
Comprehensive Profiling of Circular RNAs in Goat Dermal Papilla Cells and Prediction of Their Modulatory Roles in Hair Growth

 ,
,
Abstract
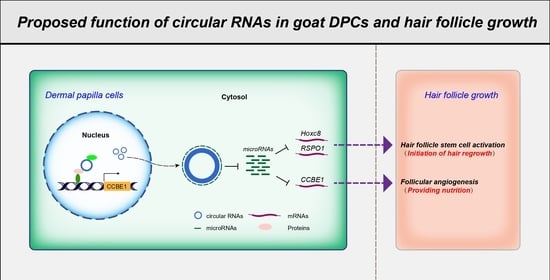
:
1. Introduction
2. Materials and Methods
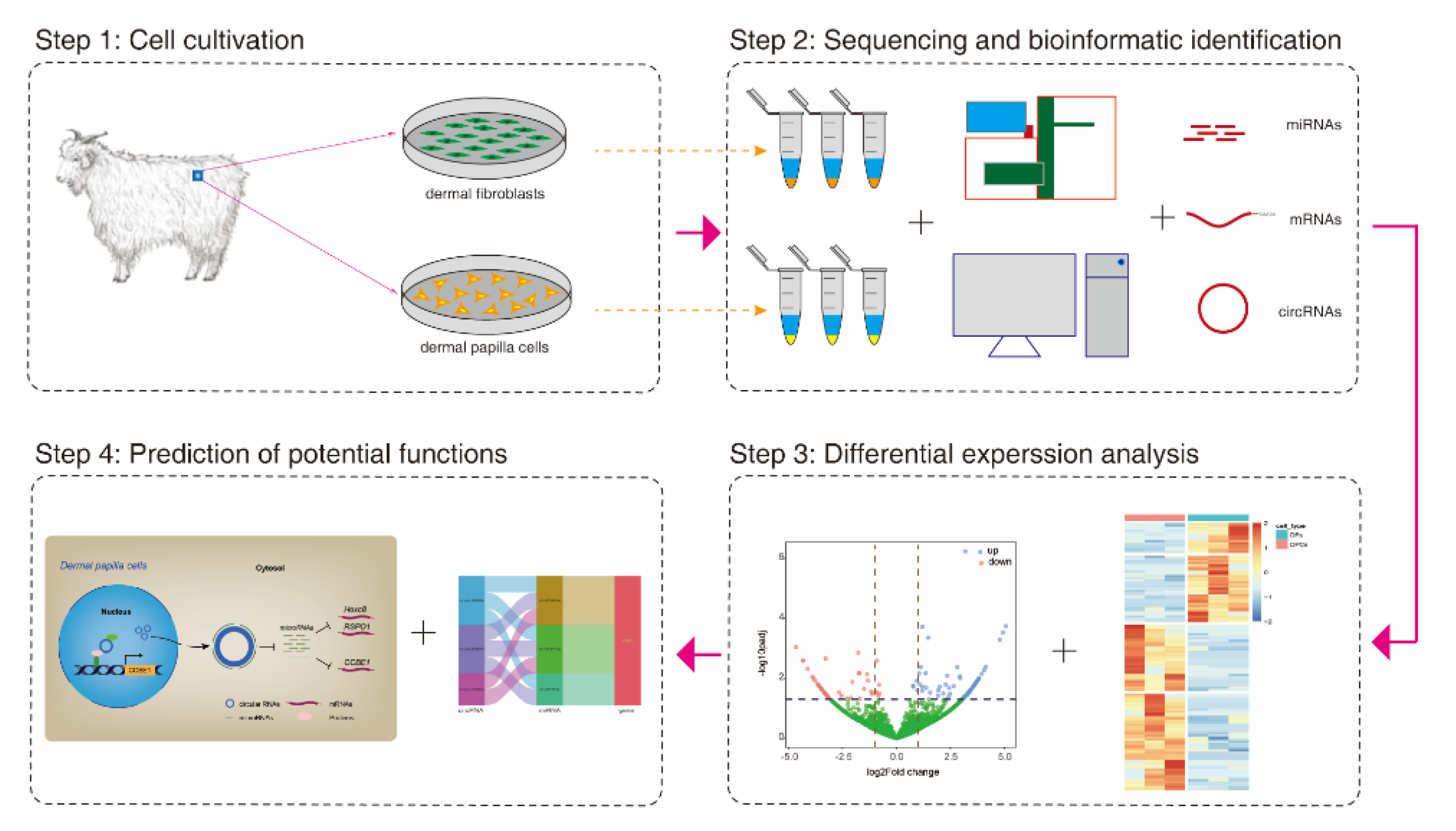
2.1. Animals and Cell Culture
2.2. Sequencing Library Construction and Reference Genome Mapping
2.3. CircRNAs Identification
2.4. Normalization of circRNAs Abundance and Differential Expression Analysis
2.5. Gene Ontology and KEGG Enrichment Analysis
2.6. Prediction of miRNA Binding Sites on circRNAs and mRNAs
2.7. CircRNAs-miRNAs-mRNAs Interplay Network Construction
3. Results
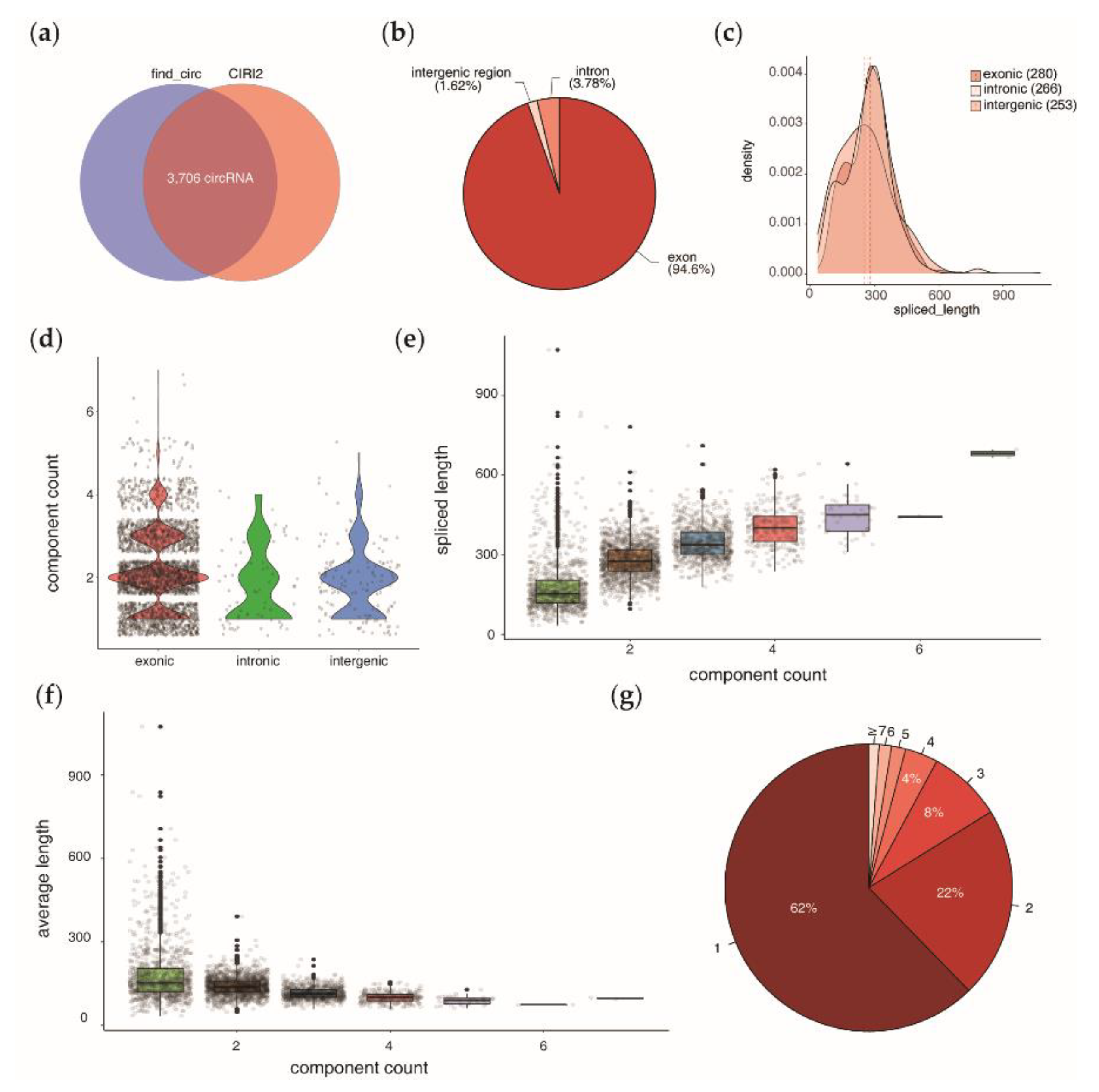
3.1. Bioinformatic Identification and Genomic Feature Characterization of Circular RNAs
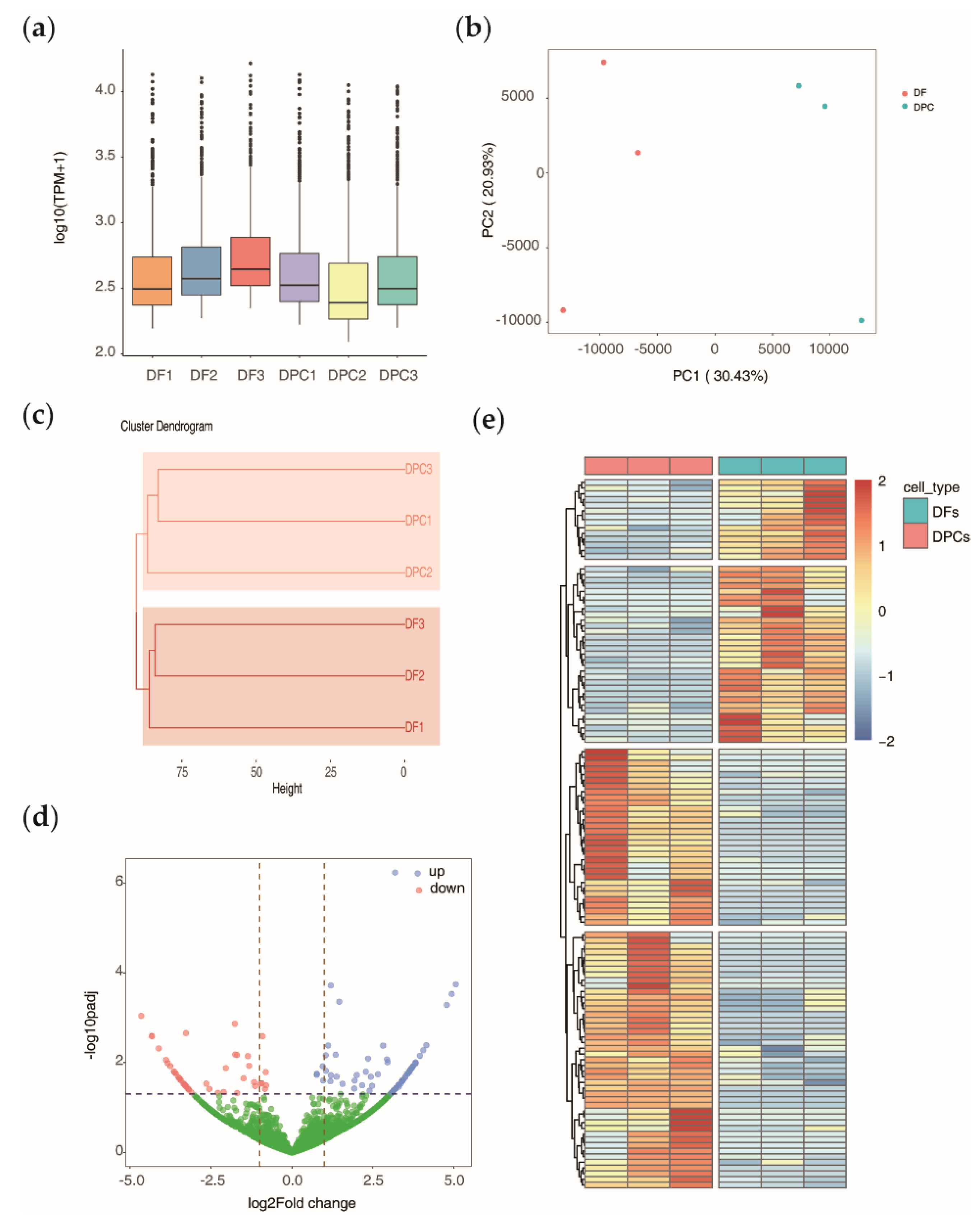
3.2. Global Expression Pattern and Differential Expression Analysis of circRNAs
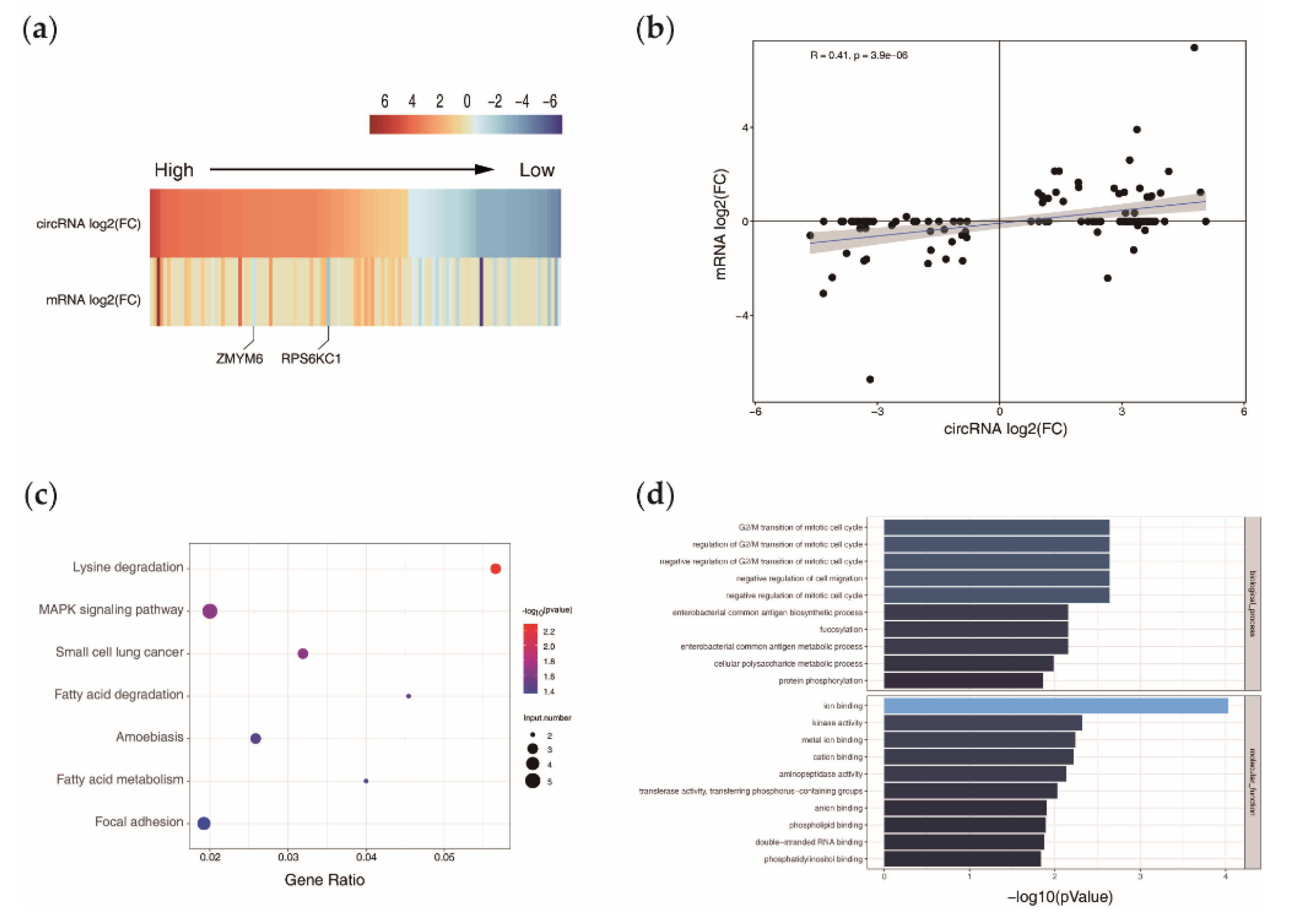
3.3. Relationship of circRNAs Expression with Their Host Genes
3.4. Functional Enrichment Analysis of the Host Genes
3.5. Screening of circRNAs Acting as ceRNAs and Their Regulatory Relationships with Functional Genes in Goat DPCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
| DPCs | dermal papilla cells |
| DFs | dermal fibroblasts |
| HOXC8 | homeobox C8 |
| RSPO1 | R-spondin 1 |
| CCBE1 | collagen and calcium-binding EGF domains 1 |
| PTEN | phosphatase and tensin homolog |
| CDK19 | cyclin-dependent kinase 19 |
| Arhgap5 | Rho GTPase activating protein 5 |
References
