S.; Lee, H.J.; Smolarek, A.K.; So, J.Y.; Simi, B.; Wang, C.X.; Janakiram, N.B. Combination of atorvastatin with sulindac or naproxen profoundly inhibits colonic adenocarcinomas by suppressing the p65/β-catenin/cyclin D1 signaling pathway in rats. Cancer Prev. Res. 2011, 4, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Ungprasert, P.; Cheungpasitporn, W.; Crowson, C.S.; Matteson, E.L. Individual non-steroidal anti-inflammatory drugs and risk of acute kidney injury: A systematic review and meta-analysis of observational studies. Eur. J. Intern. Med. 2015, 26, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; DuBois, R.N. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat. Rev. Cancer 2001, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Jüni, P.; Nartey, L.; Reichenbach, S.; Sterchi, R.; Dieppe, P.A.; Egger, M. Risk of cardiovascular events and rofecoxib: Cumulative meta-analysis. Lancet 2004, 364, 2021–2029. [Google Scholar] [CrossRef]
- Silverstein, F.E.; Faich, G.; Goldstein, J.L.; Simon, L.S.; Pincus, T.; Whelton, A.; Makuch, R.; Eisen, G.; Agrawal, N.M.; Stenson, W.F. Gastrointestinal toxicity with celecoxib vs nonsteroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: The class study: A randomized controlled trial. JAMA 2000, 284, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; McMurray, J.J.V.; Pfeffer, M.A.; Wittes, J.; Fowler, R.; Finn, P.; Anderson, W.F.; Zauber, A.; Hawk, E.; Bertagnolli, M. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N. Engl. J. Med. 2005, 352, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.F.; Quinn, A.M.; Devers, T.; Cullen, A.; Coulter, I.S.; Marison, I.W.; Loughran, S.T. In-vitro characterisation of a novel celecoxib microbead formulation for the treatment and prevention of colorectal cancer. J. Pharm. Pharmacol. 2015, 67, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Lev-Ari, S.; Strier, L.; Kazanov, D.; Madar-Shapiro, L.; Dvory-Sobol, H.; Pinchuk, I.; Marian, B.; Lichtenberg, D.; Arber, N. Celecoxib and curcumin synergistically inhibit the growth of colorectal cancer cells. Clin. Cancer Res. 2005, 11, 6738–6744. [Google Scholar] [CrossRef] [PubMed]
- Kahouli, I.; Tomaro-Duchesneau, C.; Prakash, S. Probiotics in colorectal cancer (CRC) with emphasis on mechanisms of action and current perspectives. J. Med. Microbial. 2013, 62, 1107–1123. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Zhang, X.; Covasa, M. Emerging roles of lactic acid bacteria in protection against colorectal cancer. World J. Gastroenterol. 2014, 20, 7878–7886. [Google Scholar] [CrossRef] [PubMed]
- Chong, E.S.L. A potential role of probiotics in colorectal cancer prevention: Review of possible mechanisms of action. World J. Microbiol. Biotechnol. 2014, 30, 351–374. [Google Scholar] [CrossRef] [PubMed]
- Pala, V.; Sieri, S.; Berrino, F.; Vineis, P.; Sacerdote, C.; Palli, D.; Masala, G.; Panico, S.; Mattiello, A.; Tumino, R. Yogurt consumption and risk of colorectal cancer in the italian european prospective investigation into cancer and nutrition cohort. Int. J. Cancer 2011, 129, 2712–2719. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Lin, W.-C.; Kong, M.-S.; Shi, H.N.; Walker, W.A.; Lin, C.-Y.; Huang, C.-T.; Lin, Y.-C.; Jung, S.-M.; Lin, T.-Y. Oral inoculation of probiotics lactobacillus acidophilus NCFM suppresses tumour growth both in segmental orthotopic colon cancer and extra-intestinal tissue. Br. J. Nutr. 2012, 107, 1623–1634. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Kim, Y.; Han, K.S.; You, S.; Oh, S.; Kim, S.H. Effects of lactobacillus strains on cancer cell proliferation and oxidative stress in vitro. Lett. Appl. Microbial. 