The Emerging Role of the Phosphatidylinositol 3-Kinase/ Akt/Mammalian Target of Rapamycin Signaling Network in Cancer Stem Cell Biology

 ,
, 
{kind=link}
{kind=link}
Abstract
:1. Introduction
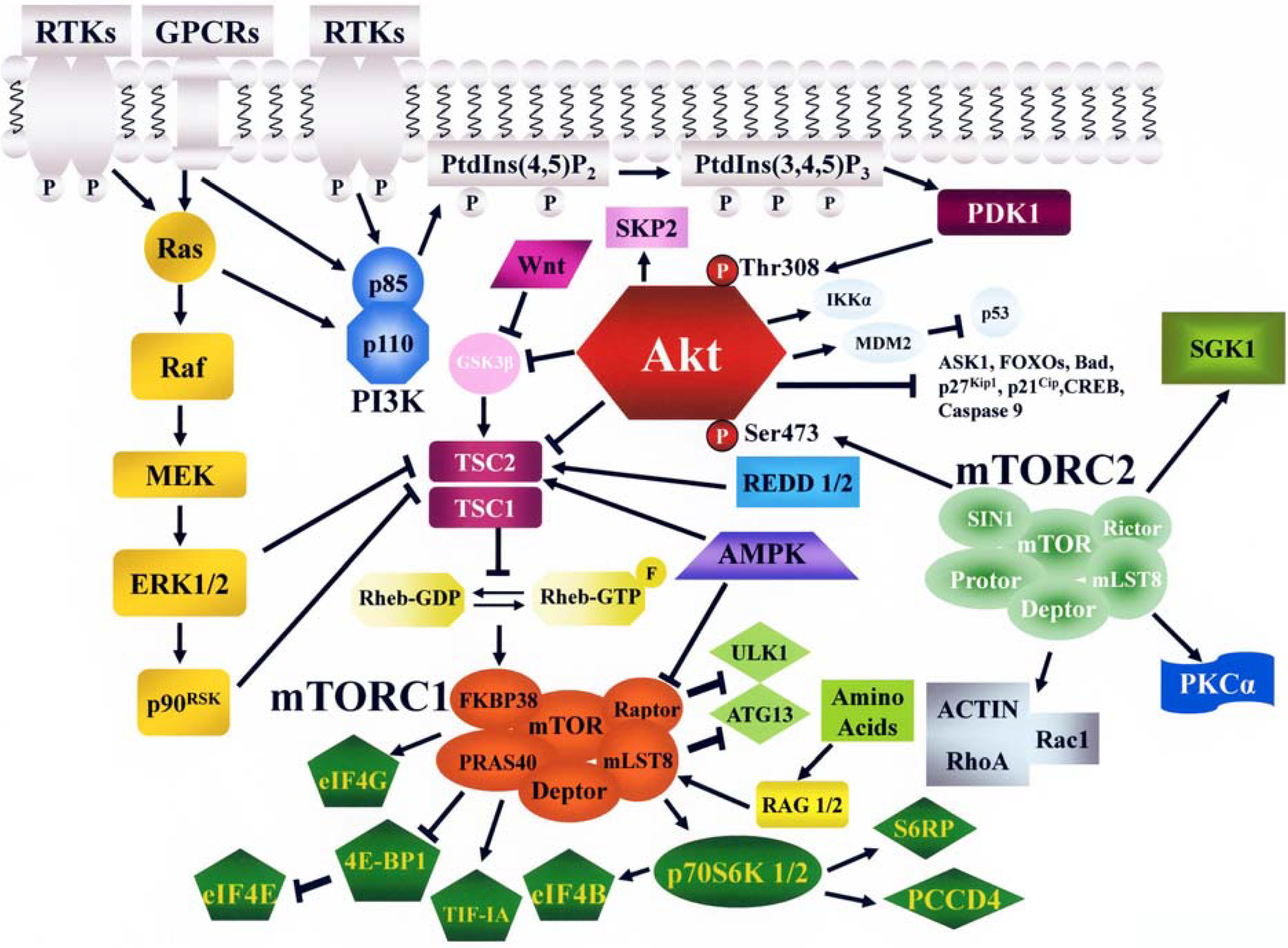
2. The PI3K/Akt/mTOR Signaling Cascade
2.1. PI3K
2.2. Akt
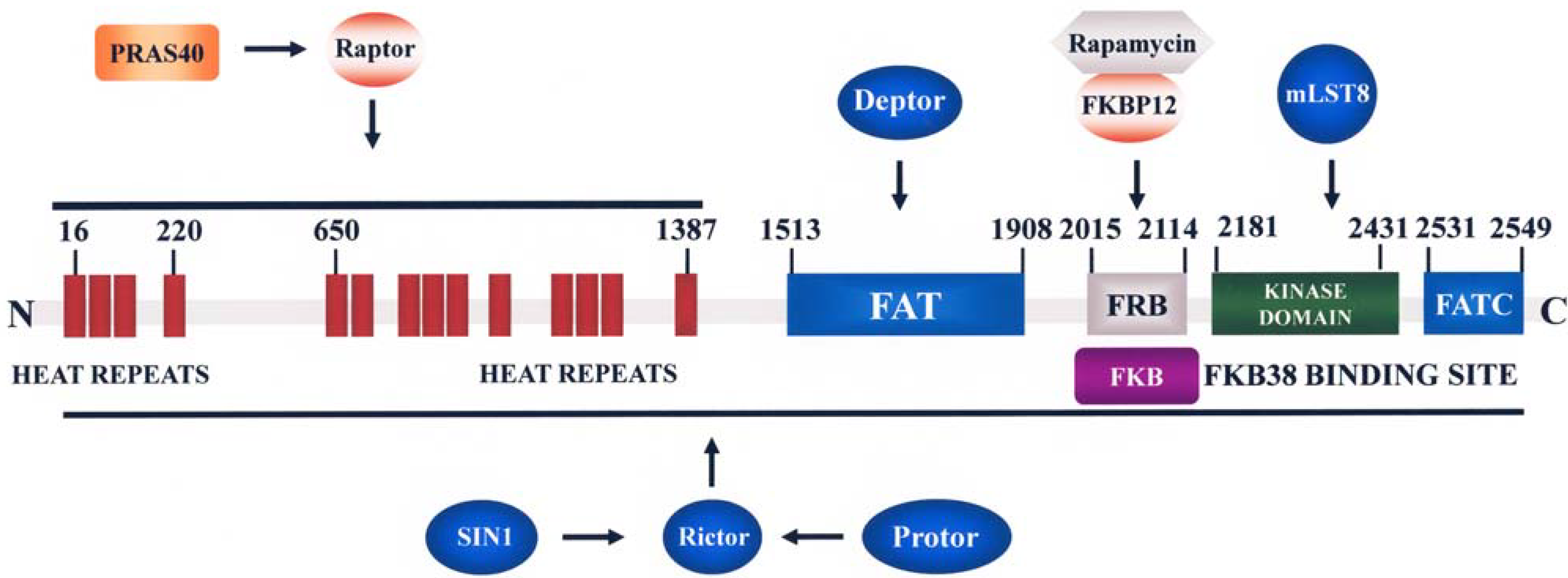
2.3. mTOR


2.3.1. Functions and Regulation of mTORC1
2.3.2. Functions and Regulation of mTORC2
2.3.3. Feedback Loops Linking mTORC1/2 with Akt and MEK/ERK Signaling
3. Negative Regulation of PI3K/Akt/mTOR Signaling
4. PI3K/Akt/mTOR Signaling in Leukemic Stem Cells (LSCs)
5. PI3K/Akt/mTOR Signaling in CSCs
6. Therapeutic Targeting of PI3K/Akt/mTOR Signaling in LSCs and CSCs
7. Conclusions and Future Perspectives
Acknowledgments
References
- Polyak, K.; Hahn, W.C. Roots and stems: stem cells in cancer. Nat. Med. 2006, 12, 296–300. [Google Scholar] [CrossRef]
- Lage, H. An overview of cancer multidrug resistance: A still unsolved problem. Cell. Mol. Life Sci. 2008, 65, 3145–3167. [Google Scholar] [CrossRef]
- Korkaya, H.; Wicha, M.S. Selective targeting of cancer stem cells: A new concept in cancer therapeutics. BioDrugs 2007, 21, 299–310. [Google Scholar] [CrossRef]
- Saito, Y.; Uchida, N.; Tanaka, S.; Suzuki, N.; Tomizawa-Murasawa, M.; Sone, A.; Najima, Y.; Takagi, S.; Aoki, Y.; Wake, A.; Taniguchi, S.; Shultz, L.D.; Ishikawa, F. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat. Biotechnol. 2010, 28, 275–280. [Google Scholar]
- Misaghian, N.; Ligresti, G.; Steelman, L.S.; Bertrand, F.E.; Basecke, J.; Libra, M.; Nicoletti, F.; Stivala, F.; Milella, M.; Tafuri, A.; Cervello, M.; Martelli, A.M.; McCubrey, J.A. Targeting the leukemic stem cell: The Holy Grail of leukemia therapy. Leukemia 2009, 23, 25–42. [Google Scholar] [CrossRef]
- Yuan, T.L.; Cantley, L.C. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef]
- Engelman, J.A. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Cancer 2009, 8, 627–644. [Google Scholar] [CrossRef]
- Franke, T.F. PI3K/Akt: getting it right matters. Oncogene 2008, 27, 6473–6488. [Google Scholar] [CrossRef]
- Jia, S.; Roberts, T.M.; Zhao, J.J. Should individual PI3 kinase isoforms be targeted in cancer? Curr. Opin.Cell Biol. 2009, 21, 199–208. [Google Scholar] [CrossRef]
- Fruman, D.A.; Bismuth, G. Fine tuning the immune response with PI3K. Immunol. Rev. 2009, 228, 253–272. [Google Scholar] [CrossRef]
- Kok, K.; Geering, B.; Vanhaesebroeck, B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem. Sci. 2009, 34, 115–127. [Google Scholar] [CrossRef]
- Falasca, M.; Maffucci, T. Rethinking phosphatidylinositol 3-monophosphate. Biochim. Biophys. Acta 2009, 1793, 1795–1803. [Google Scholar]
- Backer, J.M. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem. J. 2008, 410, 1–17. [Google Scholar] [CrossRef]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Vicencio, J.M.; Criollo, A.; Maiuri, M.C.; Kroemer, G. Anti- and pro-tumor functions of autophagy. Biochim. Biophys. Acta 1793, 1524–1532. [Google Scholar]
- Brazil, D.P.; Yang, Z.Z.; Hemmings, B.A. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem. Sci. 2004, 29, 233–242. [Google Scholar] [CrossRef]
- Chin, Y.R.; Toker, A. Function of Akt/PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cell. Signal. 2009, 21, 470–476. [Google Scholar] [CrossRef]