- Yang, C.-C.; Cotsarelis, G. Review of hair follicle dermal cells. J. Dermatol. Sci. 2010, 57, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.A. The Dermal Papilla: An Instructive Niche for Epithelial Stem and Progenitor Cells in Development and Regeneration of the Hair Follicle. Cold Spring Harb. Perspect. Med. 2014, 4, a015180. [Google Scholar] [CrossRef] [PubMed]
- Ramos, R.; Guerrero-Juarez, C.F.; Plikus, M.V. Hair Follicle Signaling Networks: A Dermal Papilla–Centric Approach. J. Investig. Dermatol. 2013, 133, 2306–2308. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Jiang, K.; Xu, Z.; Huang, H.; Qian, N.; Lu, Z.; Chen, D.; Di, R.; Yuan, T.; Du, Z.; et al. Hoxc-Dependent Mesenchymal Niche Heterogeneity Drives Regional Hair Follicle Regeneration. Cell Stem Cell 2018, 23, 487–500.e6. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, S.; Zhang, H.-S.; Deng, Z.-L.; Zhao, H.-S.; Zhao, Q.; Lei, X.-H.; Ning, L.-N.; Cao, Y.-J.; Wang, H.-B.; et al. Exogenous R-Spondin1 Induces Precocious Telogen-to-Anagen Transition in Mouse Hair Follicles. Int. J. Mol. Sci. 2016, 17, 582. [Google Scholar] [CrossRef] [PubMed]
- Yano, K.; Brown, L.F.; Detmar, M. Control of hair growth and follicle size by VEGF-mediated angiogenesis. J. Clin. Investig. 2001, 107, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Mecklenburg, L.; Tobin, D.J.; Müller-Röver, S.; Handjiski, B.; Wendt, G.; Peters, E.M.J.; Pohl, S.; Moll, I.; Paus, R. Active Hair Growth (Anagen) is Associated with Angiogenesis. J. Investig. Dermatol. 2000, 114, 909–916. [Google Scholar] [CrossRef]
- Odorisio, T.; Cianfarani, F.; Failla, C.M.; Zambruno, G. The placenta growth factor in skin angiogenesis. J. Dermatol. Sci. 2006, 41, 11–19. [Google Scholar] [CrossRef]