2006, 42, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Sah, B.N.P.; Vasiljevic, T.; McKechnie, S.; Donkor, O.N. Effect of probiotics on antioxidant and antimutagenic activities of crude peptide extract from yogurt. Food Chem. 2014, 156, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Boil. 2007, 39, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Rad. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, R.; Barros, L.; Carvalho, A.M.; Ferreira, I.C. Studies on chemical constituents and bioactivity of rosa micrantha: An alternative antioxidants source for food, pharmaceutical, or cosmetic applications. J. Agric. Food Chem. 2010, 58, 6277–6284. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.C.; Barros, L.; Abreu, R. Antioxidants in wild mushrooms. Curr. Med. Chem. 2009, 16, 1543–1560. [Google Scholar] [CrossRef] [PubMed]
- Carocho, M.; Barros, L.; Barreira, J.C.; Calhelha, R.C.; Soković, M.; Fernández-Ruiz, V.; Buelga, C.S.; Morales, P.; Ferreira, I.C. Basil as functional and preserving ingredient in “serra da estrela” cheese. Food Chem. 2016, 207, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Montero, P.; Calvo, M.; Gómez-Guillén, M.; Gómez-Estaca, J. Microcapsules containing astaxanthin from shrimp waste as potential food coloring and functional ingredient: Characterization, stability, and bioaccessibility. LWT Food Sci. Technol. 2016, 70, 229–236. [Google Scholar] [CrossRef]
- Hansen, C. Grape Seed Extract: Procyanidolic Oligomers (PCO); Healing Wisdom Publications: New York, NY, USA, 1995. [Google Scholar]
- Macheix, J.-J.; Fleuriet, A. Fruit Phenolics; CRC Press Inc.: Boca Raton, FL, USA, 1990. [Google Scholar]
- Velioglu, Y.; Mazza, G.; Gao, L.; Oomah, B. Antioxidant activity and total phenolics in selected fruits, vegetables, and grain products. J. Agric. Food chem. 1998, 46, 4113–4117. [Google Scholar] [CrossRef]
- Shi, J.; Nawaz, H.; Pohorly, J.; Mittal, G.; Kakuda, Y.; Jiang, Y. Extraction of polyphenolics from plant material for functional foods—Engineering and technology. Food Rev. Int. 2005, 21, 139–166. [Google Scholar] [CrossRef]
- Wenzel, U.; Kuntz, S.; Brendel, M.D.; Daniel, H. Dietary flavone is a potent apoptosis inducer in human colon carcinoma cells. Cancer Res. 2000, 60, 3823–3831. [Google Scholar] [PubMed]
- Pandey, K.B.; Rizvi, S.I. Plant polyphenols as dietary antioxidants in human health and disease. Oxid. Med. Cell. Longev. 2009, 2, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Shahidi, F.; Ambigaipalan, P. Phenolics and polyphenolics in foods, beverages and spices: Antioxidant activity and health effects—A review. J. Funct. Foods 2015, 18, 820–897. [Google Scholar] [CrossRef]
- Jiménez, S.; Gascón, S.; Luquín, A.; Laguna, M.; Ancin-Azpilicueta, C.; Rodríguez-Yoldi, M.J. Rosa canina extracts have antiproliferative and antioxidant effects on caco-2 human colon cancer. PLoS ONE 2016, 11, e0159136. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shi, J.-Z.; Yu, L.-M.; Goyer, R.A.; Waalkes, M.P. Mercury in traditional medicines: Is cinnabar toxicologically similar to common mercurials? Exp. Boil. Med. 2008, 233, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.C.; Rashid, R.M.; Falzon, L.; Elamin, E.M.; Zehtabchi, S. Silver sulfadiazine for the treatment of partial-thickness burns and venous stasis ulcers. J. Am. Acad. Dermatol. 2012, 66, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Liu, L.; Wang, Y.; Mao, T.; Li, J. Effects of a combination of puerarin, baicalin and berberine on the expression of proliferator-activated receptor-γ and insulin receptor in a rat model of nonalcoholic fatty liver disease. Exp. Ther. Med. 2016, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, N.; Guo, Z. Metal-based anticancer chemotherapeutic agents. Curr. Opin. Chem. Boil. 2014, 19, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Komeda, S.; Casini, A. Next-generation anticancer metallodrugs. Curr. Top. Med. Chem. 2012, 12, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Garbutcheon-Singh, K.B.; Grant, M.P.; Harper, B.W.; Krause-Heuer, A.M.; Manohar, M.; Orkey, N.; Aldrich-Wright, J.R. Transition metal based anticancer drugs. Curr. Top. Med. Chem. 2011, 11, 521–542. [Google Scholar] [CrossRef] [PubMed]