- Martelli, A.M.; Nyakern, M.; Tabellini, G.; Bortul, R.; Tazzari, P.L.; Evangelisti, C.; Cocco, L. Phosphoinositide 3-kinase/Akt signaling pathway and its therapeutical implications for human acute myeloid leukemia. Leukemia 2006, 20, 911–928. [Google Scholar] [CrossRef]
- Downward, J. PI 3-kinase, Akt and cell survival. Semin. Cell Dev. Biol. 2004, 15, 177–182. [Google Scholar] [CrossRef]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef]
- Memmott, R.M.; Dennis, P.A. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell Signal. 2009, 21, 656–664. [Google Scholar] [CrossRef]
- Dowling, R.J.; Topisirovic, I.; Fonseca, B.D.; Sonenberg, N. Dissecting the role of mTOR: lessons from mTOR inhibitors. Biochim. Biophys. Acta 1804, 433–439. [Google Scholar]
- Jiang, B.H.; Liu, L.Z. Role of mTOR in anticancer drug resistance: Perspectives for improved drug treatment. Drug Resist. Updat. 2008, 11, 63–76. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Oshiro, N.; Yoshino, K.; Hidayat, S.; Tokunaga, C.; Hara, K.; Eguchi, S.; Avruch, J.; Yonezawa, K. Dissociation of raptor from mTOR is a mechanism of rapamycin-induced inhibition of mTOR function. Genes Cells 2004, 9, 359–366. [Google Scholar] [CrossRef]
- Shor, B.; Gibbons, J.J.; Abraham, R.T.; Yu, K. Targeting mTOR globally in cancer: Thinking beyond rapamycin. Cell Cycle 2009, 8, 3831–3837. [Google Scholar] [CrossRef]
- Garcia-Echeverria, C. Allosteric and ATP-competitive kinase inhibitors of mTOR for cancer treatment. Bioorg. Med. Chem. Lett. 2010, 20, 4308–4312. [Google Scholar] [CrossRef]
- Dunlop, E.A.; Tee, A.R. Mammalian target of rapamycin complex 1: Signalling inputs, substrates and feedback mechanisms. Cell. Signal. 2009, 21, 827–835. [Google Scholar] [CrossRef]
- Rosner, M.; Hengstschlager, M. Cytoplasmic and nuclear distribution of the protein complexes mTORC1 and mTORC2: rapamycin triggers dephosphorylation and delocalization of the mTORC2 components rictor and sin1. Hum. Mol. Genet. 2008, 17, 2934–2948. [Google Scholar] [CrossRef]
- Tamburini, J.; Green, A.S.; Chapuis, N.; Bardet, V.; Lacombe, C.; Mayeux, P.; Bouscary, D. Targeting translation in acute myeloid leukemia: A new paradigm for therapy? Cell Cycle 2009, 8, 3893–3899. [Google Scholar] [CrossRef]
- Pause, A.; Belsham, G.J.; Gingras, A.C.; Donze, O.; Lin, T.A.; Lawrence, J.C., Jr.; Sonenberg, N. Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5'-cap function. Nature 1994, 371, 762–767. [Google Scholar] [CrossRef]
- De Benedetti, A.; Graff, J.R. eIF-4E expression and its role in malignancies and metastases. Oncogene 2004, 23, 3189–3199. [Google Scholar] [CrossRef]
- Yuan, X.; Zhou, Y.; Casanova, E.; Chai, M.; Kiss, E.; Grone, H.J.; Schutz, G.; Grummt, I. Genetic inactivation of the transcription factor TIF-IA leads to nucleolar disruption, cell cycle arrest, and p53-mediated apoptosis. Mol. Cell 2005, 19, 77–87. [Google Scholar] [CrossRef]
- Mayer, C.; Zhao, J.; Yuan, X.; Grummt, I. mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability. Genes Dev. 2004, 18, 423–434. [Google Scholar] [CrossRef]
- Renner, O.; Fominaya, J.; Alonso, S.; Blanco-Aparicio, C.; Leal, J.F.; Carnero, A. Mst1, RanBP2 and eIF4G are new markers for in vivo PI3K activation in murine and human prostate. Carcinogenesis 2007, 28, 1418–1425. [Google Scholar] [CrossRef]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef]
