
Shift of N-MYC Oncogene Expression in AML Patients Carrying the FLT3-ITD Mutation

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics and Experimental Design
2.1.1. Inclusion Criteria
2.1.2. Exclusion Criteria
2.2. RNA Extraction and cDNA Synthesis
2.3. DNA Extraction
2.4. Screening of Target Oncogene Mutations, including FLT3 Gene Mutations
2.5. Determination of the Allelic Load of FLT3 Gene Mutations
2.6. Quantitative Gene Expression Analysis
2.7. Cytogenetic Study
2.8. Statistical Analysis
3. Results
3.1. Analysis of Oncogene Mutations in Primary AML Patients
3.2. Determination of Allelic Load of FLT3-ITD Gene Mutation
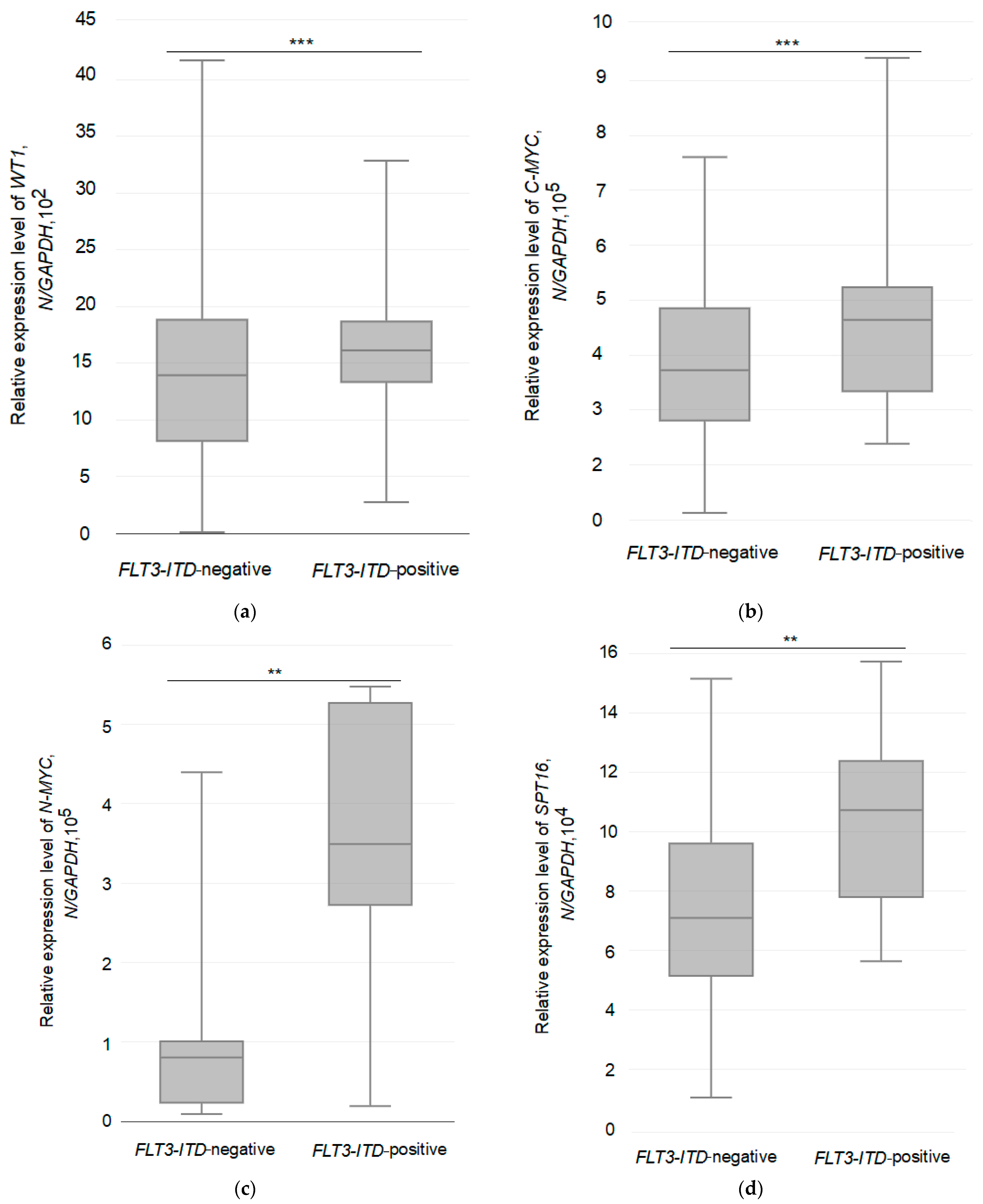
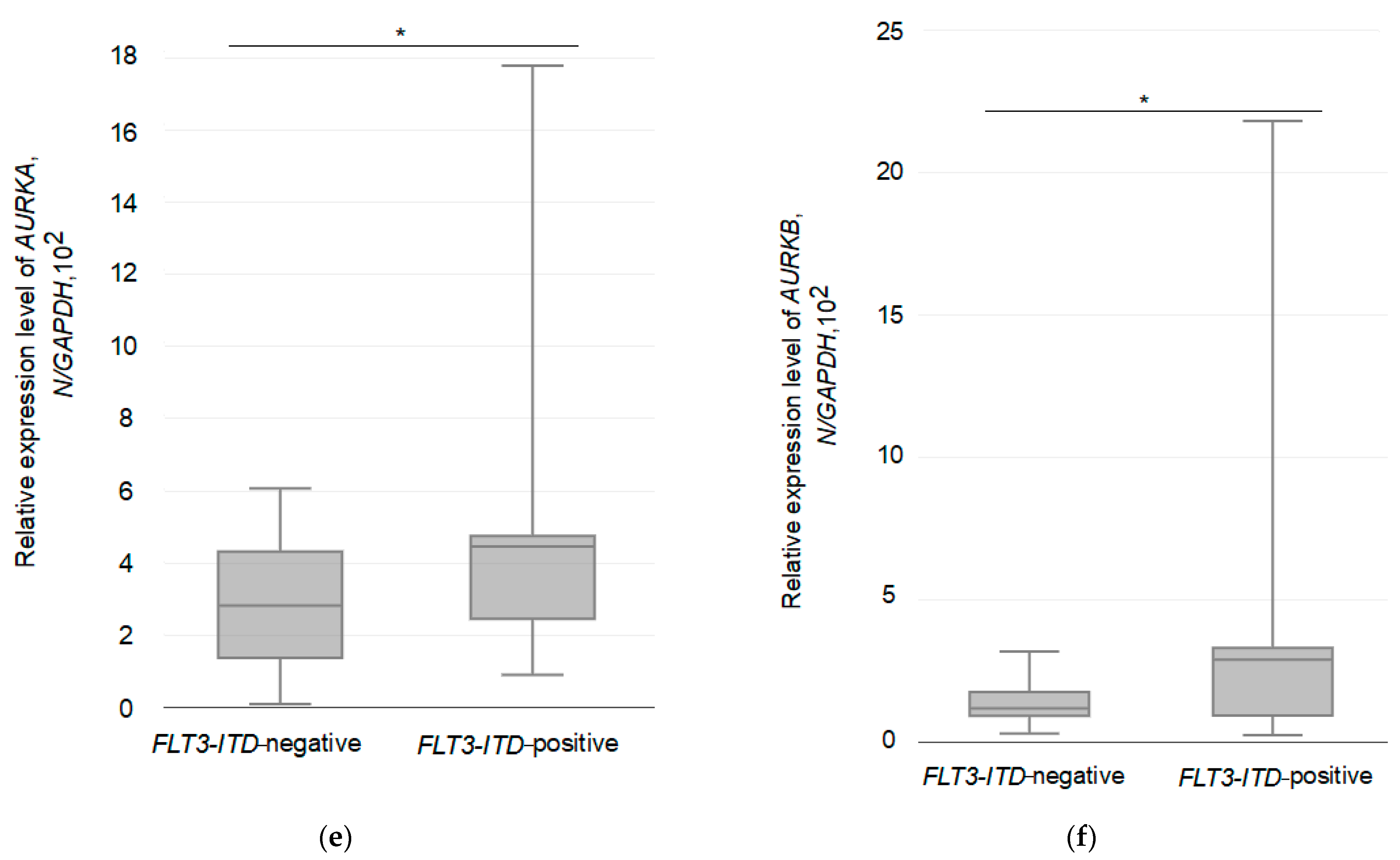
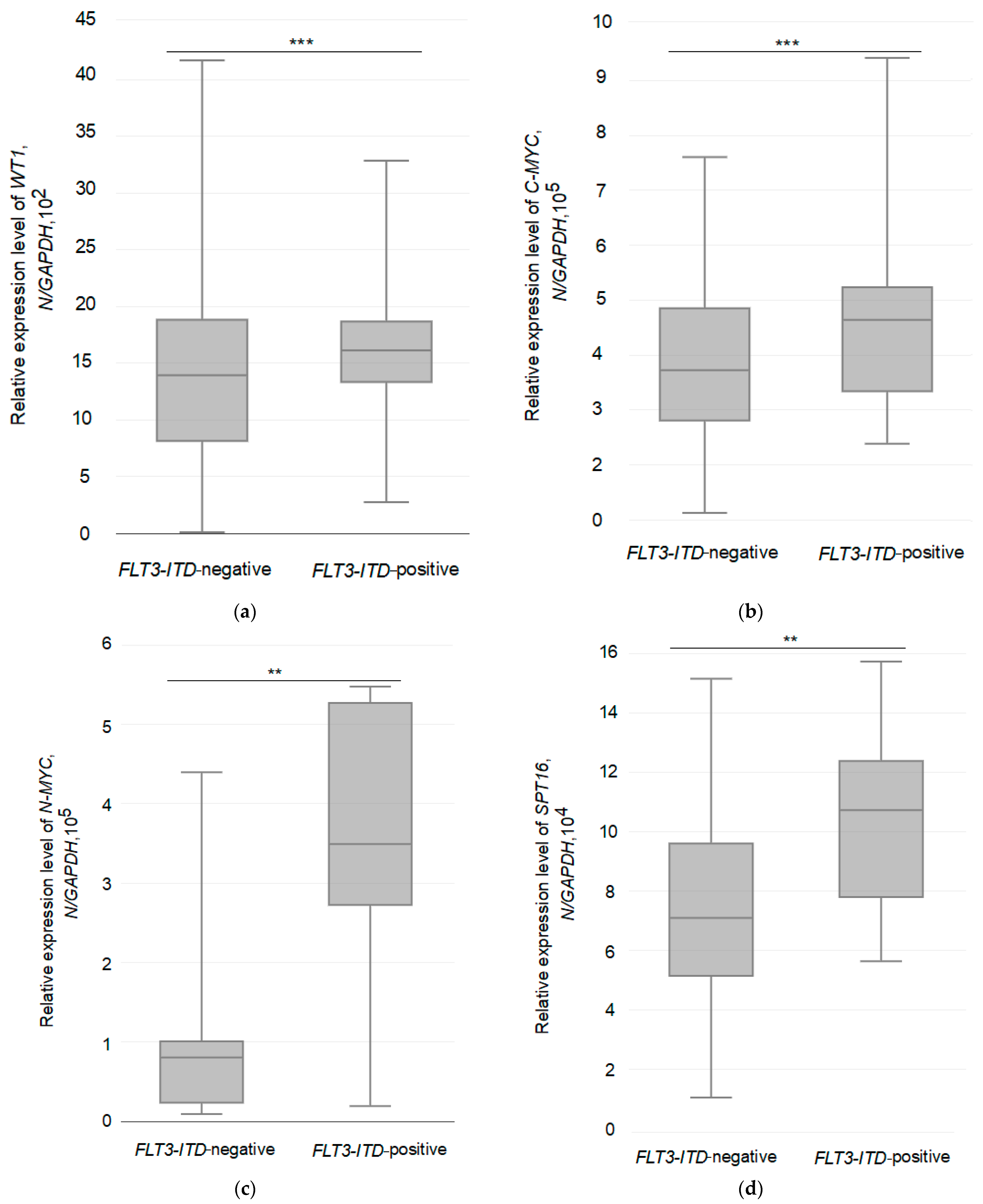
3.3. Relative Expression of the Studied Genes in Two Different Groups of AML Patients
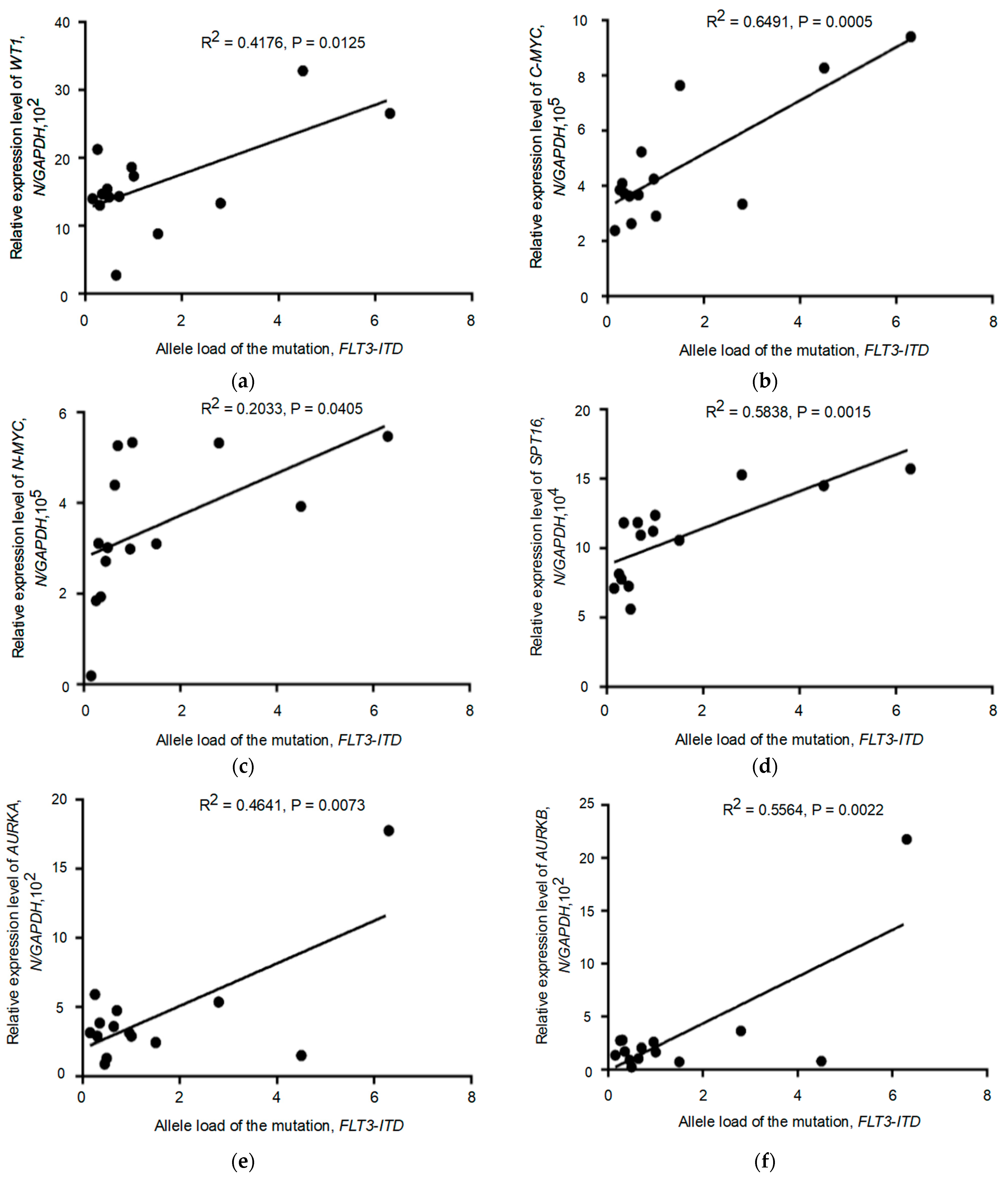
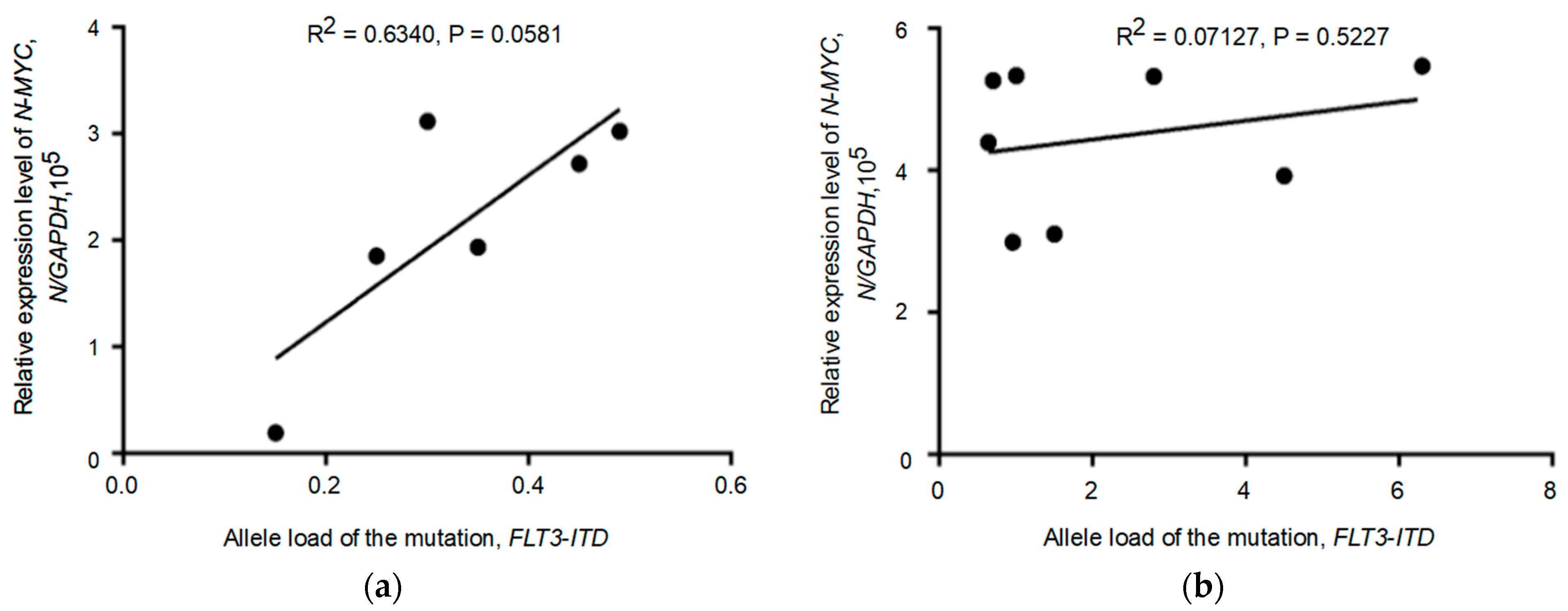
3.4. Relationship between the Expression of the Tested Genes and the Allelic Load of the FLT3-ITD Mutation
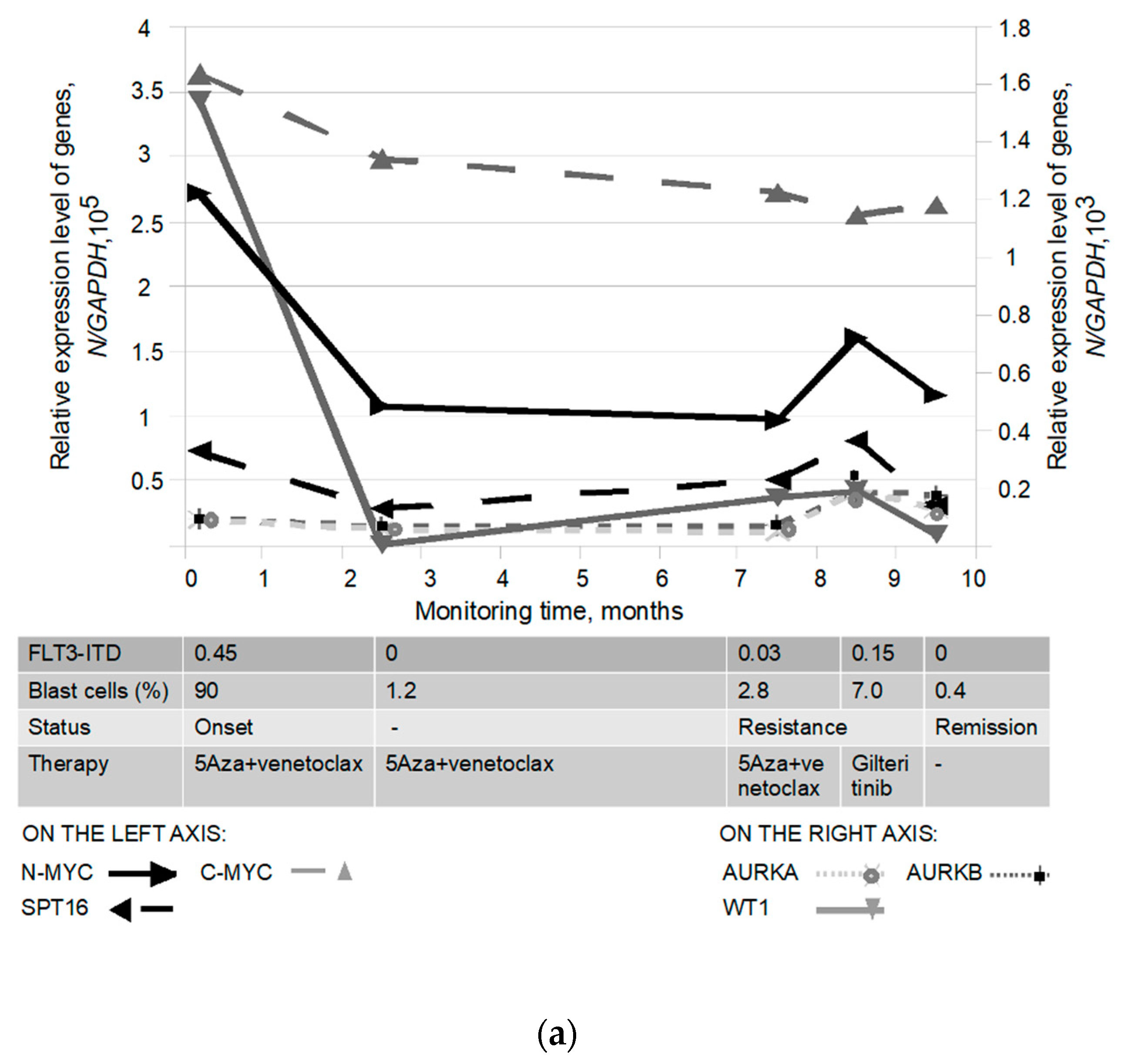
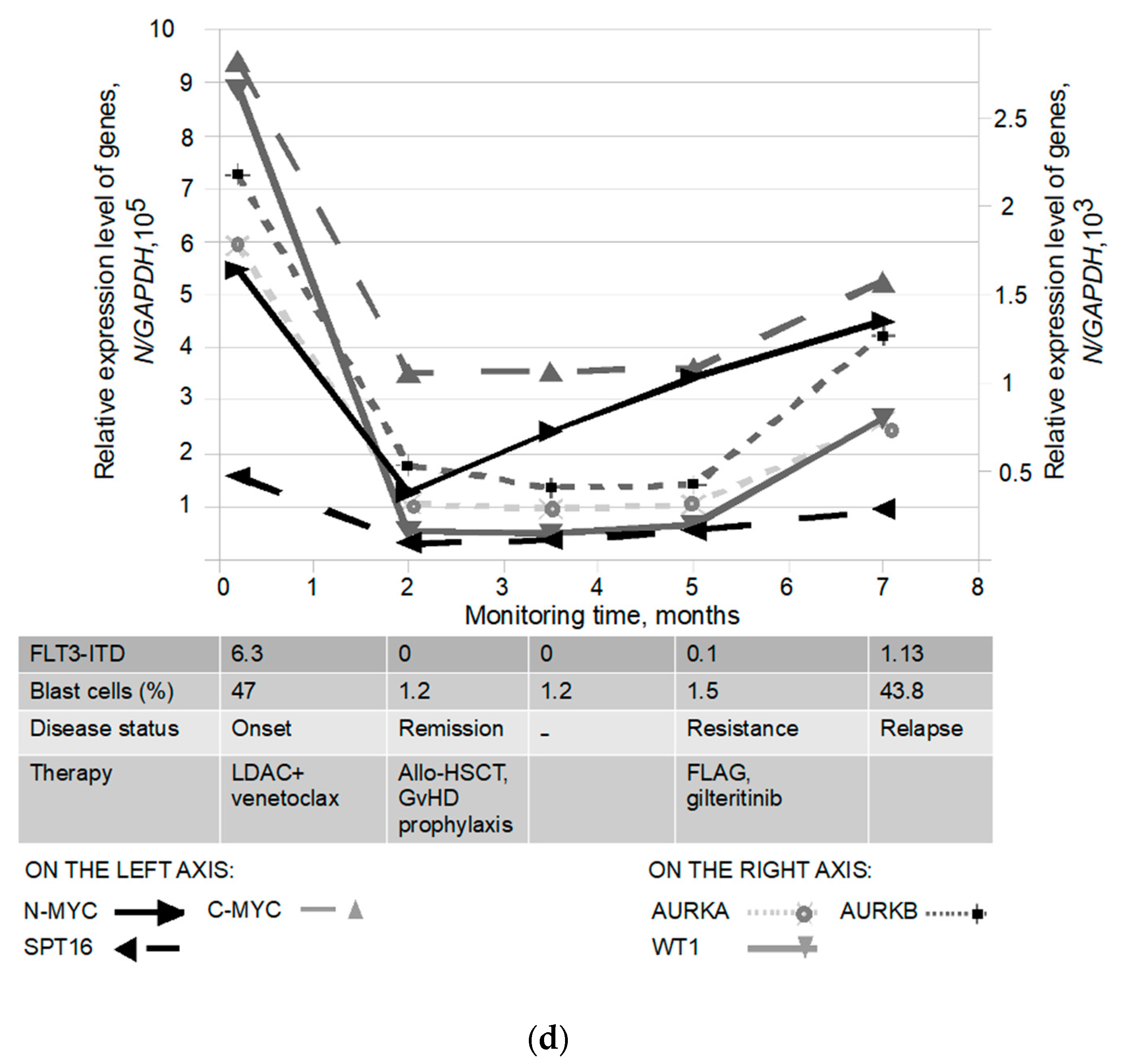
3.5. Biomarker Monitoring in FLT3-ITD Carriers Receiving Therapy
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Look, A.T. Oncogenic transcription factors in the human acute leukemias. Science 1997, 278, 1059–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caligiuri, M.A.; Strout, M.P.; Gilliland, D.G. Molecular biology of acute myeloid leukemia. Semin. Oncol. 1997, 24, 32–44. [Google Scholar] [PubMed]
- Schnittger, S.; Kohl, T.M.; Haferlach, T.; Kern, W.; Hiddemann, W.; Spiekermann, K.; Schoch, C. KIT-D816 mutations in AML1-ETO-positive AML are associated with impaired event-free and overall survival. Blood 2006, 107, 1791–1799. [Google Scholar] [CrossRef] [Green Version]
- Caligiuri, M.A.; Strout, M.P.; Lawrence, D.; Arthur, D.C.; Baer, M.R.; Yu, F.; Knuutila, S.; Mrózek, K.; Oberkircher, A.R.; Marcucci, G.; et al. Rearrangement of the ALL(MLL) in acute myeloid leukemia with normal cytogenetic. Cancer Res. 1998, 58, 55–59. [Google Scholar]
- Kim, H.G.; Kojima, K.; Swindle, C.S.; Cotta, C.V.; Huo, Y.; Reddy, V.; Clug, C.A. FLT3-ITD cooperates with inv(16) to promote progression to acute myeloid leukemia. Blood 2008, 111, 1567–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dovey, O.M.; Cooper, J.L.; Mupo, A.; Grove, C.S.; Lynn, C.; Conte, N.; Andrews, R.M.; Pacharne, S.; Tzelepis, K.; Vijayabaskar, M.S.; et al. Molecular synergy underlies the co-occurrence patterns and phenotype of NPM1-mutant acute myeloid leukemia. Blood 2017, 130, 1911–1922. [Google Scholar] [CrossRef] [Green Version]