- Frezza, M.; Hindo, S.; Chen, D.; Davenport, A.; Schmitt, S.; Tomco, D.; Dou, Q.P. Novel metals and metal complexes as platforms for cancer therapy. Curr. Pharm. Des. 2010, 16, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- Wheate, N.J.; Walker, S.; Craig, G.E.; Oun, R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010, 39, 8113–8127. [Google Scholar] [CrossRef] [PubMed]
- WoYniak, K.; Blasiak, J. Recognition and repair of DNA-cisplatin adducts. Acta Biochim. Pol. 2002, 49, 583–596. [Google Scholar]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. Third row transition metals for the treatment of cancer. Philos. Trans. R. Soc. Lond. A Math. Phys. Eng. Sci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Karasawa, T.; Steyger, P.S. An integrated view of cisplatin-induced nephrotoxicity and ototoxicity. Toxicol. Lett. 2015, 237, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Peres, L.A.B.; de Cunha, A.D. Acute nephrotoxicity of cisplatin: Molecular mechanisms. J. Bras. Nefrol. 2013, 35, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, M.S.; Silveira, A.F.; Teixeira, A.R.; Hyppolito, M.A. Mechanisms of cisplatin ototoxicity: Theoretical review. J. Laryngol. Otol. 2013, 127, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed]
- Amable, L. Cisplatin resistance and opportunities for precision medicine. Pharmacol. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Lippard, S.J. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005, 4, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Hoff, P.M.; Saad, E.D.; Costa, F.; Coutinho, A.K.; Caponero, R.; Prolla, G.; Gansl, R.C. Literature review and practical aspects on the management of oxaliplatin-associated toxicity. Clin. Colorectal Cancer 2012, 11, 93–100. [Google Scholar] [CrossRef] [PubMed]
- De Gramont, A.D.; Figer, A.; Seymour, M.; Homerin, M.; Hmissi, A.; Cassidy, J.; Boni, C.; Cortes-Funes, H.; Cervantes, A.; Freyer, G. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J. Clin. Oncol. 2000, 18, 2938–2947. [Google Scholar] [PubMed]
- Ciombor, K.K.; Wu, C.; Goldberg, R.M. Recent therapeutic advances in the treatment of colorectal cancer. Ann. Rev. Med. 2015, 66, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Li, X.; Wang, W.; Zhang, R.; Deng, L. Metal-N-heterocyclic carbene complexes as anti-tumor agents. Curr. Med. Chem. 2014, 21, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Lammer, A.D.; Cook, M.E.; Sessler, J.L. Synthesis and anti-cancer activities of a water soluble gold (III) porphyrin. J. Porphyr. Phthalocyanines 2015, 19, 398–403. [Google Scholar] [CrossRef] [PubMed]
- Lum, C.T.; Wong, A.S.-T.; Lin, M.C.M.; Che, C.-M.; Sun, R.W.-Y. A gold (III) porphyrin complex as an anti-cancer candidate to inhibit growth of cancer-stem cells. Chem. Commun. 2013, 49, 4364–4366. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, S.M.; Frezza, M.; Dou, Q.P. New applications of old metal-binding drugs in the treatment of human cancer. Front. Biosci. 2012, 4, 375. [Google Scholar] [CrossRef]
- Chaffman, M.; Brogden, R.N.; Heel, R.C.; Speight, T.M.; Avery, G.S. Auranofin. Drugs 1984, 27, 378–424. [Google Scholar] [CrossRef] [PubMed]
- Roder, C.; Thomson, M.J. Auranofin: Repurposing an old drug for a golden new age. Drugs R&D 2015, 15, 13–20. [Google Scholar]