- Eskelinen, E.L.; Saftig, P. Autophagy: A lysosomal degradation pathway with a central role in health and disease. Biochim. Biophys. Acta 2009, 1793, 664–673. [Google Scholar]
- Ganley, I.G.; Lam du, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1. ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; Guan, J.L.; Oshiro, N.; Mizushima, N. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef]
- Mavrakis, K.J.; Zhu, H.; Silva, R.L.; Mills, J.R.; Teruya-Feldstein, J.; Lowe, S.W.; Tam, W.; Pelletier, J.; Wendel, H.G. Tumorigenic activity and therapeutic inhibition of Rheb GTPase. Genes Dev. 2008, 22, 2178–2188. [Google Scholar] [CrossRef]
- Wang, L.; Harris, T.E.; Lawrence, J.C., Jr. Regulation of proline-rich Akt substrate of 40 kDa (PRAS40) function by mammalian target of rapamycin complex 1 (mTORC1)-mediated phosphorylation. J. Biol. Chem. 2008, 283, 15619–15627. [Google Scholar] [CrossRef]
- Fonseca, B.D.; Smith, E.M.; Lee, V.H.; MacKintosh, C.; Proud, C.G. PRAS40 is a target for mammalian target of rapamycin complex 1 and is required for signaling downstream of this complex. J. Biol. Chem. 2007, 282, 24514–24524. [Google Scholar] [CrossRef]
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193. [Google Scholar] [CrossRef]
- Jin, T.; George Fantus, I.; Sun, J. Wnt and beyond Wnt: multiple mechanisms control the transcriptional property of β-catenin. Cell. Signal. 2008, 20, 1697–1704. [Google Scholar] [CrossRef]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; Wang, C.Y.; He, X.; MacDougald, O.A.; You, M.; Williams, B.O.; Guan, K.L. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Lopez-Bonet, E.; Menendez, J.A. AMPK: Evidence for an energy-sensing cytokinetic tumor suppressor. Cell Cycle 2009, 8, 3679–3683. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Reiling, J.H.; Hafen, E. The hypoxia-induced paralogs Scylla and Charybdis inhibit growth by down-regulating S6K activity upstream of TSC in Drosophila. Genes Dev. 2004, 18, 2879–2892. [Google Scholar] [CrossRef] [Green Version]
- Sofer, A.; Lei, K.; Johannessen, C.M.; Ellisen, L.W. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol. Cell. Biol. 2005, 25, 5834–5845. [Google Scholar] [CrossRef]
- DeYoung, M.P.; Horak, P.; Sofer, A.; Sgroi, D.; Ellisen, L.W. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. 2008, 22, 239–251. [Google Scholar] [CrossRef]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef]
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crosby, K.; Sheen, J.H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 2009, 15, 148–159. [Google Scholar] [CrossRef]
- Garcia-Martinez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, Y.; Bersenev, A.; O'Brien, W.T.; Tong, W.; Emerson, S.G.; Klein, P.S. Pivotal role for glycogen synthase kinase-3 in hematopoietic stem cell homeostasis in mice. J. Clin. Invest. 2009, 119, 519–3529. [Google Scholar]
- Hernandez-Negrete, I.; Carretero-Ortega, J.; Rosenfeldt, H.; Hernandez-Garcia, R.; Calderon-Salinas, J.V.; Reyes-Cruz, G.; Gutkind, J.S.; Vazquez-Prado, J. P-Rex1 links mammalian target of rapamycin signaling to Rac activation and cell migration. J. Biol. Chem. 2007, 282, 23708–23715. [Google Scholar] [CrossRef]
- Huang, J.; Manning, B.D. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 2009, 37, 217–222. [Google Scholar] [CrossRef]
- Shah, O.J.; Wang, Z.; Hunter, T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol. 2004, 14, 1650–1656. [Google Scholar] [CrossRef]