- Thiede, C.; Steudel, C.; Mohr, B.; Schaich, M.; Schakel, U.; Platzbecker, U.; Wermke, M.; Bornhäuser, M.; Ritter, M.; Neubauer, A.; et al. Analysis of FLT3 activating mutations in 979 patients with acute myelogenous leukemia: Association with FAB subtypes and identifica-tion of subgroups with poor prognosis. Blood 2002, 99, 4326e35. [Google Scholar] [CrossRef] [Green Version]
- Akiki, S.; Dyer, S.A.; Grimwade, D.; Ivey, A.; Abou-Zeid, N.; Borrow, J.; Jeffries, S.; Caddick, J.; Newell, H.; Begum, S.; et al. NUP98-NSD1 fusion in association with FLT3-ITD mutation identifies a prognostically relevant subgroup of pediatric acute myeloid leukemia patients suitable for monitoring by real time quantitative PCR. Genes Chromosomes Cancer 2013, 52, 1053–1064. [Google Scholar] [CrossRef]
- Oyarzo, M.P.; Lin, P.; Glassman, A.; Bueso-Ramos, C.E.; Luthra, R.; Medeiros, L.J. Acute myeloid leukemia with t(6;9)(p23;q34) is associated with dysplasia and a high frequency of flt3 gene mutations. Am. J. Clin. Pathol. 2004, 122, 348–358. [Google Scholar] [CrossRef]
- Stirewalt, D.L.; Radich, J.P. The role of FLT in haematopoietic malignancies. Nat. Rev. Cancer 2003, 3, 650–665. [Google Scholar] [CrossRef]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Kiyoi, H.; Kawashima, N.; Ishikawa, Y. FLT3 mutations in acute myeloid leukemia: Therapeutic paradigm beyond inhibitor development. Cancer Sci. 2020, 111, 312–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, R.; Small, D. Mutant FLT3 signalling contributes to a block in myeloid differentiation. Leuk. Lymphoma 2005, 46, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- Al-Mawali, A.; Gillis, D.; Lewis, I. Immunoprofiling of leukemic stem cells CD34+/CD38−/CD123+ delineate FLT3/ITD-positive clones. J. Hematol. Oncol. 2016, 9, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiyoi, H.; Towatari, M.; Yokota, S.; Hamaguchi, M.; Ohno, R.; Saito, H.; Naoe, T. Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia 1998, 12, 1333–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Piloto, O.; Kim, K.T.; Ye, Z.; Nguyen, H.B.; Yu, X.; Levis, M.; Cheng, L.; Small, D. FLT3/ITD expression increases expansion, survival and entry into cell cycle of human haematopoietic stem/progenitor cells. Br. J. Haematol. 2007, 137, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Ghosh, J.; Ramdas, B.; Mali, R.S.; Martin, H.; Kobayashi, M.; Vemula, S.; Canela, V.H.; Waskow, E.R.; Visconte, V.; et al. Regulation of Stat5 by FAK and PAK1 in Oncogenic FLT3- and KIT-Driven Leukemogenesis. Cell Rep. 2014, 9, 1333–1348. [Google Scholar] [CrossRef] [Green Version]
- Handschuh, L.; Kaźmierczak, M.; Milewski, M.C.; Góralski, M.; Łuczak, M.; Wojtaszewska, M.; Uszczyńska-Ratajczak, B.; Lewandowski, K.; Komarnicki, M.; Figlerowicz, M. Gene expression profiling of acute myeloid leukemia samples from adult patients with AML-M1 and -M2 through boutique microarrays, real-time PCR and droplet digital PCR. Int. J. Oncol. 2018, 52, 656–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawagoe, H.; Kandilci, A.; Kranenburg, T.A.; Grosveld, G.C. Overexpression of N-Myc rapidly causes acute myeloid leukemia in mice. Cancer Res. 2007, 67, 10677–10685. [Google Scholar] [CrossRef] [Green Version]
- Murakami-Tonami, Y.; Ikeda, H.; Yamagishi, R.; Inayoshi, M.; Inagaki, S.; Kishida, S.; Komata, Y.; Jan Koster Takeuchi, I.; Kondo, Y.; Maeda, T.; et al. SGO1 is involved in the DNA damage response in MYCN-amplified neuroblastoma cells. Sci. Rep. 2016, 6, 31615. [Google Scholar] [CrossRef] [Green Version]
- Mullenders, J.; Aranda-Orgilles, B.; Lhoumaud, P.; Keller, M.; Pae, J.; Wang, K.; Kayembe, C.; Rocha, P.P.; Raviram, R.; Gong, Y.; et al. Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J. Exp. Med. 2015, 212, 1833–1850. [Google Scholar] [CrossRef]
- Yang, M.; Pan, Z.; Huang, K.; Büsche, G.; Liu, H.; Göhring, G.; Rumpel, R.; Dittrich-Breiholz, O.; Talbot, S.; Scherr, M.; et al. A unique role of p53 haploinsufficiency or loss in the development of acute myeloid leukemia with FLT3-ITD mutation. Leukemia 2022, 36, 675–686. [Google Scholar] [CrossRef]
- Bogdanov, K.V.; Nikulina, T.S.; Lomaia, E.G.; Slyadnev, M.N.; Zaritskey, A.Y. Identification of oncogene mutations in leukemia patients using microchip-based PCR analysis. Russ. J. Bioorganic Chem. 2017, 43, 544–551. [Google Scholar] [CrossRef]