- Marzano, C.; Gandin, V.; Folda, A.; Scutari, G.; Bindoli, A.; Rigobello, M.P. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Rad. Biol. Med. 2007, 42, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Ortego, L.; Cardoso, F.; Martins, S.; Fillat, M.F.; Laguna, A.; Meireles, M.; Villacampa, M.D.; Gimeno, M.C. Strong inhibition of thioredoxin reductase by highly cytotoxic gold (I) complexes. DNA binding studies. J. Inorg. Biochem. 2014, 130, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.; Machado, R.C.; Grazul, R.M.; Lopes, M.T.P.; Correa, C.C.; dos Santos, H.F.; de Almeida, M.V.; Silva, H. Novel antitumor adamantane-azole gold (I) complexes as potential inhibitors of thioredoxin reductase. J. Biol. Inorg. Chem. 2016, 21, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Galassi, R.; Burini, A.; Ricci, S.; Pellei, M.; Rigobello, M.P.; Citta, A.; Dolmella, A.; Gandin, V.; Marzano, C. Synthesis and characterization of azolate gold (I) phosphane complexes as thioredoxin reductase inhibiting antitumor agents. Dalton Trans. 2012, 41, 5307–5318. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, P.A.; Carlson, B.A.; Yoo, M.-H.; Naranjo-Suarez, S.; Xu, X.-M.; He, Y.; Asaki, E.; Seifried, H.E.; Reinhold, W.C.; Davis, C.D. The 15 kDa selenoprotein and thioredoxin reductase 1 promote colon cancer by different pathways. PLoS ONE 2015, 10, e0124487. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, D.F.D.; Abderrazak, A.; El Hadri, K.; Simmet, T.; Rouis, M. The thioredoxin system as a therapeutic target in human health and disease. Antioxid. Redox signal. 2013, 19, 1266–1303. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Papavassiliou, A.G. The potential of proteasome inhibition in the treatment of colon cancer. Expert Opin. Investig. Drugs 2006, 15, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Huang, H.; Dou, Q.P.; Liu, J. Inhibition of 19s proteasome-associated deubiquitinases by metal-containing compounds. Oncoscience 2015, 2, 457. [Google Scholar] [CrossRef] [PubMed]
- Buac, D.; Schmitt, S.; Ventro, G.; Rani Kona, F.; Dou, Q.P. Dithiocarbamate-based coordination compounds as potent proteasome inhibitors in human cancer cells. Mini Rev. Med. Chem. 2012, 12, 1193–1201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Frezza, M.; Milacic, V.; Ronconi, L.; Fan, Y.; Bi, C.; Fregona, D.; Dou, Q.P. Inhibition of tumor proteasome activity by gold-dithiocarbamato complexes via both redox-dependent and-independent processes. J. Cell. Biochem. 2010, 109, 162–172. [Google Scholar] [CrossRef] [PubMed]
- García-Moreno, E.; Gascón, S.; Rodriguez-Yoldi, M.J.; Cerrada, E.; Laguna, M. S-propargylthiopyridine phosphane derivatives as anticancer agents: Characterization and antitumor activity. Organometallics 2013, 32, 3710–3720. [Google Scholar] [CrossRef]
- García-Moreno, E.; Gascón, S.; Atrián-Blasco, E.; Rodriguez-Yoldi, M.J.; Cerrada, E.; Laguna, M. Gold (I) complexes with alkylated PTA (1,3,5-triaza-7-phosphaadamantane) phosphanes as anticancer metallodrugs. Eur. J. Med. Chem. 2014, 79, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Moreno, E.; Gascón, S.; Garcia de Jalon, J.; Romanos, E.; Jesus Rodriguez-Yoldi, M.; Cerrada, E.; Laguna, M. In vivo anticancer activity, toxicology and histopathological studies of the thiolate gold (I) complex [Au(Spyrimidine)(PTA-CH2Ph)]Br. Anti-Cancer Agents Med. Chem. 2015, 15, 773–782. [Google Scholar] [CrossRef]
- García-Moreno, E.; Tomás, A.; Atrián-Blasco, E.; Gascón, S.; Romanos, E.; Rodriguez-Yoldi, M.J.; Cerrada, E.; Laguna, M. In vitro and in vivo evaluation of organometallic gold (I) derivatives as anticancer agents. Dalton Trans. 2016, 45, 2462–2475. [Google Scholar] [CrossRef] [PubMed]
- Atrián-Blasco, E.; Gascón, S.; Rodríguez-Yoldi, M.J.; Laguna, M.; Cerrada, E. Synthesis of gold (I) derivatives bearing alkylated 1,3,5-triaza-7-phosphaadamantane as selective anticancer metallodrugs. Eur. J. Inorg. Chem. 2016, 2791–2803. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| miRNA Name | Target | Function of miRNA | References |
|---|---|---|---|