- Bhaskar, P.T.; Hay, N. The two TORCs and Akt. Dev. Cell 2007, 12, 487–502. [Google Scholar] [CrossRef]
- Lang, S.A.; Hackl, C.; Moser, C.; Fichtner-Feigl, S.; Koehl, G.E.; Schlitt, H.J.; Geissler, E.K.; Stoeltzing, O. Implication of RICTOR in the mTOR inhibitor-mediated induction of insulin-like growth factor-I receptor (IGF-IR) and human epidermal growth factor receptor-2 (Her2) expression in gastrointestinal cancer cells. Biochim. Biophys. Acta 1803, 435–442. [Google Scholar]
- Xu, X.; Sarikas, A.; Dias-Santagata, D.C.; Dolios, G.; Lafontant, P.J.; Tsai, S.C.; Zhu, W.; Nakajima, H.; Nakajima, H.O.; Field, L.J.; Wang, R.; Pan, Z.Q. The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1 for ubiquitin-dependent degradation. Mol. Cell 2008, 30, 403–414. [Google Scholar] [CrossRef]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; Papa, A.; Nardella, C.; Cantley, L.C.; Baselga, J.; Pandolfi, P.P. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Invest. 2008, 118, 3065–3074. [Google Scholar]
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004, 167, 35–41. [Google Scholar] [CrossRef]
- Dibble, C.C.; Asara, J.M.; Manning, B.D. Characterization of Rictor phosphorylation sites reveals direct regulation of mTOR complex 2 by S6K1. Mol. Cell. Biol. 2009, 29, 5657–5670. [Google Scholar] [CrossRef]
- Boulbes, D.; Chen, C.H.; Shaikenov, T.; Agarwal, N.K.; Peterson, T.R.; Addona, T.A.; Keshishian, H.; Carr, S.A.; Magnuson, M.A.; Sabatini, D.M.; Sarbassov, D.D. Rictor Phosphorylation on the Thr-1135 Site Does Not Require Mammalian Target of Rapamycin Complex 2. Mol. Cancer Res. 2010, 8, 896–906. [Google Scholar] [CrossRef] [Green Version]
- Tamburini, J.; Chapuis, N.; Bardet, V.; Park, S.; Sujobert, P.; Willems, L.; Ifrah, N.; Dreyfus, F.; Mayeux, P.; Lacombe, C.; Bouscary, D. Mammalian target of rapamycin (mTOR) inhibition activates phosphatidylinositol 3-kinase/Akt by up-regulating insulin-like growth factor-1 receptor signaling in acute myeloid leukemia: rationale for therapeutic inhibition of both pathways. Blood 2008, 111, 379–382. [Google Scholar] [CrossRef]
- Keniry, M.; Parsons, R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene 2008, 27, 5477–5485. [Google Scholar] [CrossRef]
- Stiles, B.L. Phosphatase and tensin homologue deleted on chromosome 10: Extending its PTENtacles. Int. J. Biochem. Cell Biol. 2009, 41, 757–761. [Google Scholar] [CrossRef]
- Kalesnikoff, J.; Sly, L.M.; Hughes, M.R.; Buchse, T.; Rauh, M.J.; Cao, L.P.; Lam, V.; Mui, A.; Huber, M.; Krystal, G. The role of SHIP in cytokine-induced signaling. Rev. Physiol. Biochem. Pharmacol. 2003, 149, 87–103. [Google Scholar]
- Bunney, T.D.; Katan, M. Phosphoinositide signalling in cancer: Beyond PI3K and PTEN. Nat. Cancer Rev. 2010, 10, 342–352. [Google Scholar] [CrossRef]
- Eichhorn, P.J.; Creyghton, M.P.; Bernards, R. Protein phosphatase 2A regulatory subunits and cancer. Biochim. Biophys. Acta 1795, 1–15. [Google Scholar]
- Brognard, J.; Newton, A.C. PHLiPPing the switch on Akt and protein kinase C signaling. Trends Endocrinol. Metab. 2008, 19, 223–230. [Google Scholar] [CrossRef]
- Hirano, I.; Nakamura, S.; Yokota, D.; Ono, T.; Shigeno, K.; Fujisawa, S.; Shinjo, K.; Ohnishi, K. Depletion of Pleckstrin homology domain leucine-rich repeat protein phosphatases 1 and 2 by Bcr-Abl promotes chronic myelogenous leukemia cell proliferation through continuous phosphorylation of Akt isoforms. J. Biol. Chem. 2009, 284, 22155–22165. [Google Scholar] [CrossRef]