- Gabert, J.; Beillard, E.; van der Velden, V.H.; Bi, W.; Grimwade, D.; Pallisgaard, N.; Barbany, G.; Cazzaniga, G.; Cayuela, J.M.; Cavé, H.; et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—A Europe Against Cancer program. Leukemia 2003, 17, 2318–2357. [Google Scholar] [CrossRef] [Green Version]
- Beillard, E.; Pallisgaard, N.; van der Velden, V.H.; Bi, W.; Dee, R.; van der Schoot, E.; Delabesse, E.; Macintyre, E.; Gottardi, E.; Saglio, G.; et al. Evaluation of candidate control genes for diagnosis and residual disease detection in leukemic patients using ‘real-time’ quantitative reverse-transcriptase polymerase chain reaction (RQ-PCR)—A Europe against cancer program. Leukemia 2003, 17, 2474–2486. [Google Scholar] [CrossRef] [Green Version]
- Garçon, L.; Libura, M.; Delabesse, E.; Valensi, F.; Asnafi, V.; Berger, C.; Schmitt, C.; Leblanc, T.; Buzyn, A.; Macintyre, E. DEK-CAN molecular monitoring of myeloid malignancies could aid therapeutic stratification. Leukemia 2005, 19, 1338–1344. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.; Sharma, A.; Katiyar, S.; Gupta, A.; Chaturvedi, C.P. Upfront Screening by Quantitative Real-Time PCR Assay Identifies NUP98::NSD1 Fusion Transcript in Indian AML Patients. Diagnostics 2022, 12, 3001. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.M.; Levis, M.; Hafez, M.J.; Geiger, T.; Cooper, L.C.; Smith, B.D.; Small, D.; Berg, K.D. Detection of FLT3 internal tandem duplication and D835 mutations by a multiplex polymerase chain reaction and capillary electrophoresis assay. J. Mol. Diagn. 2003, 5, 96–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döhner, K.; Schlenk, R.F.; Habdank, M.; Scholl, C.; Rücker, F.G.; Corbacioglu, A.; Bullinger, L.; Fröhling, S.; Döhner, H. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: Interaction with other gene mutations. Blood 2005, 106, 3740–4976. [Google Scholar] [CrossRef] [Green Version]
- Riera, L.; Marmont, F.; Toppino, D.; Frairia, C.; Sismondi, F.; Audisio, E.; Di Bello, C.; D’Ardia, S.; Di Celle, P.F.; Messa, E.; et al. Core binding factor acute myeloid leukaemia and c-KIT mutations. Oncol. Rep. 2013, 29, 1867–1872. [Google Scholar] [CrossRef] [Green Version]
- Østergaard, M.; Olesen, L.H.; Hasle, H.; Kjeldsen, E.; Hokland, P. WT1 gene expression: An excellent tool for monitoring minimal residual disease in 70% of acute myeloid leukaemia patients—Results from a single-centre study. Br. J. Haematol. 2004, 125, 590–600. [Google Scholar] [CrossRef]
