
A Proteinase 3 Contribution to Juvenile Idiopathic Arthritis-Associated Cartilage Damage

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. Enzyme-Linked Immunosorbent Assays (ELISA)
2.3. Collagen II Hydrolysis by Serine Proteases
2.4. Statistics
3. Results
3.1. Patient and Clinical Data
3.2. Synovial Fluid Enzyme ELISAs
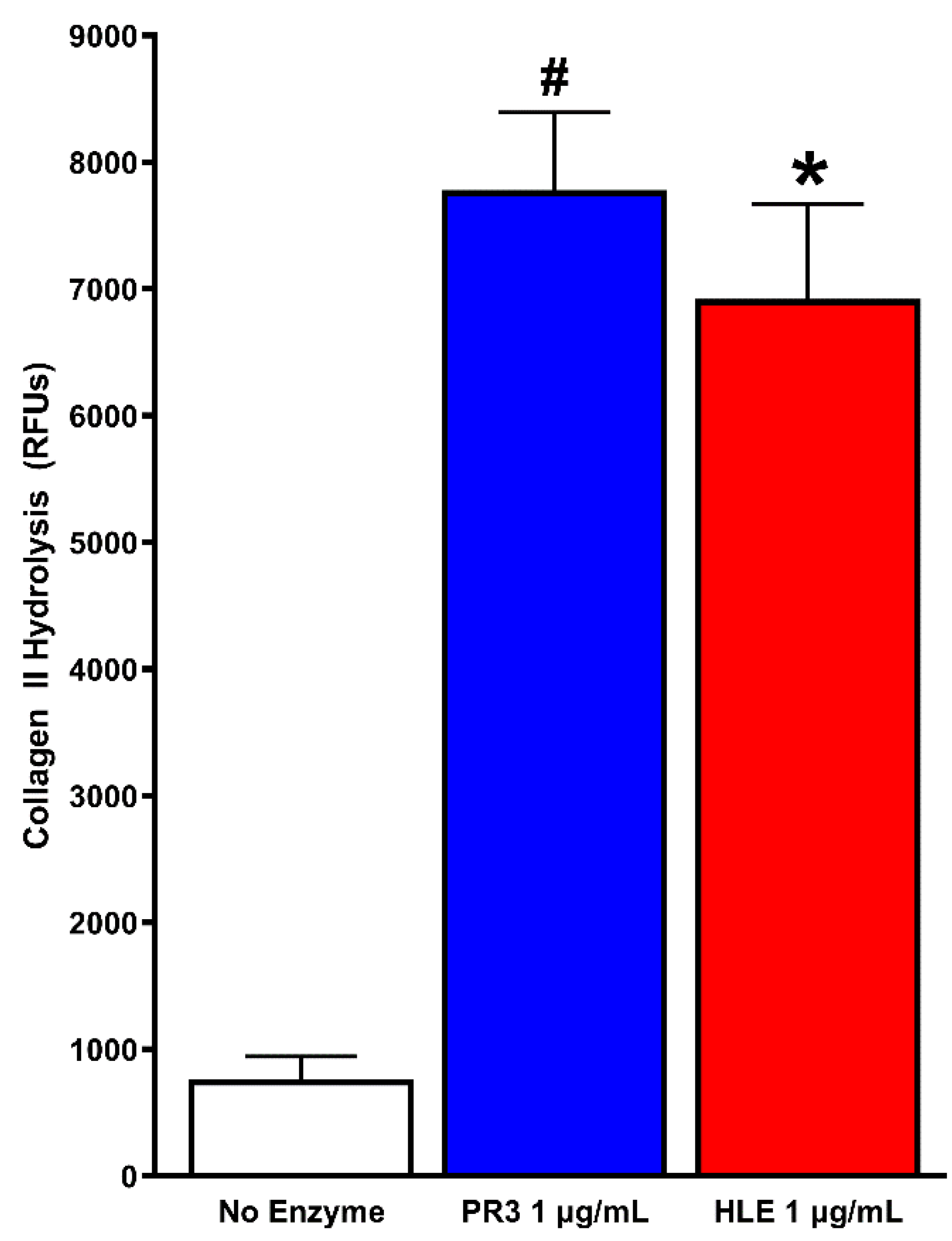
3.3. PR3 and HLE Effects on Collagen II
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Petty, R.E.; Southwood, T.R.; Manners, P.; Baum, J.; Glass, D.N.; Goldenberg, J.; He, X.; Maldonado-Cocco, J.; Orozco-Alcala, J.; Prieur, A.-M.; et al. International League of Associations for Rheumatology Classification of Juvenile Idiopathic Arthritis: Second Revision, Edmonton, 2001. J. Rheumatol. 2004, 31, 390–392. [Google Scholar]
- LeBlanc, C.M.A.; Lang, B.; Bencivenga, A.; Chetaille, A.-L.; Dancey, P.; Dent, P.; Miettunen, P.; Oen, K.; Rosenberg, A.; Roth, J.; et al. Access to Biologic Therapies in Canada for Children with Juvenile Idiopathic Arthritis. J. Rheumatol. 2012, 39, 1875–1879. [Google Scholar] [CrossRef] [PubMed]
- Glerup, M.; Rypdal, V.; Arnstad, E.D.; Ekelund, M.; Peltoniemi, S.; Aalto, K.; Rygg, M.; Toftedal, P.; Nielsen, S.; Fasth, A.; et al. Long-Term Outcomes in Juvenile Idiopathic Arthritis: Eighteen Years of Follow-Up in the Population-Based Nordic Juvenile Idiopathic Arthritis Cohort. Arthritis Care Res. 2020, 72, 507–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvis, J.N.; Jiang, K.; Petty, H.R.; Centola, M. Neutrophils: The Forgotten Cell in JIA Disease Pathogenesis. Pediatr. Rheumatol. 2007, 5, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huttenlocher, A.; Smith, J.A. Neutrophils in Pediatric Autoimmune Disease. Curr. Opin. Rheumatol. 2015, 27, 500–504. [Google Scholar] [CrossRef]