- Lane, S.W.; Scadden, D.T.; Gilliland, D.G. The leukemic stem cell niche: Current concepts and therapeutic opportunities. Blood 2009, 114, 1150–1157. [Google Scholar] [CrossRef]
- Zhang, J.; Grindley, J.C.; Yin, T.; Jayasinghe, S.; He, X.C.; Ross, J.T.; Haug, J.S.; Rupp, D.; Porter-Westpfahl, K.S.; Wiedemann, L.M.; Wu, H.; Li, L. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 2006, 441, 518–522. [Google Scholar] [CrossRef]
- Yilmaz, O.H.; Valdez, R.; Theisen, B.K.; Guo, W.; Ferguson, D.O.; Wu, H.; Morrison, S.J. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006, 441, 475–482. [Google Scholar] [CrossRef]
- Yu, H.; Li, Y.; Gao, C.; Fabien, L.; Jia, Y.; Lu, J.; Silberstein, L.E.; Pinkus, G.S.; Ye, K.; Chai, L.; Luo, H.R. Relevant mouse model for human monocytic leukemia through Cre/lox-controlled myeloid-specific deletion of PTEN. Leukemia 2010, 24, 1077–1080. [Google Scholar] [CrossRef]
- Peng, C.; Chen, Y.; Yang, Z.; Zhang, H.; Osterby, L.; Rosmarin, A.G.; Li, S. PTEN is a tumor suppressor in CML stem cells and BCR-ABL-induced leukemias in mice. Blood 2010, 115, 626–635. [Google Scholar] [CrossRef]
- Lakhanpal, G.K.; Vecchiarelli-Federico, L.M.; Li, Y.J.; Cui, J.W.; Bailey, M.L.; Spaner, D.E.; Dumont, D.J.; Barber, D.L.; Ben-David, Y. The inositol phosphatase SHIP-1 is negatively regulated by Fli-1 and its loss accelerates leukemogenesis. Blood 2010, 116, 428–436. [Google Scholar] [CrossRef]
- Munugalavadla, V.; Sims, E.C.; Borneo, J.; Chan, R.J.; Kapur, R. Genetic and pharmacologic evidence implicating the p85 α, but not p85 β, regulatory subunit of PI3K and Rac2 GTPase in regulating oncogenic KIT-induced transformation in acute myeloid leukemia and systemic mastocytosis. Blood 2007, 110, 1612–1620. [Google Scholar] [CrossRef]
- Kharas, M.G.; Okabe, R.; Ganis, J.J.; Gozo, M.; Khandan, T.; Paktinat, M.; Gilliland, D.G.; Gritsman, K. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood 2010, 115, 1406–1415. [Google Scholar] [CrossRef]
- Gan, B.; Sahin, E.; Jiang, S.; Sanchez-Aguilera, A.; Scott, K.L.; Chin, L.; Williams, D.A.; Kwiatkowski, D.J.; DePinho, R.A. mTORC1-dependent and -independent regulation of stem cell renewal, differentiation, and mobilization. Proc. Natl. Acad. Sci. USA 2008, 105, 19384–19389. [Google Scholar] [CrossRef]
- Chen, C.; Liu, Y.; Liu, R.; Ikenoue, T.; Guan, K.L.; Liu, Y.; Zheng, P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. J. Exp. Med. 2008, 205, 2397–2408. [Google Scholar] [CrossRef]
- Gan, B.; DePinho, R.A. mTORC1 signaling governs hematopoietic stem cell quiescence. Cell Cycle 2009, 8, 1003–1006. [Google Scholar] [CrossRef]
- Xu, Q.; Thompson, J.E.; Carroll, M. mTOR regulates cell survival after etoposide treatment in primary AML cells. Blood 2005, 106, 4261–4268. [Google Scholar] [CrossRef]
- Tothova, Z.; Kollipara, R.; Huntly, B.J.; Lee, B.H.; Castrillon, D.H.; Cullen, D.E.; McDowell, E.P.; Lazo-Kallanian, S.; Williams, I.R.; Sears, C.; Armstrong, S.A.; Passegue, E.; DePinho, R.A.; Gilliland, D.G. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 2007, 128, 325–339. [Google Scholar] [CrossRef]
- Naka, K.; Hoshii, T.; Muraguchi, T.; Tadokoro, Y.; Ooshio, T.; Kondo, Y.; Nakao, S.; Motoyama, N.; Hirao, A. TGF-β-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010, 463, 676–680. [Google Scholar] [CrossRef]