- Lee, E.C.S.; Elhassan, S.A.M.; Lim, G.P.L.; Kok, W.H.; Tan, S.W.; Leong, E.N.; Tan, S.H.; Chan, E.W.L.; Bhattamisra, S.K.; Rajendran, R.; et al. The roles of circular RNAs in human development and diseases. Biomed. Pharmacother. 2019, 111, 198–208. [Google Scholar] [CrossRef]
- Arcinas, C.; Tan, W.; Fang, W.; Desai, T.P.; Teh, D.C.S.; Degirmenci, U.; Xu, D.; Foo, R.; Sun, L. Adipose circular RNAs exhibit dynamic regulation in obesity and functional role in adipogenesis. Nat. Metab. 2019, 1, 688–703. [Google Scholar] [CrossRef]
- Lu, Q.; Liu, T.; Feng, H.; Yang, R.; Zhao, X.; Chen, W.; Jiang, B.; Qin, H.; Guo, X.; Liu, M.; et al. Circular RNA circSLC8A1 acts as a sponge of miR-130b/miR-494 in suppressing bladder cancer progression via regulating PTEN. Mol. Cancer 2019, 18, 111. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, L.; Chen, L.-L. The Biogenesis, Functions, and Challenges of Circular RNAs. Mol. Cell 2018, 71, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-L. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat. Rev. Mol. Cell Biol. 2020, 21, 475–490. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Luo, H.; He, J.; Huang, X.; Chen, S.; Fu, X.; Zeng, W.; Tian, Y.; Liu, S.; Li, C.-J.; et al. Comprehensive transcriptome and methylome analysis delineates the biological basis of hair follicle development and wool-related traits in Merino sheep. BMC Biol. 2021, 19, 197. [Google Scholar] [CrossRef] [PubMed]
- Shang, F.; Wang, Y.; Ma, R.; Di, Z.; Wu, Z.; Hai, E.; Rong, Y.; Pan, J.; Liang, L.; Wang, Z.; et al. Expression Profiling and Functional Analysis of Circular RNAs in Inner Mongolian Cashmere Goat Hair Follicles. Front. Genet. 2021, 12, 678825. [Google Scholar] [CrossRef]