- Pascual, E.; Jovaní, V. Synovial Fluid Analysis. Best Pract. Res. Clin. Rheumatol. 2005, 19, 371–386. [Google Scholar] [CrossRef]
- Simkin, P.A.; Bassett, J.E. Pathways of Microvascular Permeability in the Synovium of Normal and Diseased Human Knees. J. Rheumatol. 2011, 38, 2635–2642. [Google Scholar] [CrossRef]
- Medzhitov, R. Inflammation 2010: New Adventures of an Old Flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.U. An Overview of Inflammation: Mechanism and Consequences. Front. Biol. 2011, 6, 274. [Google Scholar] [CrossRef]
- Jariwala, M.P.; Laxer, R.M. NETosis in Rheumatic Diseases. Curr. Rheumatol. Rep. 2021, 23, 9. [Google Scholar] [CrossRef]
- Momohara, S.; Kashiwazaki, S.; Inoue, K.; Saito, S.; Nakagawa, T. Elastase from Polymorphonuclear Leukocyte in Articular Cartilage and Synovial Fluids of Patients with Rheumatoid Arthritis. Clin. Rheumatol. 1997, 16, 133–139. [Google Scholar] [CrossRef]
- Velvart, M.; Fehr, K. Degradation in Vivo of Articular Cartilage in Rheumatoid Arthritis and Juvenile Chronic Arthritis by Cathepsin G and Elastase from Polymorphonuclear Leukocytes. Rheumatol. Int. 1987, 7, 195–202. [Google Scholar] [CrossRef]
- Starkey, P.M.; Barrett, A.J.; Burleigh, M.C. The Degradation of Articular Collagen by Neutrophil Proteinases. Biochim. Biophys. Acta (BBA) Enzymol. 1977, 483, 386–397. [Google Scholar] [CrossRef]
- Kao, R.C.; Wehner, N.G.; Skubitz, K.M.; Gray, B.H.; Hoidal, J.R. Proteinase 3. A Distinct Human Polymorphonuclear Leukocyte Proteinase That Produces Emphysema in Hamsters. J. Clin. Investig. 1988, 82, 1963–1973. [Google Scholar] [CrossRef]