| miR-34a | SIRT1, FMNL2 and E2F5 | Inhibition and induction of p53 acetylation | [53,54] |
| miR143 | DNMT, KRAS | Induction of cell proliferation | [55] |
| miR135 | APC | Suppression of WNT pathway | [55] |
| miR-29 | DNMT 3A and 3B | Reduction of methylation | [56] |
| miR-21 | PDCD4 | Invasion and metastasis promotion | [57] |
| miR-345 | BAG | Induction of cell proliferation and invasion | [58] |
| miR-148b | CCK2R | Induction of cell proliferation | [59] |
| Let-7c | KRAS, MMP11 and PBX3 | Metastasis induction | [60] |
| Let-7a | Np95 ICBP90 RING finger | Induction of cell proliferation | [61] |
| miR-499-5p | FOXO4 and PDCD4 | Induction of metastasis | [62] |
| miR-92 | KLF4 | Promotion of cell growth and migration | [63] |
| miR-126 | SPRED1, PIK2R2/P85-β | Inhibition of cell proliferation, migration and invasion | [64] |
| miR-320 | FOXO4 and PDCD4 | Inhibition of cell proliferation | [65] |
| miR-200 family | JNK2 | Inhibition of tumour growth and metastasis and induction of sensitivity to chemotherapeutic drugs | [66] |
| miR-9 | TM4SF1 | Suppression of cell migration and invasion | [67] |
| miR-503 | calcium-sensing receptor | Induction of proliferation migration and invasion | [68] |
| miR-222 | MST3 | Induction of invasion and migration | [69] |
| miR-181b | RASSF1A | Induce proliferation and enhance cell survival | [70] |
| miR-497 | VEGFA | Inhibition of invasion and metastasis | [71] |
| miR-152 | PIK3R3 | Tumour suppressor | [72] |
| miR-187 | SOX4, NT5E and PTK6 | Inactivation of TGF-β pathway and prevention of EMT (epithelial to mesenchymal transition) | [73] |
| miR-519 | Orai1 | Tumour suppression | [74] |
| miR-155 | HMG-box transcription factor 1 | Tumour suppressor by induction of WNT/β-catenin pathway | [75] |
| miR-497 | KSR1 | Tumour growth inhibition and enhancement of chemo sensitivity | [76] |
| miR-375 | Bcl-2 | Inhibition of tumour progression | [77] |
| miR-1246 | CCNG2 | Induction of cell growth and metastasis | [78] |
| miR-140-5p | VEGFA | Inhibition of tumour progression | [78] |
| miR-144 | GSPT1 | Inhibition of proliferation and migration | [79] |
| miR-638 | Phospholipase D1 | Inhibition of cell proliferation | [80] |
| miR-99b-5p | mTOR | Inhibition of metastasis formation | [66] |
| miR-101 | SphK1 | Inhibition of cell growth and increase of paclitaxel chemo-sensitivity | [81] |
| miR-20a | TIMP-2 | Induction of epithelial-to-mesenchymal transition (EMT) | [82] |
| miR-409-3p | GAB1 | Inhibition of tumour progression and metastasis | [83] |
| LncRNA | Locus | Size (kB) | Dysfunction Type | Normal Function | Contribution to Cancer | References |
|---|---|---|---|---|---|---|
| H19 | Chr11p15.5 | 2.3 | Overexpression | Regulation of growth during development targeting Igf2 | Downregulation of the tumour suppressor RB | [88,89,90] |
| HOTAIR | Chr12q13.3 | 2.2 | Overexpression | Epigenetic silencing of gene expression | Reprogramming of chromatin state and induction of metastatic progression | [91,92] |
| MALAT1 | Chr11q13.1 | 7 | Overexpression | Alternative splicing regulation | Increase of abnormal mitosis, invasion and metastasis and induction of cell death resistance | [93] |
| HULC | Chr6p24.3 | 0.5 | Overexpression | Sponge for miR-372 and indirect upregulation of PKA and activation of CREB | Upregulation of Prkacb (catalytic subunit of PKA) | [94] |
| MEG3 | Chr14q32 | 1.6–1.8 | Downregulation | Tumour suppressor. It activates p53, inhibits cell proliferation and controls gene imprinting | Downregulation of p53, apoptosis inhibition and induction of proliferation | [95] |
| CCAT1 | Chr8q24.21 | 2.6 | Overexpression | Enhancer region for cMYC. Maintenance of the chromatin looping between MYC promoter and its enhancer | Induction of MYC expression and inhibition of G1 arrest. Enhancement cell proliferation and migration | [96,97] |