- Hui, T.; Zheng, Y.; Yue, C.; Wang, Y.; Bai, Z.; Sun, J.; Cai, W.; Zhang, X.; Bai, W.; Wang, Z. Screening of cashmere fineness-related genes and their ceRNA network construction in cashmere goats. Sci. Rep. 2021, 11, 21977. [Google Scholar] [CrossRef]
- Wang, J.; Wu, X.; Kang, Y.; Zhang, L.; Niu, H.; Qu, J.; Wang, Y.; Ji, D.; Li, Y. Integrative analysis of circRNAs from Yangtze River Delta white goat neck skin tissue by high-throughput sequencing (circRNA-seq). Anim. Genet. 2022, 53, 405–415. [Google Scholar] [CrossRef]
- Yin, R.H.; Zhao, S.J.; Jiao, Q.; Wang, Z.Y.; Bai, M.; Fan, Y.X.; Zhu, Y.B.; Bai, W.L. CircRNA-1926 Promotes the Differentiation of Goat SHF Stem Cells into Hair Follicle Lineage by miR-148a/b-3p/CDK19 Axis. Animals 2020, 10, 1552. [Google Scholar] [CrossRef]
- Stoll, L.; Rodríguez-Trejo, A.; Guay, C.; Brozzi, F.; Bayazit, M.B.; Gattesco, S.; Menoud, V.; Sobel, J.; Marques, A.C.; Venø, M.T.; et al. A circular RNA generated from an intron of the insulin gene controls insulin secretion. Nat. Commun. 2020, 11, 5611. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.-O.; Chen, T.; Xiang, J.-F.; Yin, Q.-F.; Xing, Y.-H.; Zhu, S.; Yang, L.; Chen, L.-L. Circular Intronic Long Noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. circRNA Biogenesis Competes with Pre-mRNA Splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Jahoda, C.; Oliver, R.F. The growth of vibrissa dermal papilla cells in vitro. Br. J. Dermatol. 1981, 105, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Bratka-Robia, C.B.; Mitteregger, G.; Aichinger, A.; Egerbacher, M.; Helmreich, M.; Bamberg, E. Primary cell culture and morphological characterization of canine dermal papilla cells and dermal fibroblasts. Vet. Dermatol. 