- Bories, D.; Raynal, M.-C.; Solomon, D.H.; Darzynkiewicz, Z.; Cayre, Y.E. Down-Regulation of a Serine Protease, Myeloblastin, Causes Growth Arrest and Differentiation of Promyelocytic Leukemia Cells. Cell 1989, 59, 959–968. [Google Scholar] [CrossRef]
- Kettritz, R. Neutral Serine Proteases of Neutrophils. Immunol. Rev. 2016, 273, 232–248. [Google Scholar] [CrossRef]
- Woo, M.M.H.; Patterson, E.K.; Clarson, C.; Cepinskas, G.; Bani-Yaghoub, M.; Stanimirovic, D.B.; Fraser, D.D. Elevated Leukocyte Azurophilic Enzymes in Human Diabetic Ketoacidosis Plasma Degrade Cerebrovascular Endothelial Junctional Proteins. Crit. Care Med. 2016, 44, e846–e853. [Google Scholar] [CrossRef]
- Halbwachs-Mecarelli, L.; Bessou, G.; Lesavre, P.; Lopez, S.; Witko-Sarsat, V. Bimodal Distribution of Proteinase 3 (PR3) Surface Expression Reflects a Constitutive Heterogeneity in the Polymorphonuclear Neutrophil Pool. FEBS Lett. 1995, 374, 29–33. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil Elastase, Proteinase 3, and Cathepsin G as Therapeutic Targets in Human Diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef] [Green Version]
- Harrold, L.R.; Salman, C.; Shoor, S.; Curtis, J.R.; Asgari, M.M.; Gelfand, J.M.; Wu, J.J.; Herrinton, L.J. Incidence and Prevalence of Juvenile Idiopathic Arthritis among Children in a Managed Care Population, 1996–2009. J. Rheumatol. 2013, 40, 1218–1225. [Google Scholar] [CrossRef] [Green Version]
- Thierry, S.; Fautrel, B.; Lemelle, I.; Guillemin, F. Prevalence and Incidence of Juvenile Idiopathic Arthritis: A Systematic Review. Jt. Bone Spine 2014, 81, 112–117. [Google Scholar] [CrossRef]