- Zheng, H.; Ying, H.; Yan, H.; Kimmelman, A.C.; Hiller, D.J.; Chen, A.J.; Perry, S.R.; Tonon, G.; Chu, G.C.; Ding, Z.; Stommel, J.M.; Dunn, K.L.; Wiedemeyer, R.; You, M.J.; Brennan, C.; Wang, Y.A.; Ligon, K.L.; Wong, W.H.; Chin, L.; DePinho, R.A. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 2008, 455, 1129–1133. [Google Scholar] [CrossRef]
- Rountree, C.B.; Ding, W.; He, L.; Stiles, B. Expansion of CD133-expressing liver cancer stem cells in liver-specific phosphatase and tensin homolog deleted on chromosome 10-deleted mice. Stem Cells 2009, 27, 290–299. [Google Scholar] [CrossRef]
- Korsten, H.; Ziel-van der Made, A.; Ma, X.; van der Kwast, T.; Trapman, J. Accumulating progenitor cells in the luminal epithelial cell layer are candidate tumor initiating cells in a Pten knockout mouse prostate cancer model. PLoS One 2009, 4, e5662. [Google Scholar]
- Dubrovska, A.; Kim, S.; Salamone, R.J.; Walker, J.R.; Maira, S.M.; Garcia-Echeverria, C.; Schultz, P.G.; Reddy, V.A. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc. Natl. Acad. Sci. USA 2009, 106, 268–273. [Google Scholar] [CrossRef]
- Yang, Y.; Iwanaga, K.; Raso, M.G.; Wislez, M.; Hanna, A.E.; Wieder, E.D.; Molldrem, J.J.; Wistuba, II; Powis, G.; Demayo, F.J.; Kim, C.F.; Kurie, J.M. Phosphatidylinositol 3-kinase mediates bronchioalveolar stem cell expansion in mouse models of oncogenic K-ras-induced lung cancer. PLoS One 2008, 3, e2220. [Google Scholar] [CrossRef]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E., 3rd; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163. [Google Scholar] [CrossRef]
- Xin, L.; Lawson, D.A.; Witte, O.N. The Sca-1 cell surface marker enriches for a prostate-regenerating cell subpopulation that can initiate prostate tumorigenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 6942–6947. [Google Scholar] [CrossRef]
- Lawson, D.A.; Zong, Y.; Memarzadeh, S.; Xin, L.; Huang, J.; Witte, O.N. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc. Natl. Acad. Sci. USA 2010, 107, 2610–2615. [Google Scholar] [CrossRef]
- Vermeulen, L.; Todaro, M.; de Sousa Mello, F.; Sprick, M.R.; Kemper, K.; Perez Alea, M.; Richel, D.J.; Stassi, G.; Medema, J.P. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc. Natl. Acad. Sci. USA 2008, 105, 13427–13432. [Google Scholar]
- Iliopoulos, D.; Polytarchou, C.; Hatziapostolou, M.; Kottakis, F.; Maroulakou, I.G.; Struhl, K.; Tsichlis, P.N. MicroRNAs differentially regulated by Akt isoforms control EMT and stem cell renewal in cancer cells. Science Signal. 2009, 2, ra62. [Google Scholar] [CrossRef]
- LoPiccolo, J.; Blumenthal, G.M.; Bernstein, W.B.; Dennis, P.A. Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist. Updat. 2008, 11, 32–50. [Google Scholar] [CrossRef]
- Park, S.; Chapuis, N.; Bardet, V.; Tamburini, J.; Gallay, N.; Willems, L.; Knight, Z.A.; Shokat, K.M.; Azar, N.; Viguie, F.; Ifrah, N.; Dreyfus, F.; Mayeux, P.; Lacombe, C.; Bouscary, D. PI-103, a dual inhibitor of Class IA phosphatidylinositide 3-kinase and mTOR, has antileukemic activity in AML. Leukemia 2008, 22, 1698–1706. [Google Scholar] [CrossRef]
- Ma, S.; Lee, T.K.; Zheng, B.J.; Chan, K.W.; Guan, X.Y. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008, 27, 1749–1758. [Google Scholar] [CrossRef]