2002, 13, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Kang, D.; Ma, S.; Wang, X.; Gao, Y.; Yang, Y.; Wang, X.; Chen, Y. Integrative analysis reveals ncRNA-mediated molecular regulatory network driving secondary hair follicle regression in cashmere goats. BMC Genom. 2018, 19, 222. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Ma, S.; Wang, Y.; Zhou, G.; Ding, Y.; Yang, Y.; Wang, X.; Zhang, E.; Chen, Y. Synchronous profiling and analysis of mRNAs and ncRNAs in the dermal papilla cells from cashmere goats. BMC Genom. 2019, 20, 512. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, J.; Zhao, F. CIRI: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Mouselimis, L. ClusterR: Gaussian Mixture Models, K-Means, Mini-Batch-Kmeans, K-Medoids and Affinity Propagation Clustering, version 1.2.6; R Package: 2022. Available online: https://CRAN.R-project.org/package=ClusterR (accessed on 10 April 2022).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef]
- Tian, G.-A.; Zhu, C.-C.; Zhang, X.-X.; Zhu, L.; Yang, X.-M.; Jiang, S.-H.; Li, R.-K.; Tu, L.; Wang, Y.; Zhuang, C.; et al. CCBE1 promotes GIST development through enhancing angiogenesis and mediating resistance to imatinib. Sci. Rep. 2016, 6, 31071. [Google Scholar] [CrossRef]
- Zheng, Y.; Hui, T.; Yue, C.; Sun, J.; Guo, D.; Guo, S.; Guo, S.; Li, B.; Wang, Z.; Bai, W. Comprehensive analysis of circRNAs from cashmere goat skin by next generation RNA sequencing (RNA-seq). Sci. Rep. 2020, 10, 516. [Google Scholar] [CrossRef]