- Levine, J.J.; Sherry, D.D.; Strickland, D.K.; Ilowite, N.T. Intraarticular α 2-Macroglobulin Complexes and Proteolytic Activity in Children with Juvenile Rheumatoid Arthritis. Pediatr. Res. 1993, 34, 204–207. [Google Scholar] [CrossRef] [Green Version]
- Lotito, A.P.N.; Muscará, M.N.; Kiss, M.H.B.; Teixeira, S.A.; Novaes, G.S.; Laurindo, I.M.M.; Silva, C.A.; Mello, S.B.V. Nitric Oxide-Derived Species in Synovial Fluid from Patients with Juvenile Idiopathic Arthritis. J. Rheumatol. 2004, 31, 992–997. [Google Scholar]
- Toukap, A.N.; Delporte, C.; Noyon, C.; Franck, T.; Rousseau, A.; Serteyn, D.; Raes, M.; Vanhaeverbeek, M.; Moguilevsky, N.; Nève, J.; et al. Myeloperoxidase and Its Products in Synovial Fluid of Patients with Treated or Untreated Rheumatoid Arthritis. Free Radic. Res. 2014, 48, 461–465. [Google Scholar] [CrossRef] [Green Version]
- Dolman, K.M.; van de Wiel, B.A.; Kam, C.-M.; Abbink, J.J.; Hack, C.E.; Sonnenberg, A.; Powers, J.C.; von dem Borne, A.E.G.K.; Goldschmeding, R. Determination of Proteinase 3—A1-Antitrypsin Complexes in Inflammatory Fluids. FEBS Lett. 1992, 314, 117–121. [Google Scholar] [CrossRef] [Green Version]
- Martínez del Val, E.; Rodríguez Martínez, A.; Sánchez Becerra, V.; Cruz Rojo, J.; Enríquez Merayo, E.; Barral Mena, E.; de Inocencio Arocena, J. Characteristics of Synovial Fluid in Patients with Juvenile Idiopathic Arthritis. An. Pediatr. (Engl. Ed.) 2019, 91, 244–250. [Google Scholar] [CrossRef]