- Mogi, M.; Yang, J.; Lambert, J.F.; Colvin, G.A.; Shiojima, I.; Skurk, C.; Summer, R.; Fine, A.; Quesenberry, P.J.; Walsh, K. Akt signaling regulates side population cell phenotype via Bcrp1 translocation. J. Biol. Chem. 2003, 278, 39068–39075. [Google Scholar] [CrossRef]
- An, Y.; Ongkeko, W.M. ABCG2: the key to chemoresistance in cancer stem cells? Expert Opin. Drug Metab. Toxicol. 2009, 5, 1529–1542. [Google Scholar] [CrossRef]
- Bram, E.; Ifergan, I.; Shafran, A.; Berman, B.; Jansen, G.; Assaraf, Y.G. Mutant Gly482 and Thr482 ABCG2 mediate high-level resistance to lipophilic antifolates. Cancer Chemother. Pharmacol. 2006, 58, 826–834. [Google Scholar] [CrossRef]
- Goodell, M.A.; Brose, K.; Paradis, G.; Conner, A.S.; Mulligan, R.C. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J. Exp. Med. 1996, 183, 1797–1806. [Google Scholar] [CrossRef]
- Goodell, M.A.; Rosenzweig, M.; Kim, H.; Marks, D.F.; DeMaria, M.; Paradis, G.; Grupp, S.A.; Sieff, C.A.; Mulligan, R.C.; Johnson, R.P. Dye efflux studies suggest that hematopoietic stem cells expressing low or undetectable levels of CD34 antigen exist in multiple species. Nat. Med. 1997, 3, 1337–1345. [Google Scholar] [CrossRef]
- Yamazaki, J.; Mizukami, T.; Takizawa, K.; Kuramitsu, M.; Momose, H.; Masumi, A.; Ami, Y.; Hasegawa, H.; Hall, W.W.; Tsujimoto, H.; Hamaguchi, I.; Yamaguchi, K. Identification of cancer stem cells in a Tax-transgenic (Tax-Tg) mouse model of adult T-cell leukemia/lymphoma. Blood 2009, 114, 2709–2720. [Google Scholar] [CrossRef]
- Moshaver, B.; van Rhenen, A.; Kelder, A.; van der Pol, M.; Terwijn, M.; Bachas, C.; Westra, A.H.; Ossenkoppele, G.J.; Zweegman, S.; Schuurhuis, G.J. Identification of a small subpopulation of candidate leukemia-initiating cells in the side population of patients with acute myeloid leukemia. Stem Cells 2008, 26, 3059–3067. [Google Scholar] [CrossRef]
- Salcido, C.D.; Larochelle, A.; Taylor, B.J.; Dunbar, C.E.; Varticovski, L. Molecular characterisation of side population cells with cancer stem cell-like characteristics in small-cell lung cancer. Br. J. Cancer 2010, 102, 1636–1644. [Google Scholar] [CrossRef]
- Mimeault, M.; Johansson, S.L.; Henichart, J.P.; Depreux, P.; Batra, S.K. Cytotoxic effects induced by docetaxel, gefitinib, and cyclopamine on side population and nonside population cell fractions from human invasive prostate cancer cells. Mol. Cancer Ther. 2010, 9, 617–630. [Google Scholar] [CrossRef]
- Nakanishi, T.; Chumsri, S.; Khakpour, N.; Brodie, A.H.; Leyland-Jones, B.; Hamburger, A.W.; Ross, D.D.; Burger, A.M. Side-population cells in luminal-type breast cancer have tumour-initiating cell properties, and are regulated by HER2 expression and signalling. Br. J. Cancer 2010, 102, 815–826. [Google Scholar] [CrossRef]
- Harris, M.A.; Yang, H.; Low, B.E.; Mukherjee, J.; Guha, A.; Bronson, R.T.; Shultz, L.D.; Israel, M.A.; Yun, K. Cancer stem cells are enriched in the side population cells in a mouse model of glioma. Cancer Res. 2008, 68, 10051–10059. [Google Scholar] [CrossRef]
- Chiba, T.; Miyagi, S.; Saraya, A.; Aoki, R.; Seki, A.; Morita, Y.; Yonemitsu, Y.; Yokosuka, O.; Taniguchi, H.; Nakauchi, H.; Iwama, A. The polycomb gene product BMI1 contributes to the maintenance of tumor-initiating side population cells in hepatocellular carcinoma. Cancer Res. 2008, 68, 7742–7749. [Google Scholar] [CrossRef]
- Bleau, A.M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef]