- Lv, X.; Chen, W.; Sun, W.; Hussain, Z.; Chen, L.; Wang, S.; Wang, J. Expression profile analysis to identify circular RNA expression signatures in hair follicle of Hu sheep lambskin. Genomics 2020, 112, 4454–4462. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, Q.; Bao, C.; Li, S.; Guo, W.; Zhao, J.; Chen, D.; Gu, J.; He, X.; Huang, S. Circular RNA is enriched and stable in exosomes: A promising biomarker for cancer diagnosis. Cell Res. 2015, 25, 981–984. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, X.; Pei, J.; Chu, M.; Ding, X.; Wu, X.; Liang, C.; Yan, P. CircRNA Expression Profile during Yak Adipocyte Differentiation and Screen Potential circRNAs for Adipocyte Differentiation. Genes 2020, 11, 414. [Google Scholar] [CrossRef]
- Zhao, J.; Shen, J.; Wang, Z.; Bai, M.; Fan, Y.; Zhu, Y.; Bai, W. CircRNA-0100 positively regulates the differentiation of cashmere goat SHF-SCs into hair follicle lineage via sequestering miR-153-3p to heighten the KLF5 expression. Arch. Anim. Breed. 2022, 65, 55–67. [Google Scholar] [CrossRef]
- Rybak-Wolf, A.; Stottmeister, C.; Glažar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Chen, Y.; Hu, S.; Yang, N.; Wang, M.; Liu, M.; Li, J.; Xiao, Y.; Wu, X. Systematic Analysis of Non-coding RNAs Involved in the Angora Rabbit (Oryctolagus cuniculus) Hair Follicle Cycle by RNA Sequencing. Front. Genet. 2019, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, J.; Tian, Y.; Gao, Y.; Dong, X.; Chen, W.; Yuan, X.; Yin, W.; Xu, J.; Chen, K.; et al. CircRNA inhibits DNA damage repair by interacting with host gene. Mol. Cancer 2020, 19, 128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wu, K.; Wang, L.; Wang, Z.; Han, W.; Chen, D.; Wei, Y.; Su, R.; Wang, R.; Liu, Z.; et al. Comparative study on seasonal hair follicle cycling by analysis of the transcriptomes from cashmere and milk goats. Genomics 2020, 112, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Katsuoka, K.; Schell, H.; Hornstein, O.P.; Deinlein, E.; Wessel, B. Comparative morphological and growth kinetics studies of human hair bulb papilla cells and root sheath fibroblasts in vitro. Arch. Dermatol. Res. 1986, 279, 20–25. [Google Scholar] [CrossRef]
- Chew, E.G.Y.; Tan, J.H.J.; Bahta, A.W.; Ho, B.S.-Y.; Liu, X.; Lim, T.C.; Sia, Y.Y.; Bigliardi, P.L.; Heilmann, S.; Wan, A.C.A.; et al. Differential Expression between Human Dermal Papilla Cells from Balding and Non-Balding Scalps Reveals New Candidate Genes for Androgenetic Alopecia. J. Investig. Dermatol. 2016, 136, 1559–1567. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CircRNAs ID | DPCs TPM | DFs TPM | log2 (FC) | p Value | Host Genes |
|---|---|---|---|---|---|
| chi_circ_0001237 | 27.66 | 2.60 | 3.19 | 5.83 × 10−7 | EPDR1 |
| chi_circ_0001124 | 10.42 | 0 | 5.06 | 0.00018033 | RBM33 |
| chi_circ_0005517 | 77.94 | 33.43 | 1.20 | 0.00019107 | MTCL1 |
| chi_circ_0005255 | 9.48 | 0 | 4.93 | 0.00029672 | ADAMTS9 |
| chi_circ_0003390 | 36.68 | 12.84 | 1.46 | 0.00044071 | FNDC3A |
| chi_circ_0002616 | 8.49 | 0 | 4.78 | 0.00052082 | SMOC2 |
| chi_circ_0005862 | 0 | 7.36 | −4.65 | 0.00091454 | HTRA1 |
| chi_circ_0002154 | 5.49 | 19.05 | −1.76 | 0.0013659 | PLIN2 |
| chi_circ_0004069 | 0.67 | 8.68 | −3.27 | 0.0022086 | ENAH |
| chi_circ_0003353 | 0 | 5.74 | −4.33 | 0.0025604 | CDK8 |
| chi_circ_0005056 | 56.58 | 106.67 | −0.91 | 0.0025953 | CEMIP |
| chi_circ_0004639 | 0.00 | 5.69 | −4.32 | 0.0025977 | APPBP2 |
| chi_circ_0005733 | 5.61 | 0 | 4.15 | 0.0041094 | FGFR2 |
| chi_circ_0003643 | 35.55 | 16.39 | 1.12 | 0.0041723 | TASP1 |
| chi_circ_0001957 | 9.21 | 0.94 | 2.81 | 0.0041946 | DPYSL3 |