- Punzi, L.; Ramonda, R.; Glorioso, S.; Schiavon, F.; Mariuz, S.; Gambari, P.F. Predictive Value of Synovial Fluid Analysis in Juvenile Chronic Arthritis. Ann. Rheum. Dis. 1992, 51, 522–524. [Google Scholar] [CrossRef] [Green Version]
- Taggart, C.; Cervantes-Laurean, D.; Kim, G.; McElvaney, N.G.; Wehr, N.; Moss, J.; Levine, R.L. Oxidation of Either Methionine 351 or Methionine 358 in A1-Antitrypsin Causes Loss of Anti-Neutrophil Elastase Activity. J. Biol. Chem. 2000, 275, 27258–27265. [Google Scholar] [CrossRef]
- Wu, S.M.; Pizzo, S.V. Mechanism of Hypochlorite-Mediated Inactivation of Proteinase Inhibition by A2-Macroglobulin. Biochemistry 1999, 38, 13983–13990. [Google Scholar] [CrossRef]
- Damiano, V.V.; Kucich, U.; Murer, E.; Laudenslager, N.; Weinbaum, G. Ultrastructural Quantitation of Peroxidase- and Elastase-Containing Granules in Human Neutrophils. Am. J. Pathol. 1988, 131, 235–245. [Google Scholar]
- Campbell, E.J.; Campbell, M.A.; Owen, C.A. Bioactive Proteinase 3 on the Cell Surface of Human Neutrophils: Quantification, Catalytic Activity, and Susceptibility to Inhibition. J. Immunol. 2000, 165, 3366–3374. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, B.; Poutrain, P.; Hazouard, E.; de Monte, M.; Attucci, S.; Gauthier, F.L. Competition between Elastase and Related Proteases from Human Neutrophil for Binding to A1-Protease Inhibitor. Am. J. Respir. Cell Mol. Biol. 2005, 32, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, K.; Moore, A.R.; Willoughby, D.A. Impaired Activity of Protease Inhibitors towards Neutrophil Elastase Bound to Human Articular Cartilage. Ann. Rheum. Dis. 1996, 55, 248–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, C.A.; Campbell, E.J. Extracellular Proteolysis: New Paradigms for an Old Paradox. J. Lab. Clin. Med. 1999, 134, 341–351. [Google Scholar] [CrossRef]