- Gorello, P.; Cazzaniga, G.; Alberti, F.; Dell’Oro, M.G.; Gottardi, E.; Specchia, G.; Roti, G.; Rosati, R.; Martelli, M.F.; Diverio, D.; et al. Quantitative assessment of minimal residual disease in acute myeloid leukemia carrying nucleophosmin (NPM1) gene mutations. Leukemia 2006, 20, 1103–1108. [Google Scholar] [CrossRef] [Green Version]
- Verma, R.; Babu, A. Human Chromosomes: Manual of Basic Techniques; Pergamon Press: New York, NY, USA, 1989; pp. 45–67. [Google Scholar]
- Liu, S.B.; Dong, H.J.; Bao, X.B.; Qiu, Q.C.; Li, H.Z.; Shen, H.J.; Ding, Z.X.; Wang, C.; Chu, X.L.; Yu, J.Q.; et al. Impact of FLT3-ITD length on prognosis of acute myeloid leukemia. Haematologica 2019, 104, e9–e12. [Google Scholar] [CrossRef] [Green Version]
- Sallmyr, A.; Fan, J.; Datta, K.; Kim, K.; Grosu, D.; Shapiro, P.; Small, D.; Rassool, F. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: Implications for poor prognosis in AML. Blood 2007, 111, 3173–3182. [Google Scholar] [CrossRef] [Green Version]
- Grimwade, D.; Walker, H.; Oliver, F.; Wheatley, K.; Harrison, C.; Harrison, G.; Rees, J.; Hann, I.; Stevens, R.; Burnett, A.; et al. The importance of diagnostic cytogenetics on outcome in AML: Analysis of 1612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood 1998, 92, 2322–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, H.; Marcucci, G.; Maharry, K.; Margeson, D.; Whitman, S.P.; Wu, Y.Z.; Schwind, S.; Paschka, P.; Powell, B.L.; Carter, T.H.; et al. Favorable prognostic impact of NPM1 mutations in older patients with cytogenetically normal de novo acute myeloid leukemia and associated gene- and microRNA-expression signatures: A Cancer and Leukemia Group B study. J. Clin. Oncol. 2010, 28, 596–604. [Google Scholar] [CrossRef] [Green Version]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaño-Bonilla, T.; Alonso-Dominguez, J.M.; Barragán, E.; Rodríguez-Veiga, R.; Sargas, C.; Gil, C.; Chillón, C.; Vidriales, M.B.; García, R.; Martínez-López, J.; et al. Prognostic significance of FLT3-ITD length in AML patients treated with intensive regimens. Sci Rep. 2021, 11, 20745. [Google Scholar] [CrossRef] [PubMed]
- Giudice, V.; Gorrese, M.; Vitolo, R.; Bertolini, A.; Marcucci, R.; Serio, B.; Guariglia, R.; Ferrara, I.; Pepe, R.; D’Alto, F.; et al. WT1 Expression Levels Combined with Flow Cytometry Blast Counts for Risk Stratification of Acute Myeloid Leukemia and Myelodysplastic Syndromes. Biomedicines 2021, 9, 387. [Google Scholar] [CrossRef] [PubMed]