| chi_circ_0003371 | 0 | 5.05 | −4.11 | 0.0048414 | DCLK1 |
| chi_circ_0004078 | 5.29 | 0 | 4.05 | 0.0053418 | KIF26B |
| chi_circ_0002817 | 3.91 | 13.87 | −1.75 | 0.006652 | PTGDR |
| chi_circ_0003391 | 21.04 | 8.06 | 1.35 | 0.0067238 | FNDC3A |
| chi_circ_0000379 | 4.27 | 14.53 | −1.70 | 0.0068278 | ZMAT3 |
| CircRNAs ID | DPCs_TPM | DFs_TPM | log2 (FC) | p Value | Host Gene |
|---|---|---|---|---|---|
| CiRNAs | |||||
| chi_circ_0005569 | 4.95 | 0 | 3.95 | 0.0070649 | CCBE1 |
| chi_circ_0003704 | 2.93 | 0 | 3.04 | 0.049869 | ZFAND1 |
| chi_circ_0005570 | 48.90 | 24.95 | 0.95 | 0.012148 | CCBE1 |
| Intergenic region-derived circRNAs | |||||
| chi_circ_0000835 | 8.21 | 0 | 3.30 | 0.034524 | NA |
| chi_circ_0004524 | 129.36 | 11.39 | 2.95 | 0.0098427 | NA |
| chi_circ_0006241 | 19.51 | 34.13 | −0.80 | 0.032297 | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, S.; Xu, X.; Wang, X.; Yang, Y.; Shi, Y.; Chen, Y. Comprehensive Profiling of Circular RNAs in Goat Dermal Papilla Cells and Prediction of Their Modulatory Roles in Hair Growth. Agriculture 2022, 12, 1306. https://doi.org/10.3390/agriculture12091306
Ma S, Xu X, Wang X, Yang Y, Shi Y, Chen Y. Comprehensive Profiling of Circular RNAs in Goat Dermal Papilla Cells and Prediction of Their Modulatory Roles in Hair Growth. Agriculture. 2022; 12(9):1306. https://doi.org/10.3390/agriculture12091306
Chicago/Turabian StyleMa, Sen, Xiaochun Xu, Xiaolong Wang, Yuxin Yang, Yinghua Shi, and Yulin Chen. 2022. "Comprehensive Profiling of Circular RNAs in Goat Dermal Papilla Cells and Prediction of Their Modulatory Roles in Hair Growth" Agriculture 12, no. 9: 1306. https://doi.org/10.3390/agriculture12091306