- Liou, T.G.; Campbell, E.J. Nonisotropic Enzyme-Inhibitor Interactions: A Novel Nonoxidative Mechanism for Quantum Proteolysis by Human Neutrophils. Biochemistry 1995, 34, 16171–16177. [Google Scholar] [CrossRef]
- Patterson, E.K.; Fraser, D.D.; Capretta, A.; Potter, R.F.; Cepinskas, G. Carbon Monoxide-Releasing Molecule 3 Inhibits Myeloperoxidase (MPO) and Protects against MPO-Induced Vascular Endothelial Cell Activation/Dysfunction. Free Radic. Biol. Med. 2014, 70, 167–173. [Google Scholar] [CrossRef]
- Inoue, K.; Patterson, E.K.; Capretta, A.; Lawendy, A.R.; Fraser, D.D.; Cepinskas, G. Carbon Monoxide–Releasing Molecule-401 Suppresses Polymorphonuclear Leukocyte Migratory Potential by Modulating F-Actin Dynamics. Am. J. Pathol. 2017, 187, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
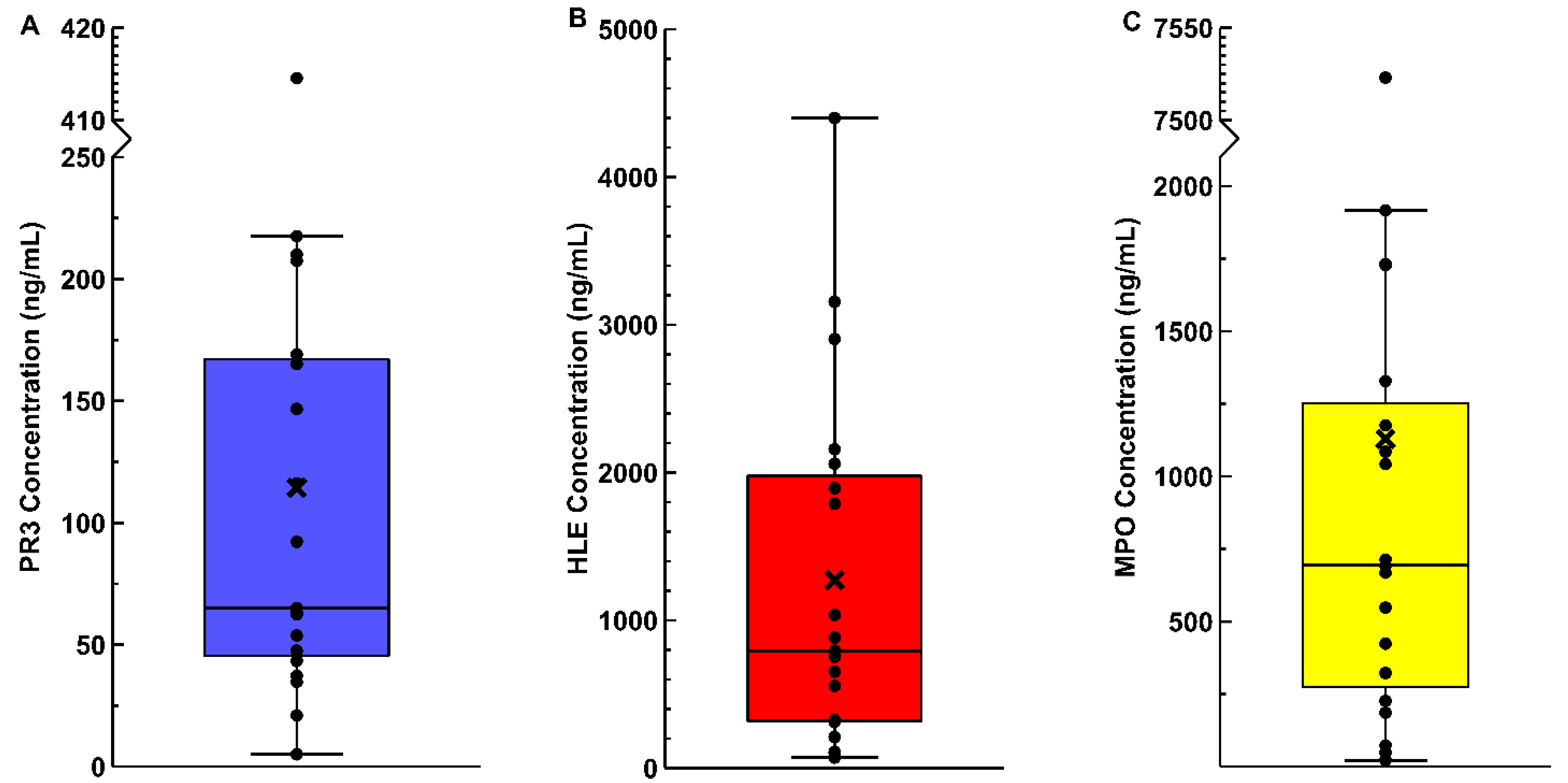

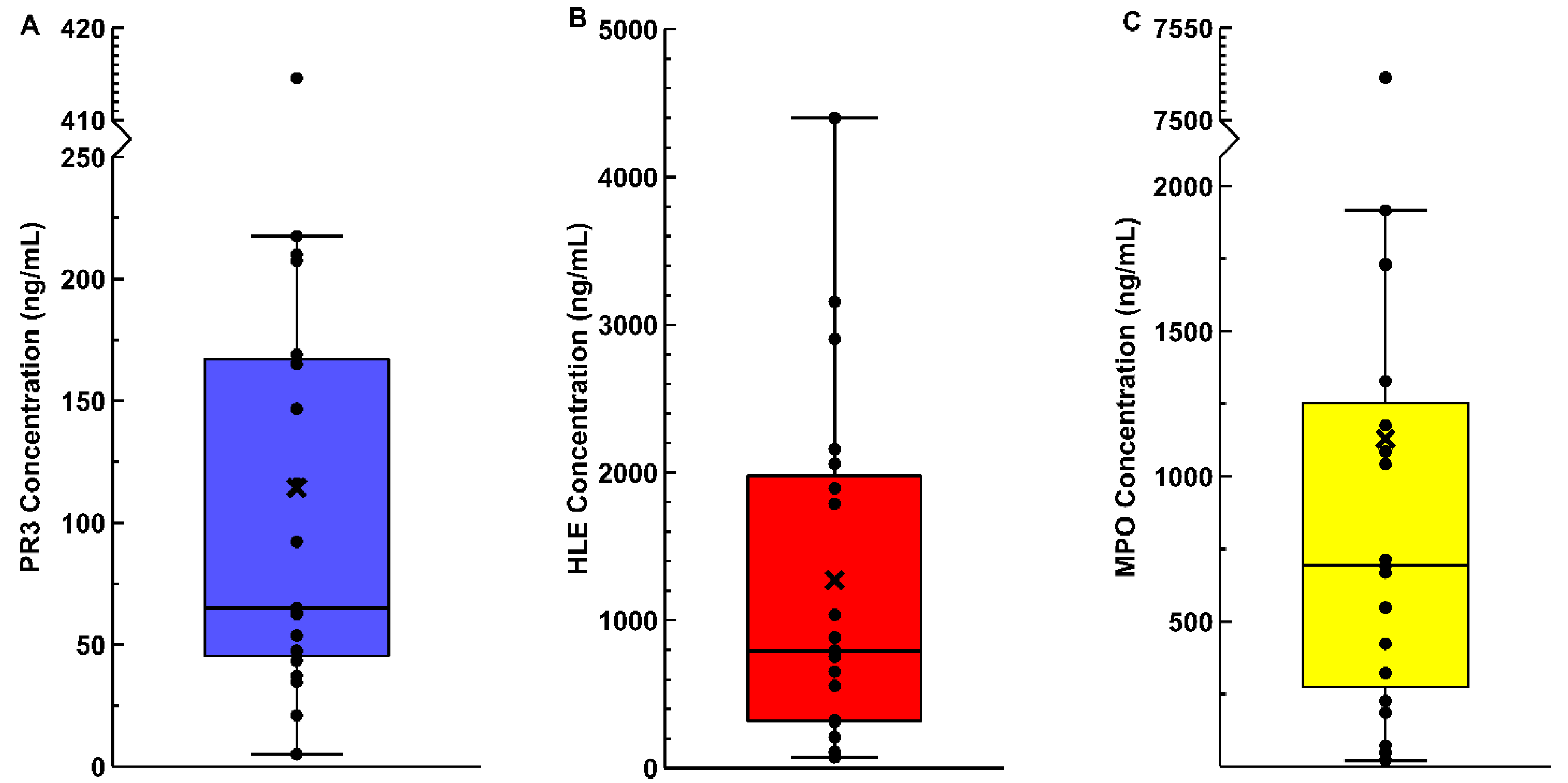{kind=link}
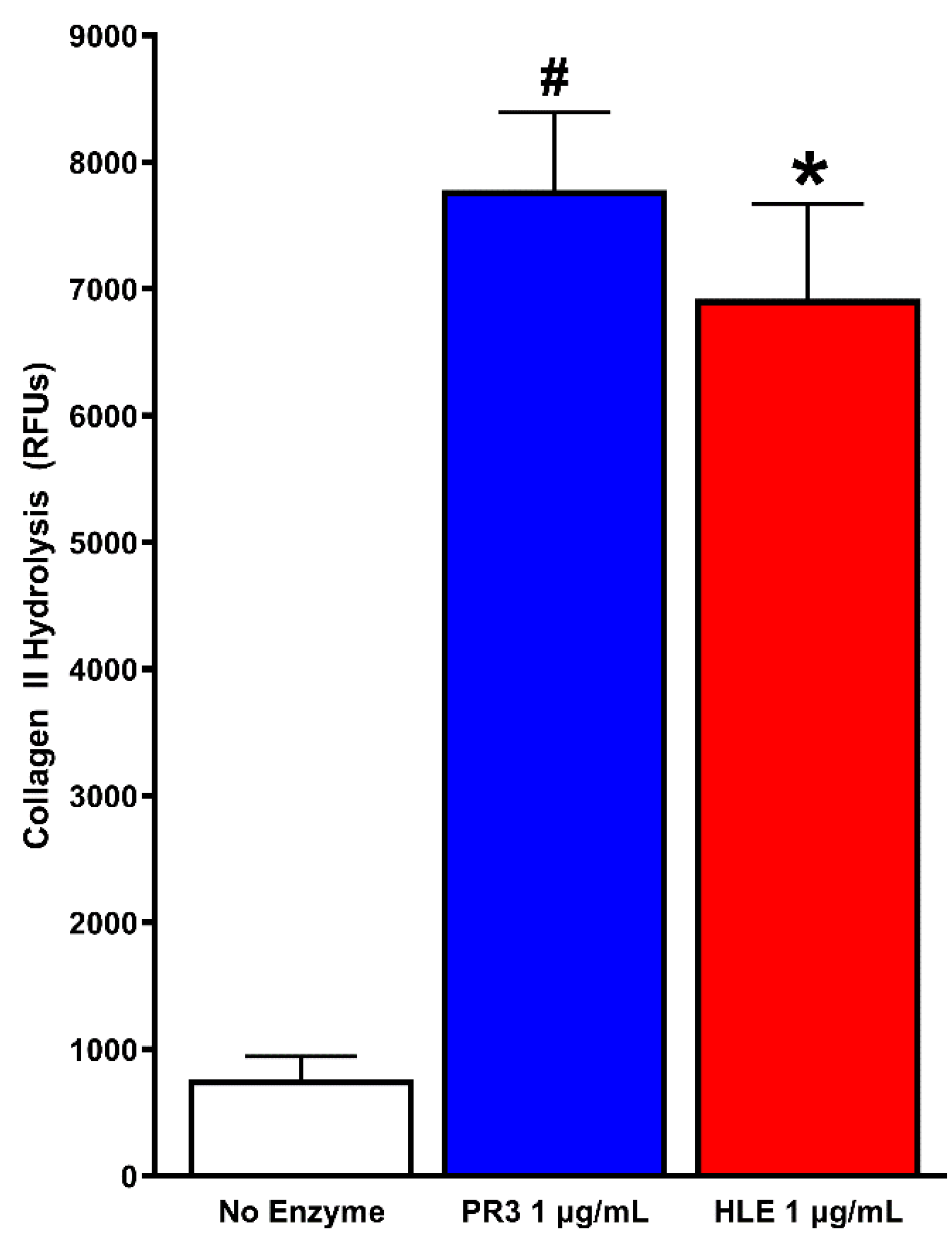{kind=link}
| Age (Years) | Sex | JIA Diagnosis | Knee Aspirated | Synovial Fluid WBC (×106/mL) | Synovial Fluid PMN (×106/mL) |
|---|---|---|---|---|---|
| 2 | F | Polyarthritis RF-neg | Right | NA | NA |
| 5 | F | Oligoarthritis JIA | Right | 13.26 | 8.36 |
| 6 | F | Polyarthritis RF-neg JIA | Right | NA | NA |
| 2 | F | Psoriatic JIA | Right | 25.66 | 22.33 |
| 7 | F | Oligoarthritis JIA | Right | NA | NA |
| 17 | F | Oligoarthritis-extended JIA | Right | NA | NA |
| 10 | M | Oligoarthritis JIA | Right | NA | NA |
| 8 | F | Oligoarthritis JIA | Right | 7.35 | 4.41 |
| 17 | F | Polyarthritis RF-neg JIA | Left | 6.22 | 3.36 |