- Spassov, B.V.; Stoimenov, A.S.; Balatzenko, G.N.; Genova, M.L.; Peichev, D.B.; Konstantinov, S.M. Wilms’ tumor protein and FLT3-internal tandem duplication expression in patients with de novo acute myeloid leukemia. Hematology 2011, 16, 37–42. [Google Scholar] [CrossRef]
- Cilloni, D.; Renneville, A.; Hermitte, F.; Hills, R.K.; Daly, S.; Jovanovic, J.V.; Gottardi, E.; Fava, M.; Schnittger, S.; Weiss, T.; et al. Real-time quantitative polymerase chain reaction detection of minimal residual disease by standardized WT1 assay to enhance risk stratification in acute myeloid leuke 511 mia: A European Leukemia Net study. J. Clin. Oncol. 2009, 27, 5195–5201. [Google Scholar] [CrossRef]
- Hsiao, T.H.; Wang, R.C.; Lu, T.J.; Shih, C.H.; Su, Y.C.; Tsai, J.R.; Jhan, P.P.; Lia, C.S.; Chuang, H.N.; Chang, K.H.; et al. Chemoresponse of de novo Acute Myeloid Leukemia to “7+3” Induction can Be Predicted by c-Myc-facitated Cytogenetics. Front. Pharmacol. 2021, 12, 649267. [Google Scholar] [CrossRef]
- Shi, J.; Whyte, W.A.; Zepeda-Mendoza, C.J.; Milazzo, J.P.; Shen, C.; Roe, J.S.; Minder, J.L.; Mercan, F.; Wang, E.; Eckersley-Maslin, M.A.; et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013, 27, 2648–2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulikkan, J.A.; Hegde, M.; Ahmad, H.M.; Belaghzal, H.; Illendula, A.; Yu, J.; O’Hagan, K.; Ou, J.; Muller-Tidow, C.; Wolfe, S.A.; et al. CBFβ-SMMHC Inhibition Triggers Apoptosis by Disrupting MYC Chromatin Dynamics in Acute Myeloid Leukemia. Cell 2018, 174, 172–186.e21. [Google Scholar] [CrossRef] [Green Version]
- Krygier, A.; Szmajda-Krygier, D.; Sałagacka-Kubiak, A.; Jamroziak, K.; Żebrowska-Nawrocka, M.; Balcerczak, E. Association between the CEBPA and c-MYC genes expression levels and acute myeloid leukemia pathogenesis and development. Med. Oncol. 2020, 37, 109. [Google Scholar] [CrossRef] [PubMed]
- Astolfi, A.; Vendemini, F.; Urbini, M.; Melchionda, F.; Masetti, R.; Franzoni, M.; Libri, V.; Serravalle, S.; Togni, M.; Paone, G.; et al. MYCN is a novel oncogenic target in pediatric T-cell acute lymphoblastic leukemia. Oncotarget 2014, 5, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Basit, F.; Andersson, M.; Hultquist, A. The Myc/Max/Mxd Network Is a Target of Mutated Flt3 Signaling in Hematopoietic Stem Cells in Flt3-ITD-Induced Myeloproliferative Disease. Stem Cells Int. 2018, 2018, 3286949. [Google Scholar] [CrossRef] [PubMed]