| CCAT2 | Chr8q24 | 0.4 | Overexpression SNP rs6983267 | Upregulation of MYC and enhancement of WNT signalling pathway through TCF7L2 | Induction of cell proliferation, invasion and chromosomal instability | [98] |
| CRNDE | Chr16:hCG_1815491 | 10 | Overexpression | Scaffold for regulatory complexes | Contribution to Warburg effect. Increase of CRC risk | [99] |
| LOC285194 | Chr3q13.31 | 2.1 | Downregulation | Unknown | Decrease of cell migration and metastasis | [100] |
| OCC-1 | Chr12121.1 | 1.2–1.3 | Overexpression | Unknown | Induction of cell proliferation and apoptosis resistance | [101] |
| lincRNA-p21 | - | 3.1 | Downregulation | Binds to hnRNP-K and repress genes transcriptionally regulated by p53. It is necessary for p53-dependent apoptotic induction but not for cell-cycle arrest | Induction of apoptosis evasion, invasion and enhancement of Warburg effect | [102] |
| LIT1 | Chr11q15.5 | 91 | Loss of imprinting | Organization of a tissue/lineage-specific nuclear domain involved in epigenetic silencing of the Kcnq1 imprinting control region | Unknown | [103] |
| PTENP1 | Chr9q13.3 | 3.9 | Downregulation | Decoy for miRNA targeting PTEN | Reduction of PTEN level and enhancement of cell growth | [104] |
| MYLKP1 | Chr3p12.3 | 106 | Overexpression | Pseudogen | Induction of proliferation | [105] |
| pou5f1p1 (OCT4) | Chr8q24 | 0.4 | Overexpression | Pseudogen | Increased of risk of CRC | [106] |
| UCA1 | Chr19p13.12 | 1.4, 2.2, 2.7 | Overexpression | Embryonic development | Induction of resistance to drug-induced apoptosis | [107] |
| PCAT1 | Chr8p24 | 1.9 | Overexpression | BRCA2 inhibition | Regulation of cell response to genotoxic stress and impairing of DNA damage repair. High levels are associated with poorer survival rate | [108,109,110] |
| PRNCR1 | Chr8p24 | 13 | Overexpression | Binding to the androgen receptor and enhancement of both androgen-receptor-mediated gene activation and proliferation | Increase of cell proliferation | [72,111,112] |
| LET | Chr15q24.1 | 2.3 | Downregulation | Downregulation of hypoxia signalling by decreasing HIF1 stability. Induction of NF90 ubiquitination and Go/G1 arrest | Induction of metastasis | [113] |
| ncRAN | Chr17q25.1 | 2.3 | Overexpression | Unknown | Enhancement of cell migration and invasion | [114] |
| PVT1 | Chr8p24.21 | >300 | Overexpression | Regulation of C-MYC | Anti-apoptotic activity. Increase of cell proliferation and cell-cycle progression | [115,116] |
| Molecular Marker Type | Biomarker | Contribution to Cancer | Predictive Use | Samples Used for the Test | Status | References |
|---|---|---|---|---|---|---|
| DNA | Microsatellite instability (MSI) test. Panel of mononucleotide marker (Bat-25, Bat-26, NR-21, NR-24, MONO-27), ≥30% of unstable loci are considered MSI tumours. | Accumulation of alteration in highly repeated DNA sequences | For MSI+ tumours: Prognosis: good, aggressively: low, treatment: lack of response to 5-FU, good response to irinotecan | Tumour-based samples | In use | [183,184,185] |
| - | KRAS, NRAS | Proliferation enhancement through EGFR-signalling activation | If mutated: Prognosis: bad and poor survival (codon 12 and 13). Treatment: limited response to EGFR | Tumour-based samples, stool | In use for tumour-based samples and under evaluation for stool | [186,187,188] |
| BRAF | Proliferation enhancement through EGFR-signalling activation | If mutated: Classification of CRC: sporadic, Prognosis: poor, Treatment: limited response to EGFR-targeted therapy. | Tumour-based samples | In use | [189,190,191] | |
| CpG Island Methylator Phenotype. e.g., Vimentin methylation. | Transcriptional regulation which lead to colorectal carcinogenesis | Classification of CRC in CIMP, Presence of BRAF mutations | Tumour-based samples, stool, blood samples | Under evaluation in tumour samples and in use for stool | [192,193] | |