| 3 | F | Oligoarthritis JIA | Right | 9.91 | 0.59 |
| 5 | F | Oligoarthritis JIA | Right | 9.53 | 3.43 |
| Left | |||||
| 15 | F | Polyarthritis RF-pos JIA | Right | 28.56 | 24.56 |
| 14 | F | Oligoarticular JIA | Right | 8.51 | 4.08 |
| 14 | M | Enthesitis-related JIA | Right | 8.62 | 3.02 |
| 8 | M | Oligoarthritis JIA | Left | 0.48 | 0.08 |
| 13 | F | Oligoarticular JIA | Right | 5.08 | 1.27 |
| 17 | F | Oligoarthritis JIA | Right | 6.23 | 1.62 |
| 14 | F | Oligoarthritis JIA | Left | 7.57 | 1.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patterson, E.K.; Vanin Moreno, N.; Fraser, D.D.; Cepinskas, G.; Iida, T.; Berard, R.A. A Proteinase 3 Contribution to Juvenile Idiopathic Arthritis-Associated Cartilage Damage. Pathophysiology 2021, 28, 320-327. https://doi.org/10.3390/pathophysiology28030021
Patterson EK, Vanin Moreno N, Fraser DD, Cepinskas G, Iida T, Berard RA. A Proteinase 3 Contribution to Juvenile Idiopathic Arthritis-Associated Cartilage Damage. Pathophysiology. 2021; 28(3):320-327. https://doi.org/10.3390/pathophysiology28030021
Chicago/Turabian StylePatterson, Eric K., Nicolas Vanin Moreno, Douglas D. Fraser, Gediminas Cepinskas, Takaya Iida, and Roberta A. Berard. 2021. "A Proteinase 3 Contribution to Juvenile Idiopathic Arthritis-Associated Cartilage Damage" Pathophysiology 28, no. 3: 320-327. https://doi.org/10.3390/pathophysiology28030021
APA StylePatterson, E. K., Vanin Moreno, N., Fraser, D. D., Cepinskas, G., Iida, T., & Berard, R. A. (2021). A Proteinase 3 Contribution to Juvenile Idiopathic Arthritis-Associated Cartilage Damage. Pathophysiology, 28(3), 320-327. https://doi.org/10.3390/pathophysiology28030021