- Alaminos, M.; Mora, J.; Cheung, N.K.; Smith, A.; Qin, J.; Chen, L.; Gerald, W.L. Genome-wide analysis of gene expression associated with MYCN in human neuroblastoma. Cancer Res. 2003, 63, 4538–4546. [Google Scholar] [PubMed]
- Carter, D.R.; Murray, J.; Cheung, B.B.; Gamble, L.; Koach, J.; Tsang, J.; Sutton, S.; Kalla, H.; Syed, S.; Gifford, A.J.; et al. Therapeutic targeting of the MYC signal by inhibition of histone chaperone FACT in neuroblastoma. Sci. Transl. Med. 2015, 7, 312ra176. [Google Scholar] [CrossRef] [Green Version]
- Orphanides, G.; Wu, W.H.; Lane, W.S.; Hampsey, M.; Reinberg, D. The chromatin-specifictranscription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 1999, 577, 284–288. [Google Scholar] [CrossRef]
- Belotserkovskaya, R.; Oh, S.; Bondarenko, V.A.; Orphanides, G.; Studitsky, V.M.; Reinberg, D. FACT facilitates transcription-dependent nucleosome alteration. Science 2003, 301, 1090–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otto, T.; Horn, S.; Brockmann, M.; Eilers, U.; Schüttrumpf, L.; Popov, N.; Kenney, A.M.; Schulte, J.H.; Beijersbergen, R.; Christiansen, H.; et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 2009, 15, 67–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, M.; Yamaguchi, H.; Najima, Y.; Usuki, K.; Ueki, T.; Oh, I.; Mori, S.; Kawata, E.; Uoshima, N.; Kobayashi, Y.; et al. Prognostic impact of low allelic ratio FLT3-ITD and NPM1 mutation in acute myeloid leukemia. Blood Adv. 2018, 2, 2744–2754. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Group of Patients and the Frequency of Mutations | |
|---|---|
| FLT3-ITD-Negative | FLT3-ITD-Positive |
| t(8;21)(q22;q22) AML1-ETO †, n ′ = 13, 29.5%; inv16(p13;q21) CBFβ-MYH11 ‡, n = 8, 18.2%; normal karyotype, 46XX, n = 4, 46XY, n = 5, 20.4% | FLT3-ITD *≠, n = 14, 31.8%; |
| Number of Patients in the Group, n | FLT3-ITD Mutation | χ2 Test, Fisher’s Test, p-Value | |
|---|---|---|---|
| Achievement of complete hematological remission | |||
| Yes | No | Insertion length | χ2, F, p |
| 3 | 3 | <39 bp | 0.000, 1.0000, p > 0.05 |
| 4 | 4 | >39bp | |
| Survival | |||
| Alive | Dead | Insertion length | χ2, F, p |