| Integrity of cell-free DNA (cfDNA) | Apoptosis | Diagnosis and monitoring | Blood sample | Under evaluation | [194] | |
| RNA | gene microarray and gene panels of RNA | Unknown | CRC diagnosis evaluation of relapse risk | Tumour-based samples, stool, blood | Clinical validation | [195,196] |
| miRNA biomarker panel. e.g., miR-21, miR-106a | Unknown | Diagnosis and prognosis | Tumour-based samples, stool, blood | Clinical validation | [197,198] | |
| EGFR ligand biomarker panel (amphiregulin, epiregulin, DUSP6 and SLC26A3) | Proliferation enhancement through EGFR-signalling activation | Response to EGFR-targeted therapy | Tumour-based samples | Under evaluation | [199,200,201] | |
| Protein | Tumour-specific protein determination. e.g., Calprotectin, CEA, DAF, CA19-9 | Unknown | Diagnosis, prognosis, monitoring | Stool, blood | Clinical validation | [202,203,204,205,206] |
| Others | Circulating nucleic acids, proteins and tumour cells | Unknown | Diagnosis, monitoring | Blood | Clinical validation | [207,208,209] |
| Assay | Name of the Assay | DNA Markers Used | Type of Test | References |
|---|---|---|---|---|
| ColonSentry® (GeneNews, (Toronto, ON, Canada)) | Determination of relative risk to suffer CRC | ANXA3, CLECD4, LMNB1, PRRG4, TNFAIP6, VNN1, IL2RB | qRT-PCR | [211,212] |
| Oncotype DX® Colon Cancer Assay (Genomic Health, Inc., Redwood City, CA, USA) | Prediction of recurrence in individuals with stage II CRC following surgery | 7 Genes associated with CRC recurrence (Ki-67, C-MYC, MYBL2, FAP, BGN, INHBA, GADD45B,) and 5 reference genes (ATPSE, PGK1, GPX1, UBB, VDAC2) | qRT-PCR | [213,214] |
| ColoPrint® (Agendia, BV, Amsterdam, The Netherland) | Determination of risk of distant recurrence of the disease in individuals with stage II and III colon cancer | MCTP1, LAMA3, CTSC, PYROXD1, EDEM1, IL2RB, ZNF697, SLC6A11, IL2RA, CYFIP2, PIM3, LIF, PLIN3, HSD3B1, ZBED4, PPARA, THNSL2, CA4388O2 | Microarray | [215,216] |
| Colorectal Cancer DSA® (Almac Diagnostics, Craigavon, UK) | Risk of CRC recurrence within 5 years | ABCC3, FGF1, ISG15, OXNAD1, PPP2CA, PRKACB, TP53INP1, ARHGAP18, BEST1, FKBP5, KITLG, LAMP3, MRPS31, NPM3 | Microarray | [217] |
| GeneFx Colon® (Precision Therapeutics, Pittsburgh, PA, USA) | Risk of CRC recurrence within 5 years | - | 634-transcript DNA microarray-based gene signature | [218] |
| OncoDefender-CRC® (Everist Genomics, Ann Arbor, MI, USA) | Risk of recurrence of cancer in individuals of stage I or II colon cancer or stage I rectal cancer. | BMI1, ETV6, H3F3B, RPS10 | qRT-PCR | [219,220] |
| Previstage (DiagnoCure, Quebec City, QC, Canada) | Identification of patients with low risk of recurrence | Quantification of GCC mRNA | qRT-PCR | [221] |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mármol, I.; Sánchez-de-Diego, C.; Pradilla Dieste, A.; Cerrada, E.; Rodriguez Yoldi, M.J. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. Int. J. Mol. Sci. 2017, 18, 197. https://doi.org/10.3390/ijms18010197
Mármol I, Sánchez-de-Diego C, Pradilla Dieste A, Cerrada E, Rodriguez Yoldi MJ. Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. International Journal of Molecular Sciences. 2017; 18(1):197. https://doi.org/10.3390/ijms18010197
Chicago/Turabian StyleMármol, Inés, Cristina Sánchez-de-Diego, Alberto Pradilla Dieste, Elena Cerrada, and María Jesús Rodriguez Yoldi. 2017. "Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer" International Journal of Molecular Sciences 18, no. 1: 197. https://doi.org/10.3390/ijms18010197
APA StyleMármol, I., Sánchez-de-Diego, C., Pradilla Dieste, A., Cerrada, E., & Rodriguez Yoldi, M. J. (2017). Colorectal Carcinoma: A General Overview and Future Perspectives in Colorectal Cancer. International Journal of Molecular Sciences, 18(1), 197. https://doi.org/10.3390/ijms18010197