- Hu, C.; Li, H.; Li, J.; Zhu, Z.; Yin, S.; Hao, X.; Yao, M.; Zheng, S.; Gu, J. Analysis of ABCG2 expression and side population identifies intrinsic drug efflux in the HCC cell line MHCC-97L and its modulation by Akt signaling. Carcinogenesis 2008, 29, 2289–2297. [Google Scholar] [CrossRef]
- Gallia, G.L.; Tyler, B.M.; Hann, C.L.; Siu, I.M.; Giranda, V.L.; Vescovi, A.L.; Brem, H.; Riggins, G.J. Inhibition of Akt inhibits growth of glioblastoma and glioblastoma stem-like cells. Mol. Cancer Ther. 2009, 8, 386–393. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Becher, O.J.; Rosenblum, M.K.; Pandolfi, P.P.; Manova-Todorova, K.; Holland, E.C. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008, 22, 436–448. [Google Scholar] [CrossRef]
- Martelli, A.M.; Evangelisti, C.; Chiarini, F.; Grimaldi, C.; Cappellini, A.; Ognibene, A.; McCubrey, J.A. The emerging role of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in normal myelopoiesis and leukemogenesis. Biochim. Biophys. Acta 2010, 1803, 991–1002. [Google Scholar]
- Eyler, C.E.; Foo, W.C.; LaFiura, K.M.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells 2008, 26, 3027–3036. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Becher, O.J.; Holland, E.C. Cancer stem cells and survival pathways. Cell Cycle 2008, 7, 1371–1378. [Google Scholar] [CrossRef]
- Pannuti, A.; Foreman, K.; Rizzo, P.; Osipo, C.; Golde, T.; Osborne, B.; Miele, L. Targeting Notch to Target Cancer Stem Cells. Clin. Cancer Res. 2010, 16, 3141–3152. [Google Scholar] [CrossRef]
- Merchant, A.A.; Matsui, W. Targeting Hedgehog -- a Cancer Stem Cell Pathway. Clin. Cancer Res. 2010, 16, 3130–3140. [Google Scholar] [CrossRef]
- Takebe, N.; Ivy, S.P. Controversies in Cancer Stem Cells: Targeting Embryonic Signaling Pathways. Clin. Cancer Res. 2010, 16, 3106–3112. [Google Scholar] [CrossRef]
- Mueller, M.T.; Hermann, P.C.; Witthauer, J.; Rubio-Viqueira, B.; Leicht, S.F.; Huber, S.; Ellwart, J.W.; Mustafa, M.; Bartenstein, P.; D'Haese, J.G.; Schoenberg, M.H.; Berger, F.; Jauch, K.W.; Hidalgo, M.; Heeschen, C. Combined targeted treatment to eliminate tumorigenic cancer stem cells in human pancreatic cancer. Gastroenterology 2009, 137, 1102–1113. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Martelli, A.M.; Evangelisti, C.; Chiarini, F.; Grimaldi, C.; McCubrey, J.A. The Emerging Role of the Phosphatidylinositol 3-Kinase/ Akt/Mammalian Target of Rapamycin Signaling Network in Cancer Stem Cell Biology. Cancers 2010, 2, 1576-1596. https://doi.org/10.3390/cancers2031576
Martelli AM, Evangelisti C, Chiarini F, Grimaldi C, McCubrey JA. The Emerging Role of the Phosphatidylinositol 3-Kinase/ Akt/Mammalian Target of Rapamycin Signaling Network in Cancer Stem Cell Biology. Cancers. 2010; 2(3):1576-1596. https://doi.org/10.3390/cancers2031576
Chicago/Turabian StyleMartelli, Alberto M., Camilla Evangelisti, Francesca Chiarini, Cecilia Grimaldi, and James A. McCubrey. 2010. "The Emerging Role of the Phosphatidylinositol 3-Kinase/ Akt/Mammalian Target of Rapamycin Signaling Network in Cancer Stem Cell Biology" Cancers 2, no. 3: 1576-1596. https://doi.org/10.3390/cancers2031576
APA StyleMartelli, A. M., Evangelisti, C., Chiarini, F., Grimaldi, C., & McCubrey, J. A. (2010). The Emerging Role of the Phosphatidylinositol 3-Kinase/ Akt/Mammalian Target of Rapamycin Signaling Network in Cancer Stem Cell Biology. Cancers, 2(3), 1576-1596. https://doi.org/10.3390/cancers2031576