| 2 | 4 | <39 bp | 0.389, 0.67204, p > 0.05 |
| 4 | 4 | >39 bp | |
| Achievement of complete hematological remission | |||
| Yes | No | Allelic load | χ2, F, p |
| 6 | 0 | <0.5 | 10.500, 0.00233, p < 0.05 |
| 1 | 7 | >0.5 | |
| Survival | |||
| Alive | Dead | Allelic load | χ2, F, p |
| 5 | 1 † | <0.5 | 7.024, 0.02564, p < 0.05 |
| 1 | 7 | >0.5 | |
| Group of Patients | Number of Patients, n | Average Level of Gene Expression (N */GAPDH) | ANOVA with Bonferroni’s Post-Test: F-Stat, p-Value |
|---|---|---|---|
| WT1 | |||
| FLT3-ITD-negative | 30 | 1442 | 8.1005, 0.008042 |
| FLT3-ITD-positive | 14 | 1625 | |
| C-MYC | |||
| FLT3-ITD-negative | 30 | 380,538 | 9.2243, 0.005011 |
| FLT3-ITD-positive | 14 | 465,038 | |
| N-MYC | |||
| FLT3-ITD-negative | 30 | 80,509 | 4.651, 0.03946 |
| FLT3-ITD-positive | 14 | 347,762 | |
| SPT16 | |||
| FLT3-ITD-negative | 30 | 77,468 | 4.6288, 0.0399 |
| FLT3-ITD-positive | 14 | 107,399 | |
| AURKA | |||
| FLT3-ITD-negative | 30 | 286 | 1.8963, 0.179 |
| FLT3-ITD-positive | 14 | 425 | |
| AURKB | |||
| FLT3-ITD-negative | 30 | 132 | 0.04112, 0.8407 |
| FLT3-ITD-positive | 14 | 316 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogdanov, K.; Kudryavtseva, E.; Fomicheva, Y.; Churkina, I.; Lomaia, E.; Girshova, L.; Osipov, Y.; Zaritskey, A. Shift of N-MYC Oncogene Expression in AML Patients Carrying the FLT3-ITD Mutation. Pathophysiology 2023, 30, 296-313. https://doi.org/10.3390/pathophysiology30030024
Bogdanov K, Kudryavtseva E, Fomicheva Y, Churkina I, Lomaia E, Girshova L, Osipov Y, Zaritskey A. Shift of N-MYC Oncogene Expression in AML Patients Carrying the FLT3-ITD Mutation. Pathophysiology. 2023; 30(3):296-313. https://doi.org/10.3390/pathophysiology30030024
Chicago/Turabian StyleBogdanov, Konstantin, Ekaterina Kudryavtseva, Yulia Fomicheva, Irina Churkina, Elza Lomaia, Larisa Girshova, Yuri Osipov, and Andrey Zaritskey. 2023. "Shift of N-MYC Oncogene Expression in AML Patients Carrying the FLT3-ITD Mutation" Pathophysiology 30, no. 3: 296-313. https://doi.org/10.3390/pathophysiology30030024
APA StyleBogdanov, K., Kudryavtseva, E., Fomicheva, Y., Churkina, I., Lomaia, E., Girshova, L., Osipov, Y., & Zaritskey, A. (2023). Shift of N-MYC Oncogene Expression in AML Patients Carrying the FLT3-ITD Mutation. Pathophysiology, 30(3), 296-313. https://doi.org/10.3390/pathophysiology30030024